Introduction
Over the last decades, a plethora of experimental
studies and clinical trials investigating cardioprotective
strategies such as ischemic or pharmacological conditioning to
protect against or minimize the extent of ischemia/reperfusion
(I/R) injury have been published (1). The demand for this kind of protection
is high, because I/R injury such as myocardial infarction are among
the leading causes of death worldwide (2). Unfortunately, attempts to transfer
cardioprotective strategies from bench to bedside have not been
successful thus far (1). This
appears to be due to the presence of confounding factors, including
comorbidities such as diabetes mellitus or associated pathological
changes which inhibit cardioprotective mechanisms, for example in
signal transduction (3). Therefore,
cardioprotective strategies have to be investigated not only under
physiological but also under pathological conditions.
Endothelial dysfunction (ED) promotes or is closely
associated with most cardiovascular diseases (CVD) and their
typical comorbidities such as diabetes mellitus (4). In the presence of ED, insufficient
bioavailability of nitric oxide (NO) in the vascular system causes
an imbalance in vascular tone with reduced endothelium-dependent
relaxation in response to vasodilatory stimuli (5,6). In
light of the high number of patients affected by both CVD and ED
and the potential of a loss of efficacy of cardioprotective
strategies under these pathological conditions, cardioprotective
strategies should be examined under ED.
Dexmedetomidine (DEX), a highly selective
α2-adrenergic receptor agonist, has cardioprotective properties
against I/R injury regardless of whether DEX is used in a setting
of pre- or postconditioning (7,8).
Furthermore, this protection appears to be effective even under
pathological conditions, as He et al (9) have shown that cardioprotection against
I/R injury by preconditioning with DEX can be maintained under ED
using the in vitro model of an isolated perfused rat heart.
In line with this, it has been previously demonstrated by the
authors that protection by preconditioning with DEX persists even
under hyperglycaemia, a further pathological condition associated
with CVD (10). However, this was
not the case for protection induced by postconditioning with DEX.
Whether the protective effect of DEX postconditioning is maintained
under ED is, to the best of our knowledge, not known.
In addition to its cardioprotective properties, DEX
also has an influence on the hemodynamic response of the
vasculature. Infusion of DEX induces an initial vasoconstriction by
activation of postsynaptic α2B adrenoreceptors (AR), which in turn
is reversed by activation of α2A AR in the central nervous system
mediated by a reduction in blood pressure and heart rate (11). As isolated-perfused hearts lack a
neural connection, only vasoconstriction is caused by DEX,
potentially resulting in increased vascular tone and higher
perfusion pressure. This is particularly relevant in the context of
ED, where impaired NO bioavailability leads to an inability to
counteract vasoconstriction effectively. Thus, DEX may exacerbate
vascular damage by further increasing resistance and pressure in
the coronary vessels, which can negatively influence its
cardioprotective effects against I/R injury. In our previous
studies investigating conditioning with DEX, a constant pressure
system was used, as has been recommended for the investigation of
cardioprotective strategies against I/R injury (7,8,12). In
constant pressure mode, the vascular system of the heart can react
individually to the perfusion pressure through vasomotion and thus
during phases of high pressure induce dilation to reduce vascular
damage. In this mode, the perfusion flow is also regulated via the
vascular resistance. By contrast, in constant flow mode, which was
used by He et al (9) to
study preconditioning with DEX under ED, a pre-specified pump flow
rate is maintained against any vascular resistance, leading to
higher pressures and therefore potentially greater vascular damage
during vasoconstriction. He et al (9) perfused a buffer containing high
amounts of potassium for ED induction, whereas for successful
damage of endothelial function by such solutions, a constant flow
mode is mandatorily (13). In order
to be able to use this protocol as well, the concept of treatment
duration and concentration for conditioning with DEX from our
former studies with a constant pressure system was transferred to
the constant flow mode (7).
However, due to the vasoconstrictive properties of DEX, the choice
of perfusion mode could influence the cardioprotective effectivity
of DEX. When DEX is used in constant pressure mode, the
vasoconstrictive effect of DEX could increase resistance and
elevate perfusion pressures, resulting in a reduction of perfusion
flow. Otherwise, in a flow-constant system, the combination of a
lack of adaptability to an increased vascular pressure via a
reduction in perfusion flow and DEX-induced vasoconstriction can
lead to increased shear stress, which in turn can be responsible
for severe damage to the vessels and thus also to the tissue.
The aim of the present study was to investigate: i)
if 5 min pre-treatment with 3 nM DEX has cardioprotective effects
in the setting of ED in a constant flow Langendorff system; ii) if
ED influences effects of post-treatment with DEX and iii) if
different experimental modes (constant pressure vs. constant flow)
modulate protective effects.
Materials and methods
Animals
All experiments were performed in accordance with
the Guide for the Care and Use of Laboratory Animals, published by
the U.S. National Institute of Health (NIH publication no. 85-23,
revised 1996), after approval (approval no. O27/12, 10.05.2012) by
the Center for Animal Experiments and Scientific Animal Welfare
(ZETT) Heinrich-Heine University (Düsseldorf, Germany). The animals
were under the care of the ZETT. The rats had ad libitum
access to food and water during a 12/12-h light-dark cycle at a
temperature of 22±3˚C and a relative humidity of 55±10%. The
animals' health and behaviour were monitored daily. Humane
endpoints were not defined, as this in vitro study was based
on organ removal from healthy animals following euthanasia by
decapitation under sedation. In the case of a change of location,
the animals were given sufficient acclimatization time to minimize
stress caused by transport and the new environment. The cages
contained sufficient bedding and enrichment for hiding to meet the
rats' need for safety. Every effort was made to minimize the number
and suffering of the animals in the present study. A total of 42
animals were randomized into six groups. No animal had to be
euthanized or was found dead prior to the start of the experiments.
Two animals were excluded from analysis because their hearts did
not fulfil the minimum criterion for the left ventricular developed
pressure of 80 mmHg at the end of the adaption phase in the
Langendorff system. Therefore, a total of 40 animals were included
in the analysis.
Langendorff system
Male Wistar rats aged 2-3 months were randomized
into the respective groups. Prior to decapitation for euthanasia,
the animals were sedated by intraperitoneal injection of 100 mg/kg
body weight sodium-pentobarbital (Narcoren; Boehringer Ingelheim)
to ensure pain- and stress-free euthanasia. Agglutination was
prevented by injection of 3,330 IU/kg heparin sodium (Braun SE).
Following confirmation of an adequate depth of sedation, indicated
by the absence of the toe pinch reflex (after ~5 min), the animals
were decapitated with a guillotine. After chest opening, hearts
were isolated and mounted on a constant flow Langendorff system (12
ml/min) and perfused with a modified Krebs-Henseleit buffer (KHB;
118 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM
KH2PO4, 24.9 mM NaHCO3, 2.5 mM
CaCl2, 1 mM lactate, 0.5 mM EDTA; and 11 mM glucose at
37˚C). For the induction of global ischemia, perfusion with KHB was
interrupted and the heart was then immersed in KHB aerated with
nitrogen. Reperfusion was induced by restoring the heart perfusion
with KHB while the ischemic buffer bath around the heart was
removed. For treatment with DEX, a solution of 300 nM
dexmedetomidine hydrochloride (cat. no. SML0956; Merck KGaA)
dissolved in KHB was introduced into the perfusate via a syringe
driver with 1% of the flow rate of the Langendorff system to treat
the heart with an effective concentration of 3 nM DEX. For the
induction of ED, hearts were perfused with KHB containing 60 mM KCl
(K+) for 10 min. In this buffer variant, the amount of NaCl was
reduced from 116 to 63 mM to maintain equal osmotic conditions. The
buffer composition and treatment duration was adapted from a
protocol published by He et al (9). In their study, successful ED induction
was demonstrated by the inability of K+-treated hearts
to respond adequately with vasodilation to the application of the
vasodilator histamine. The perfusion with K+ was
implemented via a separate circuit in the Langendorff system to
ensure an exact duration of the perfusion and to avoid buffer
mixing. A water-filled balloon placed in the left ventricle (LV)
was used for continuous LV pressure measurements, which were
digitized using an analogue to digital converter (PowerLab/8SP,
ADInstruments Pty Ltd.) at a sampling rate of 500 Hz and recorded
on a Personal Computer using Labchart 8.0 for Windows
(ADInstruments Pty Ltd.). During the adaptation phase, the left
ventricular diastolic pressure (LVP min) was set at 3 to 5 mmHg.
The following hemodynamic variables were determined: heart rate,
LVP min, left ventricular systolic pressure (LVP max), left
ventricular developed pressure (LVDP=LVP max-LVP min), maximum rate
of pressure change in the LV (dP/dt max), and minimum rate of
pressure change in the LV (dP/dt min). A pressure transducer
connected near the heart measured coronary perfusion pressure
(CPP).
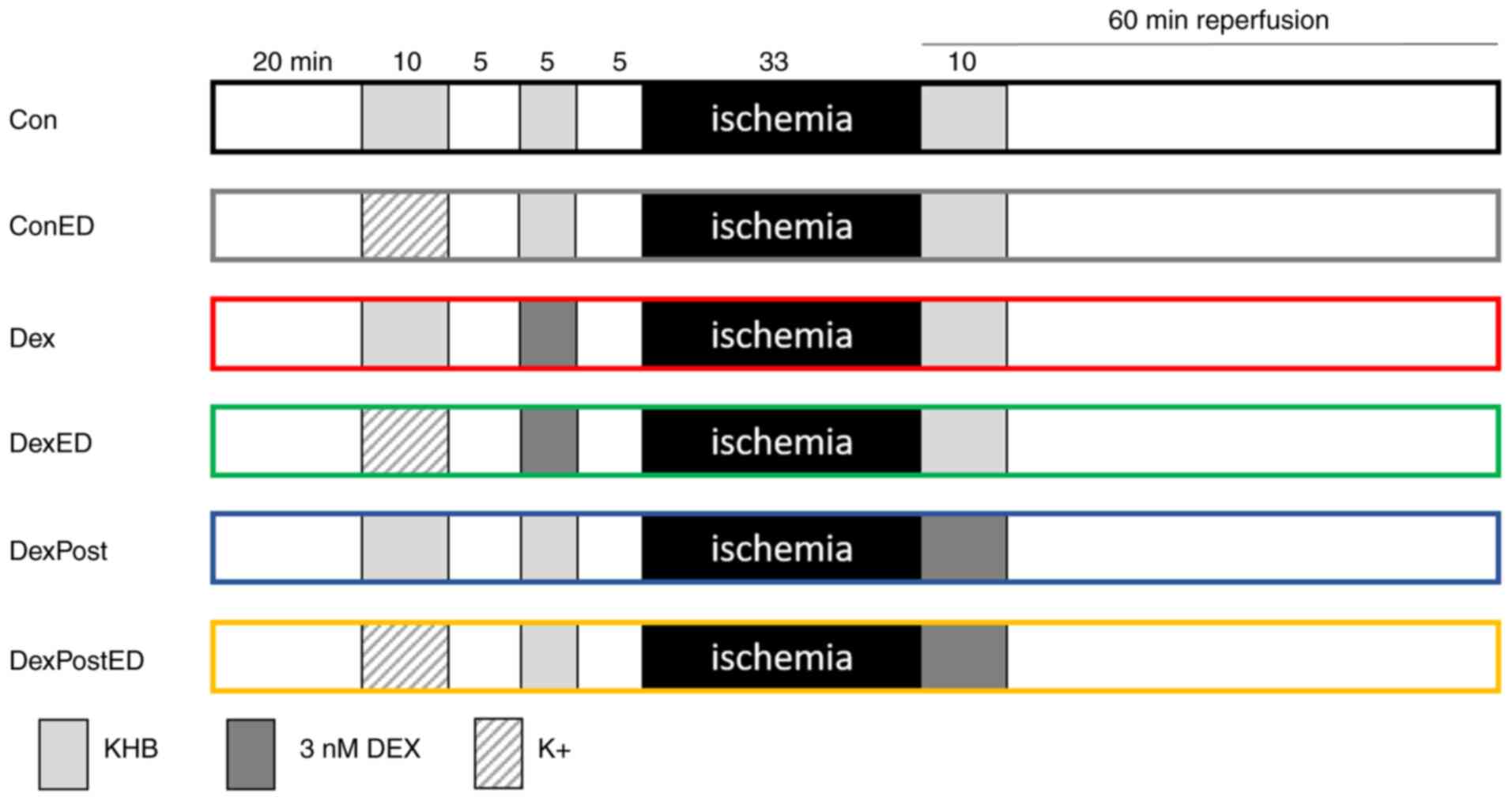
Experimental setup
All hearts underwent the following protocol: 20 min
adaption phase, 10 min induction of ED, 5 min washout, 5 min first
treatment phase (1st TP), 5 min washout, 33 min ischemia, and 60
min reperfusion, including a second treatment phase of 10 min at
its beginning (Fig. 1). Hearts were
randomized into 6 groups. Hearts in control group Con received only
KHB, while those in group ConED were perfused with K+
for 10 min to induce ED. In the DEX pre-treatment groups, hearts
were treated with 3 nM DEX for 5 min during the first treatment
phase after they had been previously perfused with either
K+ for the induction of ED (DexED) or with KHB as a
control (Dex). In a previous study, a pre-treatment duration of 5
min and a concentration of 3 nM DEX had revealed cardioprotective
effects on infarct size (7).
Increasing the concentration to 30 nM DEX or extending the
treatment duration to 10 or 25 min did not improve the extent of
infarct size reduction by pre-treatment with DEX. To investigate a
potential impact of ED on post-treatment with DEX, hearts were
perfused with 3 nM DEX in the second treatment phase for 10 min,
which had cardioprotective effects on infarct size in previous
studies, whereas shortening the treatment phase to 5 min for
post-treatment has not yet been investigated (8,10,14).
After 20 min adaption phase, these two groups were perfused with
K+ for induction of ED (DexPostED) or with KHB for
control conditions (DexPost).
Infarct size determination
To determine infarct size, a staining with 0.75%
triphenyl-tetrazolium chloride (cat. no. 37130.02; SERVA
Electrophoresis GmbH) was performed on trimmed 1-mm thick heart
slices. For quantification, an investigator blinded to group
assignment measured the total area of the LV and the infarcted
areas using planimetry, from which infarct size was calculated as
the infarcted area expressed as a percentage of the LV area
(SigmaScan Pro5; Systat Software, Inc.).
Statistical analysis
A-priori sample size analysis with infarct size
chosen as the primary endpoint was performed with G*Power
3.1.9.7(15); all other statistical
analyses statistical analyses were performed using GraphPad Prism
10 (Dotmatics). The impact of ED on pre- or post-treatment with DEX
was analysed using a one-way analysis of variance (ANOVA) followed
by Tukey's multiple comparisons tests. The calculated total sample
size needed to detect differences on infarct size between 6 groups
was 42 (effect size 0.65, power 80%, α=0.05). For variables
measured longitudinally over time, baseline measurements were
compared with ensure equivalent starting points. In addition, for
comprehensive assessment, measurements received after 60 min of
reperfusion were analysed. To analyse vasoconstriction induced by
DEX before ischemia, two-way ANOVA (conditioning x endothelial
function) at time point 1st TP 4 min on values of LVDP was
performed. The level of significance was defined as P<0.05,
otherwise the effect was declared not significant. Data are shown
as the mean ± standard deviation (SD).
Results
The cardioprotective effect of 3 nM DEX
pre-treatment for 5 min or post-treatment for 10 min under control
conditions or ED using a constant flow Langendorff system was
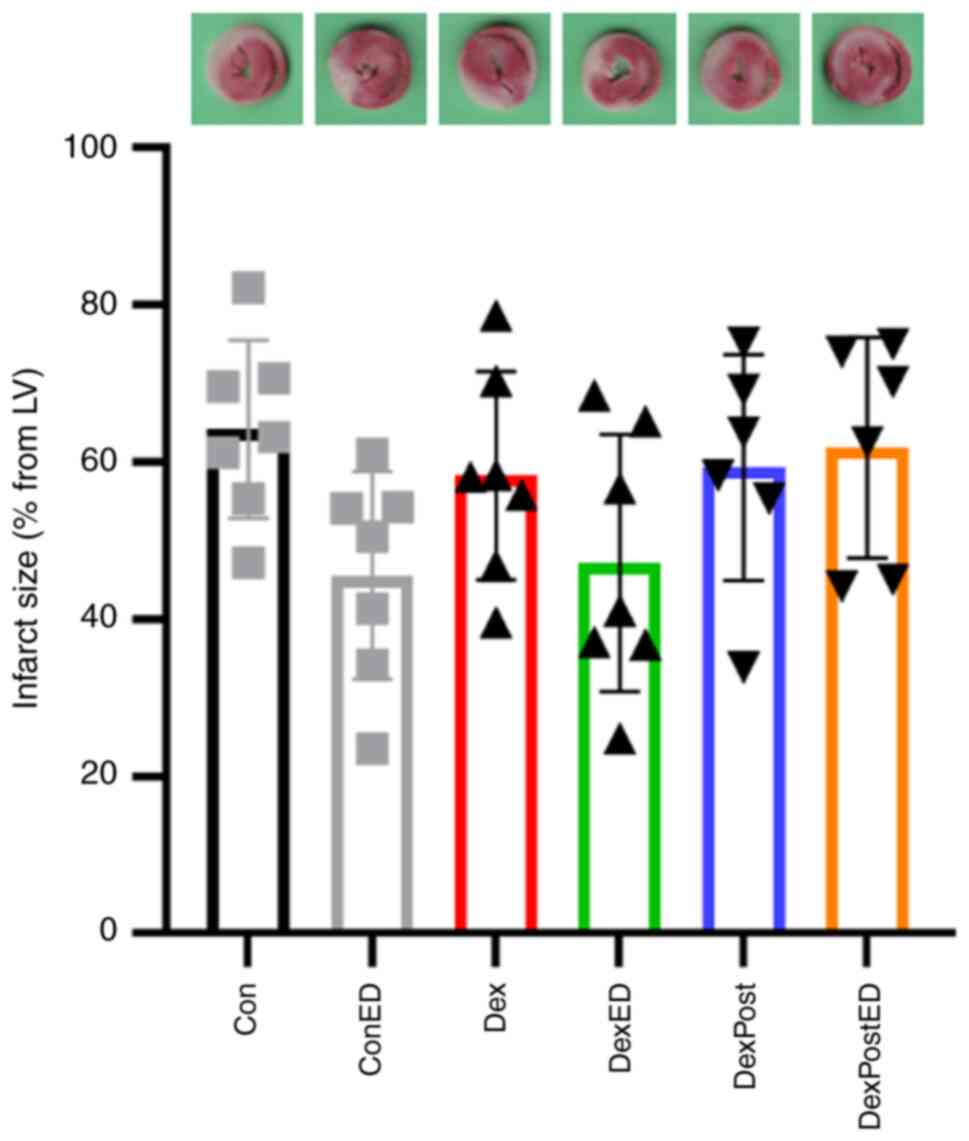
examined. As shown in Fig. 2,
infarct sizes are not different between groups, meaning neither
treatment with DEX nor ED induction has an significant influence on
infarct size (Con: 64±11%, ConED: 46±13%, Dex: 58±13%, DexED:
47±16%, DexPost: 59±14%, DexPostED: 62±14%, P=0.0796, F=2.179).
Body weights, wet and dry heart weights are not
different between groups (Table I).
Peak pressure during ischemia (ischemia peak) is reduced by
K+ perfusion, independent of pre-treatment with DEX or
KHB. Variables of heart function such as heart rate, LVP min, LVP
max, LVDP, dP/dt max, dP/dt min, and CPP are not different at
baseline between the six groups ensuring equal starting conditions
(Fig. S1). After 60 min of
reperfusion, the comparison of groups pre-treated with DEX reveals
that perfusion with K+ increases LVDP. Furthermore,
dP/dt max is elevated and dP/dt min is diminished by perfusion with
K+ in groups pre-treated with DEX.
 | Table IPeak pressure during ischemia
(ischemia peak), body and heart weight. Data of experiments with a
5 min pre-treatment or a 10 min post-treatment (Post) with
dexmedetomidine (DEX) or vehicle as control (Con) under
physiological conditions or ED. Data are presented as the mean ±
SD, n=6-7. |
Table I
Peak pressure during ischemia
(ischemia peak), body and heart weight. Data of experiments with a
5 min pre-treatment or a 10 min post-treatment (Post) with
dexmedetomidine (DEX) or vehicle as control (Con) under
physiological conditions or ED. Data are presented as the mean ±
SD, n=6-7.
| | Con | ConED | Dex | DexED | DexPost | DexPostED |
|---|
| Body weight, g | 299±15 | 299±31 | 303±24 | 300±19 | 284±19 | 288±13 |
| Ischemia peaks,
mmHg | 76±9 | 61±6a | 76±7 | 62±9b | 68±7 | 63±10 |
| Heart weight wet,
g | 1.08±0.12 | 1.07±0.14 | 1.15±0.09 | 1.00±0.09 | 1.05±0.08 | 1.08±0.05 |
| Heart weight dry,
mg | 138±17 | 150±20 | 144±9 | 145±30 | 137±8 | 147±14 |
These data suggest that treatment with 3 nM DEX has
no cardioprotective effect, neither on infarct size nor on heart
function, in the constant flow Langendorff-system, under
physiological conditions or ED, while perfusion with K+
in combination with pre-treatment with DEX improves heart
function.
Discussion
In the present study, it was revealed that by using
a constant flow Langendorff system, neither pre- nor post-treatment
with 3 nM DEX has any cardioprotective effect on infarct size or
heart function; this is independent of induction of ED via
perfusion with K+. In DEX pre-treated hearts, however,
an improvement of in heart function after 60 min of reperfusion was
detectable under ED. Results from constant pressure mode
Langendorff systems are not transferable to constant flow
experiments.
In the present study, treatment with 3 nM DEX for 5
min failed to reduce infarct size after 30 min of ischemia and 60
min of reperfusion in a constant flow Langendorff system. By
contrast, a similar treatment protocol for preconditioning using a
constant pressure system approximately halved the infarct size from
49 to 24% (7). Even lower
concentrations of DEX, namely 0.03 and 1 nM DEX, were still able to
reduce infarct sizes (reduction by ~10 %), but not to the extent of
3 nM DEX. Interestingly, in a study by He et al (9) using a constant flow mode system, a
longer pre-treatment duration of 30 min and a slightly higher
concentration of 10 nM DEX resulted in an infarct size reduction in
hearts of male Sprague-Dawley rats following the same time protocol
of I/R injury. Wang et al (16) used a comparable protocol to He et
al (9) and also showed a
cardioprotective effect with reduced infarct sizes by treatment
with 10 nM DEX for 30 min in a constant flow Langendorff system
after a slightly prolonged ischemia time of 40 and 60 min of
reperfusion. Thus, both studies underline the protective effect of
30 min treatment with 10 nM DEX in constant flow mode, whereby the
small variations in the time protocols for I/R probably have only a
minor influence. Therefore, different perfusion conditions appear
to modulate cardioprotection by DEX. One possible explanation is
that DEX needs a minimal time interval to induce a cardioprotective
effect. In constant flow mode, DEX is transported rapidly through
the vessels independently of the onset of vasoconstriction, while
in constant pressure mode, the beginning of vasoconstriction would
reduce the flow, allowing a longer retention time of DEX in the
vessels. On one hand, this theory is supported by the observation
that in constant pressure mode, a treatment duration of 5 min is
sufficient to induce cardioprotection (7). On the other hand, this theory is
contradicted by the finding that in these experiments coronary
flow, acting as a surrogate for coronary pressure in a constant
pressure system, is only slightly reduced by DEX and thus only
minimally increased coronary vasoconstriction is shown. In the
present study, however, DEX induces vasoconstriction, which is
exemplified by the increased CPP after application of DEX (1st TP 4
min; two-way ANOVA, P=0.0018, F=2.502), which persists until onset
of ischemia. This could also lead to higher shear stress in
constant flow mode, which might in turn lead to more damage to the
heart. This could be another explanation for the lack of protection
by DEX. Thus, during ischemia, the heart is in ischemic
contraction, which means maximal vasoconstriction that slowly
starts to abate with the onset of reperfusion. By contrast, in
constant flow mode a specific volume is transported per unit of
time, regardless of the high vascular pressure at this time point,
thus severely damaging the vessels. DEX-induced vasoconstriction
could slow down the relaxation or inhibit sufficient relaxation and
thus contribute to even greater damage. A further explanation for
the lack of cardioprotection by 3 nM DEX in the constant pressure
mode could also be differences, for example in sensitivity to DEX,
between Wistar (used in our group) and Sprague Dawley rats [used by
He et al (9) and Wang et
al (16)], although only very
small differences would probably be expected here.
Interestingly, pre-treatment with 3 nM DEX does not
lead to cardioprotection in the present study, but increases CPP
and thus has a vasoconstrictive effect suggesting that DEX in
principle is able to induce its signalling receptors/pathways under
the given conditions. However, both functions are mediated via the
α2-AR, as the α2-AR antagonist yohimbine can inhibit DEX-induced
cardioprotection and also vasoconstriction (17-19).
This suggests that successful cardioprotection by DEX requires
either a stronger activation of the α2-AR signalling than
vasoconstriction or the activation of additional signalling
receptors/pathways. One such possible pathway is the binding of DEX
to I1-imidazoline receptors (I1R). Yoshikawa et al (19) reported that efaroxan, an α2-AR and
I1R antagonist, can abolish the reduction in infarct size by
preconditioning with DEX in hypertrophied hearts, whereas yohimbine
has no effect here. This suggests a role of I1R in the
cardioprotective signalling effect of DEX. Whether I1R plays a role
in the signalling under the conditions of the present study must be
analysed in further studies.
An unexpected finding was the protective effect on
heart function in hearts pre-treated with DEX under ED, observed as
an increase in LVDP and improved contractility compared with the
respective control hearts. In addition, a slight reduction of
infarct size under ED is detectable in control and DEX pre-treated
groups, whereas this reduction is not observed in the DexPostED
group. The protocol for the induction of ED with respect to buffer
composition and treatment duration was based on the protocol from
the study of He et al (9),
which has shown no influence on infarct size and heart function by
this protocol. The different setup of the Langendorff systems could
be responsible for these contradictory results. For the present
study, the Langendorff system was modified with a separate circuit
for perfusion of K+ to ensure exact perfusion durations
and to avoid mixing of the buffers, whereas He et al
(9) did not describe details of the
construction of their Langendorff system. Another explanation for
the slight protective effect could be the modulation of perfusion
of hearts by K+. Specifically, compared with hearts
perfused with KHB alone, perfusion with K+ induces a
sustained reduction in LVP max, which, with an unchanged LVP min,
results in a decrease in LVDP until the onset of ischemia (Fig. S1A-C). After 60 min of reperfusion,
the pressure situation is equal for all groups except an increased
LVDP for group DexED, indicating improved cardiac function. A
possible explanation for the protection induced by perfusion with
K+ could be that hearts with lower LVDP before ischemia
could reduce their energy demand, which might result in an improved
performance in the reperfusion phase. Furthermore, independently of
the modulation of hemodynamic variables by K+,
cardioprotective signalling might be activated. The
hypercontraction due to perfusion with K+, as evidenced
by a CPP of ~150 mmHg and a decrease in heart rate, could mimic
ischemic preconditioning (Fig. S1D
and G). However, short, repetitive
I/R cycles are normally required for the induction of a
cardioprotective effect. Interestingly, Okada et al
(17) also discussed that DEX
induced vasoconstriction may trigger ischemic preconditioning
signalling as a possible mechanism of its cardioprotective effect.
In summary, a protocol for ED induction without slight additional
cardioprotective effects would be more meaningful to study effects
on cardioprotection and an optimization of the protocol would be
helpful.
Like pre-treatment, post-treatment with DEX also has
no influence on IS or heart function regardless of presence of ED
or physiological conditions. Furthermore, the DEX post-treatment
attenuates improvement of LVDP by K+ after 60 min of
reperfusion (Fig. S1C), which was
observed in the group DexED. Negative effects induced by
post-treatment with DEX on isolated perfused rat hearts using a
constant flow Langendorff system were also reported by Mimuro et
al (20). The authors measured
an increase of infarct size compared with control hearts after I/R
by applying 1 or 10 nM DEX immediately after ischemia with the
onset of reperfusion. This increase was inhibited by yohimbine,
indicating an α2-AR dependent mechanism.
In future studies, a possible inhibition of
DEX-mediated cardioprotection by ED should be further investigated,
as the existence of endothelial cells appears to be essential for
successful cardioprotection by DEX. Riquelme et al (21) showed that IR-induced cardiomyocyte
cell death could only be reduced by DEX when they were co-cultured
with endothelial cells. Furthermore, these authors showed in
isolated rat hearts that DEX activates endothelial nitric oxide
(NO) synthase (eNOS) to produce NO using NOS inhibitor L-NAME
(N-Nitroarginine methyl ester) or the NO scavenger PTIO
(2-Phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl 3-oxide). It should
be taken into account that eNOS is not only present in endothelial
cells, but also in cardiomyocytes. Therefore, cardiomyocytes could
serve as additional source for NO production required for
cardioprotection (22). A role for
eNOS in cardioprotection against I/R injury by DEX preconditioning
was also shown by Ibacache et al (23). In rat hearts pre-treated with 10 nM
DEX for 25 min in vivo or ex vivo, phosphorylation of
eNOS, extracellular signal-regulated kinase (ERK) 1/2 and Akt was
increased. Furthermore, inhibition of the phosphatidylinositol
3-kinase (PI3K)/Akt/ERK/eNOS signalling cascade by the PI3K
inhibitor LY-294002 abolished the infarct size-reducing effect of
DEX preconditioning. To further unravel the signalling transduction
of DEX preconditioning, this group analysed protein expression of
the α2-AR isoforms A, B, C in extracts of whole adult rat hearts.
All isoforms were detected, although the isotypes are
differentially expressed in cardiomyocytes, endothelial cells and
other cell types. Previously, Takahashi et al (18) analysed single cell RNA sequence data
and showed that in human hearts all three isotypes are present, but
none of them is predominantly expressed in cardiomyocytes. Whereas
in endothelial cells, primarily α2-AR isoforms A and B are
expressed (18). Therefore, the
detailed interplay between cardiomyocytes and endothelial cells in
the context of DEX preconditioning should be investigated in future
studies to maintain the protective effect in the heart of patients
with an ED. Not only PI3K/Akt/ERK/eNOS signalling but also other
known mechanisms involved in the cardioprotective signalling of the
cardioprotective effect of DEX preconditioning, such as the
activation of large-conductance Ca2+-activated
K+ channel or the involvement of miRNAs, could play a
role here (7,18). Before further investigating the
cardioprotective properties of DEX under ED, it is essential to
establish a standardized cardioprotective protocol, defining the
optimal concentration and treatment duration under constant flow
conditions. Furthermore, translation to an in vivo model for
I/R injury, for example left anterior descending occlusion in a
mouse model, is imperative, as ex vivo models such as the
Langendorff system do not fully reflect physiological conditions.
This limitation is particularly relevant given that DEX also exerts
effects via the central nervous system, which is not accounted for
in the Langendorff system. Lastly, there are protocols for ED
induction in mice, for example by chronic intraperitoneal
administration of angiotensin II, allowing to study DEX
preconditioning under ED in vivo (24).
A few limitations of the present study should be
addressed. Additional modifications of the protocol for a
successful cardioprotection by DEX were not implemented. For
example, using higher concentrations of DEX or extending the
perfusion time to 30 min could have led to a protective effect of
DEX on IS and heart function. In addition, the slight protective
effect by ED induction was not further analysed. For example, lower
buffer temperatures, that is. hypothermia, could have contributed
to this effect. Khaliulin et al (25) demonstrated that temperature
conditioning is cardioprotective in isolated-perfused rat hearts by
inhibition of opening probability of mitochondrial
permeability-transition pore (mPTP) and improvement of heart
function during reperfusion. A further limitation is the moderate
standard deviation (SD) observed in infarct sizes. However, all
groups exhibit a similar range of SDs regardless of treatment,
suggesting that the variability reflects biological variation
rather than treatment-specific effects. Another limitation of the
present study is the exclusive use of male rats, even though this
is the usual model in cardiovascular research. In the field of
myocardial infarction and cardioprotection, numerous studies have
already shown sex-specific differences (26). Sex-specific differences have also
been identified for the effect of DEX. For example, Vincent et
al (27) were able to show in
female rats that the time to awaken from DEX anaesthesia varied
depending on the female cycle, whereas awakening from anaesthesia
with isoflurane, sevoflurane or propofol was independent of the
female cycle. In humans, the sexes also appear to be differently
susceptible to DEX, as a clinical study showed a significant
dependence between sex and DEX dosage for sedation (28). Although these studies investigated
the sedative properties of DEX with regard to sex-specific
differences, it can be assumed that the cardioprotective or
vasoconstrictive properties are also subject to such sex-specific
influences. Thus, cardioprotective doses of DEX studied in male
individuals should not be directly transferred to female
individuals but should be explicitly analysed.
In conclusion, studies investigating
cardioprotective effects of DEX should take into account its
vasoconstrictive effect. Different hemodynamic conditions in in
vitro models can modulate the cardioprotective properties of
DEX.
Supplementary Material
Hemodynamic variables. Quantification
of hemodynamic variables for pre-treatment (yellow box) or
post-treatment (blue box) with 3 nM Dex or vehicle (Con) under
physiological conditions or under ED induced by perfusion with
Krebs-Henseleit buffer containing 60 mM KCl (K+). (A) LVP max; (B)
LVP min; (C) LVDP; (D) heart rate; (E) dP/dt max; (F) dP/dt min;
(G) CPP. 1st TP: first treatment phase. Data are presented as the
mean +/- SD. Statistical tests were only performed for baseline and
at 60 min of reperfusion (Rep 60 min). One-way ANOVA followed by
Tukey’s multiple comparisons tests for all comparisons at the
respective time point. *P<0.05 Dex vs. DexED (n=6-7).
Dex, dexmedetomidine; ED, endothelial dysfunction; LVP max, maximal
left ventricular pressure; LVP min, minimal LVP; LVDP, developed
LVP; dP/dt max, maximal rate of rise of LVP; dP/dt min, minimal
rate of rise of LVP; CPP, coronary perfusion pressure; ns, not
significant (P>0.05).
Acknowledgements
The authors thank Mr Jan Olligs, (Städtische
Kliniken Mönchengladbach, Germany) for supporting statistical
analysis and proofreading. This article partially fulfills the
requirements of an MD thesis by Miss Sophia De Luca-Rhoner.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
SDLR performed all Langendorff experiments. MS
performed the planimetry to determine infarct size. AR designed the
study, performed statistical analysis, and wrote the manuscript. AR
and AH interpreted the data. AH revised critically the manuscript.
SDLR and AR confirm the authenticity of all the raw data. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
All animal experiments were performed in accordance
with the Guide for the Care and Use of Laboratory Animals,
published by the U.S. National Institute of Health (NIH publication
No. 85-23, revised 1996), after approval by the local Animal Care
and Use Committee of the Heinrich-Heine University, Düsseldorf,
Germany (approval no. O27/12).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Use of artificial intelligence tools
During the preparation of this work, an artificial
intelligence tool (www.deepl.com)
was used to improve the readability and language of the manuscript,
and subsequently, the authors revised and edited the content
produced by the artificial intelligence tool as necessary, taking
full responsibility for the ultimate content of the present
manuscript.
References
|
1
|
Heusch G: Cardioprotection and its
translation: A need for new paradigms? Or for new pragmatism? An
opinionated retro- and perspective. J Cardiovasc Pharmacol Ther.
28(10742484231179613)2023.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Virani SS, Alonso A, Benjamin EJ,
Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR,
Cheng S, Delling FN, et al: Heart disease and stroke
statistics-2020 update: A report from the American heart
association. Circulation. 141:e139–e596. 2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Kleinbongard P, Botker HE, Ovize M,
Hausenloy DJ and Heusch G: Co-morbidities and co-medications as
confounders of cardioprotection-Does it matter in the clinical
setting? Br J Pharmacol. 177:5252–5269. 2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Segers VFM, Bringmans T and De Keulenaer
GW: Endothelial dysfunction at the cellular level in three
dimensions: Severity, acuteness, and distribution. Am J Physiol
Heart Circ Physiol. 325:H398–H413. 2023.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Roy R, Wilcox J, Webb AJ and O'Gallagher
K: Dysfunctional and dysregulated nitric oxide synthases in
cardiovascular disease: Mechanisms and therapeutic potential. Int J
Mol Sci. 24(15200)2023.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Cyr AR, Huckaby LV, Shiva SS and
Zuckerbraun BS: Nitric oxide and endothelial dysfunction. Crit Care
Clin. 36:307–321. 2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Behmenburg F, Pickert E, Mathes A, Heinen
A, Hollmann MW, Huhn R and Berger MM: The cardioprotective effect
of dexmedetomidine in rats is dose-dependent and mediated by BKCa
channels. J Cardiovasc Pharmacol. 69:228–235. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bunte S, Behmenburg F, Majewski N,
Stroethoff M, Raupach A, Mathes A, Heinen A, Hollmann MW and Huhn
R: Characteristics of dexmedetomidine postconditioning in the field
of myocardial ischemia-reperfusion injury. Anesth Analg. 130:90–98.
2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
He L, Hao S, Wang Y, Yang W, Liu L, Chen H
and Qian J: Dexmedetomidine preconditioning attenuates
ischemia/reperfusion injury in isolated rat hearts with endothelial
dysfunction. Biomed Pharmacother. 114(108837)2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Torregroza C, Feige K, Schneider L, Bunte
S, Stroethoff M, Heinen A, Hollmann MW, Huhn R and Raupach A:
Influence of hyperglycemia on dexmedetomidine-induced
cardioprotection in the isolated perfused rat heart. J Clin Med.
9(1445)2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Castillo RL, Ibacache M, Cortinez I,
Carrasco-Pozo C, Farías JG, Carrasco RA, Vargas-Errázuriz P, Ramos
D, Benavente R, Torres DH and Méndez A: Dexmedetomidine Improves
Cardiovascular and ventilatory outcomes in critically Ill patients:
Basic and clinical approaches. Front Pharmacol.
10(1641)2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Skrzypiec-Spring M, Grotthus B, Szelag A
and Schulz R: Isolated heart perfusion according to
Langendorff-still viable in the new millennium. J Pharmacol Toxicol
Methods. 55:113–126. 2007.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Cartier R, Pellerin M, Hollmann C and
Pelletier LC: Effects of pressure and duration of hyperkalemic
infusions on endothelial function. Ann Thorac Surg. 55:700–705.
1993.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Raupach A, Karakurt E, Torregroza C, Bunte
S, Feige K, Stroethoff M, Brandenburger T, Heinen A, Hollmann MW
and Huhn R: Dexmedetomidine provides cardioprotection during early
or late reperfusion mediated by different mitochondrial
K+-channels. Anesth Analg. 132:253–260. 2021.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Faul F, Erdfelder E, Lang AG and Buchner
A: G*Power 3: A flexible statistical power analysis program for the
social, behavioral, and biomedical sciences. Behav Res Methods.
39:175–191. 2007.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wang L, Liu J, Wang Z, Qian X, Zhao Y,
Wang Q, Dai N, Xie Y, Zeng W, Yang W, et al: Dexmedetomidine abates
myocardial ischemia reperfusion injury through inhibition of
pyroptosis via regulation of miR-665/MEF2D/Nrf2 axis. Biomed
Pharmacother. 165(115255)2023.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Okada H, Kurita T, Mochizuki T, Morita K
and Sato S: The cardioprotective effect of dexmedetomidine on
global ischaemia in isolated rat hearts. Resuscitation. 74:538–545.
2007.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Takahashi K, Yoshikawa Y, Kanda M, Hirata
N and Yamakage M: Dexmedetomidine as a cardioprotective drug: A
narrative review. J Anesth. 37:961–970. 2023.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Yoshikawa Y, Hirata N, Kawaguchi R,
Tokinaga Y and Yamakage M: Dexmedetomidine maintains its direct
cardioprotective effect against ischemia/reperfusion injury in
hypertensive hypertrophied myocardium. Anesth Analg. 126:443–452.
2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Mimuro S, Katoh T, Suzuki A, Yu S, Adachi
YU, Uraoka M, Sano H and Sato S: Deterioration of myocardial injury
due to dexmedetomidine administration after myocardial ischaemia.
Resuscitation. 81:1714–1717. 2010.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Riquelme JA, Westermeier F, Hall AR,
Vicencio JM, Pedrozo Z, Ibacache M, Fuenzalida B, Sobrevia L,
Davidson SM, Yellon DM, et al: Dexmedetomidine protects the heart
against ischemia-reperfusion injury by an endothelial eNOS/NO
dependent mechanism. Pharmacol Res. 103:318–327. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Ghasemi A and Jeddi S: Quantitative
aspects of nitric oxide production in the heart. Mol Biol Rep.
49:11113–11122. 2022.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ibacache M, Sanchez G, Pedrozo Z, Galvez
F, Humeres C, Echevarria G, Duaso J, Hassi M, Garcia L, Díaz-Araya
G and Lavandero S: Dexmedetomidine preconditioning activates
pro-survival kinases and attenuates regional ischemia/reperfusion
injury in rat heart. Biochim Biophys Acta. 1822:537–545.
2012.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Trejo-Moreno C, Jimenez-Ferrer E,
Castro-Martinez G, Méndez-Martínez M, Santana MA, Arrellín-Rosas G,
Pedraza-Chaverri J, Medina-Campos ON, Hernández-Téllez B,
Ramírez-Pliego O, et al: Characterization of a murine model of
endothelial dysfunction induced by chronic intraperitoneal
administration of angiotensin II. Sci Rep. 11(21193)2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Khaliulin I, Clarke SJ, Lin H, Parker J,
Suleiman MS and Halestrap AP: Temperature preconditioning of
isolated rat hearts-a potent cardioprotective mechanism involving a
reduction in oxidative stress and inhibition of the mitochondrial
permeability transition pore. J Physiol. 581:1147–1161.
2007.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Querio G, Geddo F, Antoniotti S, Gallo MP
and Penna C: Sex and response to cardioprotective conditioning
maneuvers. Front Physiol. 12(667961)2021.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Vincent KF, Mallari OG, Dillon EJ, Stewart
VG, Cho AJ, Dong Y, Edlow AG, Ichinose F, Xie Z and Solt K:
Oestrous cycle affects emergence from anaesthesia with
dexmedetomidine, but not propofol, isoflurane, or sevoflurane, in
female rats. Br J Anaesth. 131:67–78. 2023.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Ding Y, Liu A, Wang Y, Zhao S, Huang S,
Zhu H, Ma L, Han L, Shu S, Zheng L and Chen X: Genetic
polymorphisms are associated with individual susceptibility to
dexmedetomidine. Front Genet. 14(1187415)2023.PubMed/NCBI View Article : Google Scholar
|