Introduction
Hyperpigmentation disorders are conditions
characterized by the darkening of the skin due to excessive melanin
production or deposition. Such disorders are categorized into three
types: Diffuse, linear and reticulated (1). Diffuse hyperpigmentation refers to a
condition characterized by the widespread darkening of the skin,
often affecting multiple areas of the body, including acquired
universal melanosis (AUM), erythema dyschromicum perstans (EDP),
lichen planus pigmentosus (LPP), familial progressive
hyperpigmentation (FPH), post-inflammatory hyperpigmentation and
drug-induced hyperpigmentation (2-4).
Linear hyperpigmentation presents darkened streaks or lines on the
skin that follow a linear pattern. This includes flagellate
pigmentation, pigmentary demarcation lines, linear and whorled
nevoid hypermelanosis and incontinentia pigmenti (5). Reticulated hyperpigmentation refers to
a pattern of darkened patches on the skin that form a net- or
lace-like appearance and includes dyskeratosis congenita, prurigo
pigmentosa, dermatopathia pigmentosa reticularis, Dowling-Degos
disease and reticulate acropigmentation of Kitamura (6). The causes of hyperpigmentation are
diverse, encompassing genetic factors, hormonal changes, sun
exposure, inflammation, certain medications, and underlying medical
conditions (7,8).
Although many hyperpigmentation disorders are rare,
available epidemiological data provide varying estimates of
prevalence across the three clinical types: diffuse, linear, and
reticulated. Among diffuse hyperpigmentation disorders, Addison's
disease, an acquired condition caused by adrenal insufficiency, has
a reported prevalence of 100-140 cases per million in Europe and
North America (9,10). In contrast, genetic causes of
diffuse hyperpigmentation are much rarer. For example,
dyschromatosis universalis hereditaria, which may be inherited in
an autosomal dominant or recessive manner and is associated with
mutations in ATP binding cassette subfamily B member 6
(ABCB6), SAM and SH3 domain containing 1 (SASH1), or
KIT ligand (KITLG), has an estimated prevalence of
approximately 0.3 per 100,000 individuals (11,12).
Another rare diffuse condition, FPH, has been reported in only 9
cases worldwide (13). Reticulated
hyperpigmentation is seen in disorders such as DC, which is
associated with mutations in genes involved in telomere
maintenance, including dyskerin pseudouridine synthase 1,
telomerase RNA component (TERC), and regulator of telomere
elongation helicase 1 and is estimated to occur in fewer than 1 per
1,000,000 individuals (14). DPR
has only 20 cases reported globally, while reticulate
acropigmentation of Kitamura has 130 documented cases worldwide
(13). Linear hyperpigmentation
disorders are also rare. IP, a well-characterized example, is an
X-linked dominant disorder caused by pathogenic variants in the
inhibitor of nuclear factor kappa B kinase regulatory subunit gamma
(IKBKG) gene. It primarily affects females and is typically
lethal in males. In European populations, the estimated birth
prevalence of IP is ~1.2 per 100,000 live births (13). Given the rarity and genetic
complexity of these conditions, especially those with suspected
Mendelian inheritance, advanced genomic approaches are essential
for elucidating their molecular basis.
Next-generation sequencing technologies have
revolutionized genomic research by enabling high-throughput
sequencing and comprehensive DNA sequence analysis. Among these,
long-read whole-genome sequencing is advantageous owing to its
ability to generate long contiguous sequences of DNA (15). This technology excels in resolving
complex genomic regions, including structural variants (SVs),
repetitive sequences and intricate genomic rearrangements that
short-read sequencing methods often miss or inaccurately
characterize (16,17). The present study used PacBio
long-read whole-genome sequencing to ensure a complete and accurate
genome representation.
The aim of the present study was to investigate the
genetic basis of a rare congenital hyperpigmentation disorder in
monozygotic twins.
Materials and methods
Skin biopsy and histopathological
study
The present study was approved by the Institutional
Review Board of the Faculty of Medicine, Chulalongkorn University
(Bangkok, Thailand (approval no. 264/62) and conducted in
accordance with the 1964 Helsinki Declaration and its later
amendments, as well as all relevant ethical guidelines and
regulations. The rights and interests of all participants were
fully protected. Fifteen-year-old monozygotic twin girls with
generalized hyperpigmentation presented in April 2019 at Khanu
Woralaksaburi Hospital, Kamphaeng Phet, Thailand were referred to
Naresuan University Hospital, Phitsanulok, Thailand. Both parents
of the patients were aged 56 years. Skin biopsy was performed on
the abdomen of both twins for histopathological study, diagnosis
and fibroblast culture. Written informed consent was obtained from
the parents of the patients. Tissue was fixed in 10% formaldehyde
at room temperature for 24 h, followed by tissue processing which
included dehydration through a graded ascending ethanol series,
clearing in xylene, and embedding in paraffin. Paraffin-embedded
tissues were then sectioned at 4-5 µm thickness for histological
analysis. Hematoxylin and eosin staining was performed at room
temperature following deparaffinization in xylene, rehydration
through graded descending ethanol, staining with Harris's
hematoxylin for 7.5 min, bluing with lithium carbonate,
counterstaining with eosin for 1 min, followed by dehydration and
clearing. Sections were imaged using an Olympus BX50 light
microscope (Olympus Corporation), with image capture via Olympus
cellSens Standard software v1.3 (evidentscientific.com/en/products/software/cellsens).
Clinical laboratory tests
As part of the clinical work-up, both twins
underwent a physical examination, electrocardiogram (ECG), and
routine laboratory testing, including a complete blood count (CBC)
and morning serum cortisol assessment, performed at Naresuan
University Hospital, Phitsanulok, Thailand. A standard-dose (250
µg) adrenocorticotropic hormone (ACTH) stimulation test was
performed in the morning between 08:00 and 09:00 to account for
diurnal variation in cortisol secretion. A baseline blood sample
was collected for measurement of serum cortisol and
17-hydroxyprogesterone prior to ACTH administration. Subsequently,
250 µg of cosyntropin was administered intravenously, followed by
blood sampling at 30 and 60 min post-injection to assess stimulated
cortisol and 17-OHP levels. Serum cortisol levels were analyzed
using the electrochemiluminescence immunoassay method. The
reference range was 6.2-19.4 µg/dl in the morning and 2.3-11.9
µg/dl in the afternoon. Serum 17-hydroxyprogesterone levels were
analyzed using the radioimmunoassay (RIA) method, with a reference
range of 0.10-1.50 ng/ml.
Whole genome long-read sequencing and
data processing
Genomic DNA was obtained from both twins and their
parents, extracted from leucocytes using a Gentra Puregene Blood
kit (Qiagen GmbH) and sheared using a Megaruptor 2 long hydropore
to obtain DNA fragments of 15 kb. DNA fragments were analyzed using
a Qubit fluorometer and pulsed-field gel electrophoresis. DNA
fragments (2-5 µg) were prepared using the SMRTbell Express
Template Prep kit 2.0 (cat. no. P/N 100-938-900) and SMRTbell
Enzyme Clean-up kit 2.0 (cat. no. P/N 101-932-600; both Pacific
Biosciences). Fragments <10 kb were eliminated using BluePippin
(Sage Science). SMRTbell libraries were sequenced on the Sequel II
system using Sequel II sequencing kit 2.0 (cat. no. P/N
101-820-200; Pacific Biosciences) and raw subreads were processed
through the circular consensus sequencing (CCS) workflow using
PacBio SMRTLink version 10.0 (pacb.com/products-and-services/analytical-software/smrt-analysis)
to generate high fidelity (HiFi) long reads, in which both DNA
strands were sequenced multiple times due to the circular nature of
the template, achieving a minimum estimated quality value of 20
(phred-scaled, corresponding to an accuracy of 99%). All samples
were aligned to the GRCh38 human reference genome using pbmm2
v1.9.0 (github.com/PacificBiosciences/pbmm2). The aligned CCS
binary alignment map data were used for small variant, SV and copy
number variant (CNV) detection.
Short tandem repeat (STR)
analysis
FASTQ files, generated from long-read whole-genome
sequencing, were aligned to GRCh38 using LAST v1418 tool (18), yielding a MAF file. The MAF file was
then used by tandem-genotypes v1.1.0(19) to predict STR unit change of each
read in relative to repeat unit in the reference genome. The repeat
unit change was estimated in the twins, parents and 236 unaffected
controls from the in-house database at Excellence Center for
Genomics and Precision Medicine, King Chulalongkorn Memorial
Hospital, Bangkok, Thailand. This database contains long-read
sequencing data from patients with various genetic conditions, as
well as from parents or siblings of certain probands. Importantly,
the 236 samples designated as controls did not include individuals
with pigmentation disorder. Next, STRs were identified using an
in-house algorithm. Repeat patterns with 2-6 bases were selected to
calculate distributions of repeat unit using kernel density
estimation. Gaussian kernel with a bandwidth of five was selected
to smooth the estimated density curve. Minimum points were used at
the curve to cluster the data. For autosomal dominance mode, STRs
exhibiting expansion only in the twins but not in their parents and
controls, were searched. For autosomal recessive mode, expansion in
both twins and parents but not in controls were searched. STRs were
further reviewed for their relevance to the disease by examining
genomic context, literature on disease association and predicted
effects on gene function or expression. Analysis of single
nucleotide variants (SNVs), insertions and deletions (Indels), SV,
CNV and methylation profile is described in the Data S1.
Kinship analysis
Kinship coefficient was estimated using
kinship-based inference from the KING package v2.3.2, based on
genotype data derived from whole-genome sequencing (20). Relatedness was inferred based on
estimated kinship coefficients using the following classification:
Kinship coefficient >0.354 indicates duplicates or monozygotic
twins, where (0.177, 0.354), (0.0884, 0177) and (0.0442, 0.0884)
indicate first-second- and third-degree relatives,
respectively.
Cell culture
Fibroblast cells were acquired from skin biopsies of
two affected twins carrying the TYMS variant and four
unaffected individuals who were not in the in-house database. The
controls included three males and one female, all pediatric donors
aged 1-5 years, with no history of pigmentation disorder or related
skin conditions. The cells were cultured in DMEM supplemented with
10% fetal bovine serum (both Cytiva; HyClone) and 1X
antibiotic-antimycotic (Gibco; Thermo Fisher Scientific, Inc.) and
then incubated at 37˚C with 5% CO2 (21). The use of primary human cells in
cell culture experiments was approved by the Institutional Review
Board of the Faculty of Medicine, Chulalongkorn University
(approval no. 813/63).
Reverse transcription-quantitative
(RT-q)PCR-based mRNA quantification
RNA was extracted from fibroblasts and peripheral
blood mononuclear cells (PBMCs) using the QIAamp RNA Blood Mini kit
(Qiagen GmbH). PBMC controls included both parents of the twins and
one unrelated 45-year-old adult female, all without any history of
pigmentation disorders or related skin conditions. These PBMC
controls were independent from the aforementioned fibroblast
controls. cDNA was prepared from 1 µg RNA using the ImProm-II™ RT
System (Promega Corporation) according to the manufacturer's
protocol. The input RNA from fibroblasts was primed with an equal
mix of anchored dT oligonucleotides for reverse-transcribing RNA to
cDNA. TaqMan probes specific for TYMS (cat. no.
Hs00426586_m1) and β-actin (Assay ID: Hs01060665_g1) were used, and
all reactions were set up with Luna® Universal Probe qPCR Master
Mix (New England Biolabs). A total of three replicates/sample were
run for both TYMS and β-actin on the Applied Biosystems
StepOnePlus Real-Time PCR system (Thermo Fisher Scientific, Inc.)
and the results were analyzed using StepOne Software v2.3 (Thermo
Fisher Scientific, Inc.). qPCR thermocycling conditions were as
follows: Initial denaturation at 95˚C for 60 sec, followed by 40
cycles of denaturation at 95˚C for 15 sec and extension at 60˚C for
30 sec. The expression of the gene of interest (TYMS) was
normalized against the control gene (β-actin), and the relative
change between affected and controls was analyzed using the
2-ΔΔCq method (22).
Protein expression analysis by western
blotting
To assess TYMS expression, western blot
analysis was performed. Briefly, protein lysate was prepared from
fibroblast cells using radioimmunoprecipitation assay buffer
supplemented with protease inhibitors (Cell Signaling Technology,
Inc.). The protein concentration was determined using a
Bicinchoninic Acid Protein Assay kit (Thermo Fisher Scientific,
Inc.). Equal amounts of protein (50 µg/lane) were separated by 12%
SDS-PAGE. The proteins were transferred onto a PVDF membrane using
iBlot™ 2 Transfer Stacks (Invitrogen; Thermo Fisher Scientific,
Inc.). The membrane was blocked with 5% non-fat dry milk and 1% BSA
in TBS for 1 h at room temperature to prevent non-specific binding.
Subsequently, the membrane was incubated overnight at 4˚C with the
primary antibody against TYMS (1:500; cat. no. HL1236; Invitrogen;
Thermo Fisher Scientific, Inc.). After washing with 0.1% TBST, the
membrane was incubated with the corresponding anti-rabbit IgG,
horseradish peroxidase-conjugated secondary antibody (1:2,000; cat.
no. 7074; Cell Signaling Technology, Inc.) for 3 h at room
temperature. TYMS protein bands were visualized at 34 kDa using an
enhanced chemiluminescence detection SuperSignal™ West Femto
Maximum Sensitivity Substrate (Thermo Fisher Scientific, Inc.), and
chemiluminescent signals were visualized and quantified using the
ImageQuant™ LAS 4000 system (version 1.3; GE Healthcare Life
Sciences). The relative protein expression levels were normalized
to the housekeeping gene β-actin (13E5) Rabbit mAb (1:1,000, Cat.
No. 7087; Cell Signaling Technology, Inc.).
Results
Clinical characterization of
twins
The twins had a kinship coefficient of 0.4934,
confirming their monozygosity and ensuring that the sequencing data
accurately corresponded to them. Both twins exhibited progressive
skin darkening since birth, with strikingly similar pigmentation
patterns. Both twins were healthy without any other symptoms. They
were born to non-consanguineous parents with normal skin color and
no family history of hyperpigmentation disorder. No history of drug
intake or urine discoloration or photosensitivity was reported. The
date of admission was April 2019 for adrenal insufficiency workup,
which yielded a negative result. The patients were referred to a
dermatologist. The first skin biopsy was performed in September
2019 for histopathological examination, followed by a second biopsy
~6 months later to obtain fibroblasts for cell culture. There was
no subsequent follow-up.
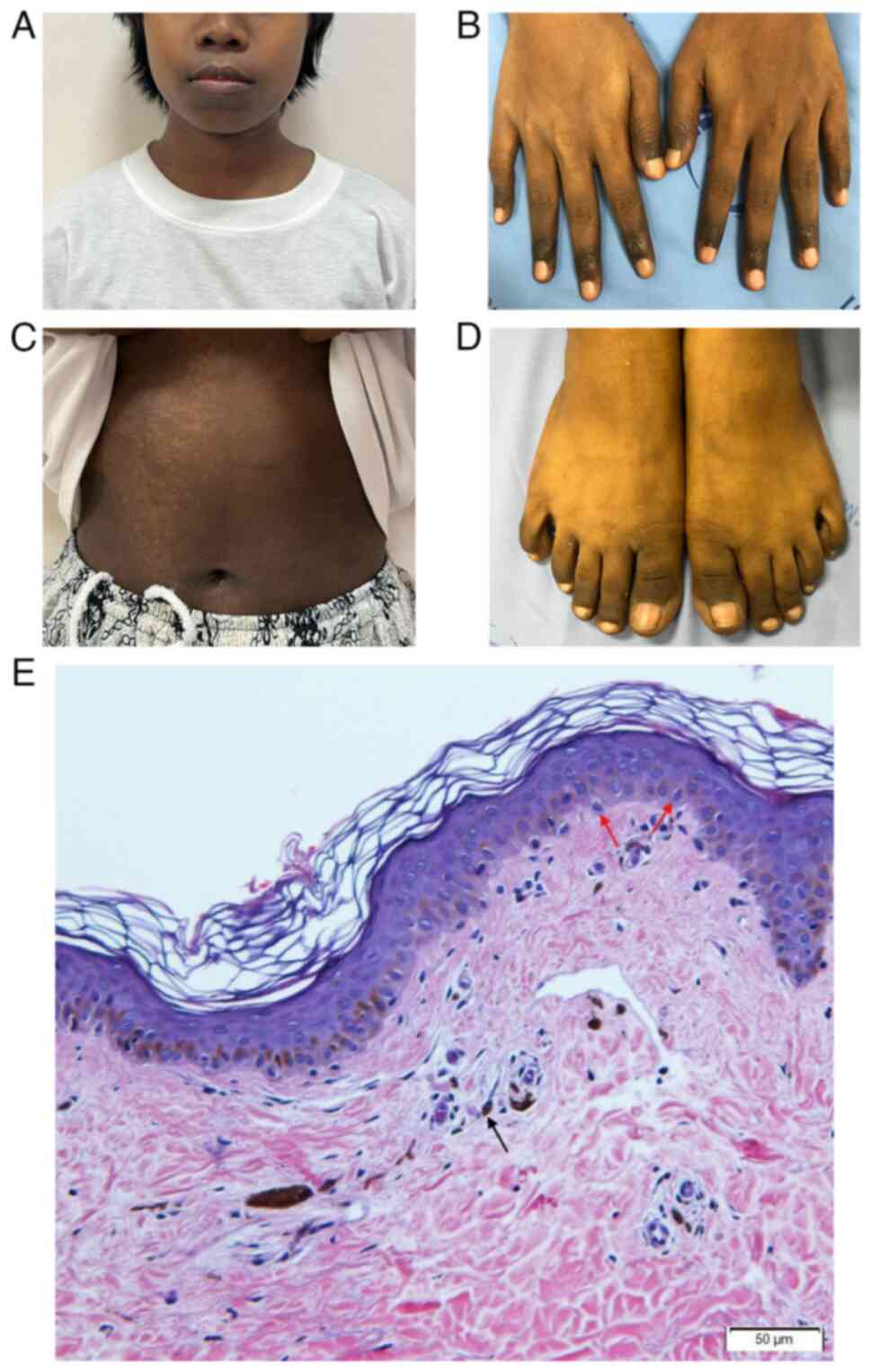
On examination, both twins had generalized, diffuse,
dark-black hyperpigmentation of the skin on the face, neck, body,
hands and feet (Fig. 1A-D) and some
reticulate hypopigmentation and hyperpigmentation were found on the
abdomen. Hyperpigmentation was prominent on the distal finger and
feet. Tongue and buccal mucosa also exhibited some
hyperpigmentation. The hair and nails were normal.
Skin biopsy from the right trunk revealed increased
melanin pigmentation in basal and suprabasal layers and presence of
melanophages in upper dermis. These melanophages were identified as
large, round or oval cells with abundant cytoplasmic melanin
granules, giving them a dark brown/black appearance under the
microscope. Additionally, no melanocyte proliferation was observed,
based on the absence of increased melanocyte density or atypical
melanocytic nests in the basal epidermis (Fig. 1E). Physical examination of both
twins revealed no abnormality aside from hyperpigmentation.
Electrocardiogram results were normal. Complete blood count showed
mild anemia with hematocrit 31.7% and no macrocytosis. Cortisol
levels in the morning and ACTH stimulation test results were
normal, thus, excluding Addison's disease.
Sequencing output
HiFi reads were mapped on the GRCh38 human reference
genome. The mean breadth of coverage of the four samples was 97.19%
(range, 95.08-98.94%). The average sequencing depth of the four
samples was 28X (range, 26-31X).
Variant analysis. SNV/Indel
analysis
Variants were filtered based on inheritance pattern.
The number of concordant SNVs (twins have the same genotype) and
Indels between twins from DeepVariant was 7,030,746. There were
29,137 autosomal dominant de novo variants, 236,591
autosomal recessive variants and 28,108,264 pairs of compound
heterozygous variants. In the first approach, two de novo
and two pairs of compound heterozygous variants were classified as
likely pathogenic and frameshift mutations, according to Franklin
by Genoox. However, those variants were regarded as false positives
after visually inspecting BAM files with Integrative Genomics
Viewer v2.15.4(23).
In the second approach, one autosomal recessive was
identified in phosphatidylinositol glycan anchor biosynthesis class
B (PIGB) (c.981T>G), which was a missense variant. This
variant was absent in the in-house database but present in controls
in gnomAD database. The gene constraint score of the c.981T>G
variant indicated a tolerance to variation. Based on the guidelines
of the American College of Medical Genetics and Genomics (ACMG),
PIGB (c.981T>G) variant was classified as benign.
Moreover, eight compound heterozygous variants which were missense
variants in myosin heavy chain 13 (MYH13) (c.53G>A and
c.791A>G), serpin family H member 1 (SERPINH1)
(c.293G>A and c.1066G>A), myosin VIIA (MYO7A)
(c.781G>A and c.4349A>G), UDP glucuronosyltransferase family
1 member A1 (UGT1A1) (c.1459C>T and c.964A>G), keratin
36 (KRT36) (c.122G>A and c.677G>T), tectonin
β-propeller repeat containing 1 (TECPR1) (c.2270C>A and
c.577G>A), tripartite motif containing 10 (TRIM10)
(c.842G>A and c.977C>T), and missense and intronic variants
in cholinergic receptor nicotinic epsilon subunit (CHRNE)
(c.1375T>C and c.917+562T>C) were identified (Table SI). Despite their absence in the
in-house database and controls in gnomAD database, the gene
constraint score of all variants indicated tolerance to variation.
Based on ACMG guidelines, compound heterozygous variants were
classified as variants of uncertain significance.
SV analysis. The number of concordant SVs
between twins from pbsv was 73,948 variants, of which 69,106 passed
quality control and were filtered based on inheritance pattern,
yielding 157 de novo and 2,267 autosomal recessive variants.
No compound heterozygous variants were retained. In the first
approach, no pathogenic or likely pathogenic variants were found.
In the second approach, two autosomal recessive variants in the
solute carrier family 22 member 31 (SLC22A31) and a region
from sialic acid binding Ig like lectin 14 (SIGLEC14) to
SIGLEC5 were obtained. After inspecting BAM files, these SVs
were true positives; however, they were not rare (frequency >1%
in the in-house genome database) in Thai individuals. Therefore, no
SVs were considered to be associated with the phenotype.
CNV analysis. No CNVs were
phenotype-associated. Copy numbers that were different from both
parents were identified in 13 regions. After annotating the regions
with ClassifyCNV, eight regions were predicted as uncertain
significance (four of which were located within or overlapped
protein-coding genes) and five were predicted as benign. After
checking the copy number in these regions in the in-house database,
all regions were ruled out since allele frequency was >1%.
Methylation profile analysis
The present study identified 211 DMRs, of which 161
resided in 143 genes and 50 DMRs were not in genes. The annotation
showed that 51.66, 18.01, 12.32, 10.90, 5.69 and 1.42% were in the
promoter (≤2kb), distal intergenic, intron, exon, promoter (2-3 kb)
and 3' untranslated region. respectively. After performing
biological function prediction, 35 significant terms were
identified (Table SII). However,
the significant enrichments were not related to skin pigmentation
or melanogenesis.
STR analysis
After starting with 718,245 repeat patterns,
filtering for STR motifs 2-6 base pairs in length, resulting in
240,677 patterns. Results from the in-house algorithm returned
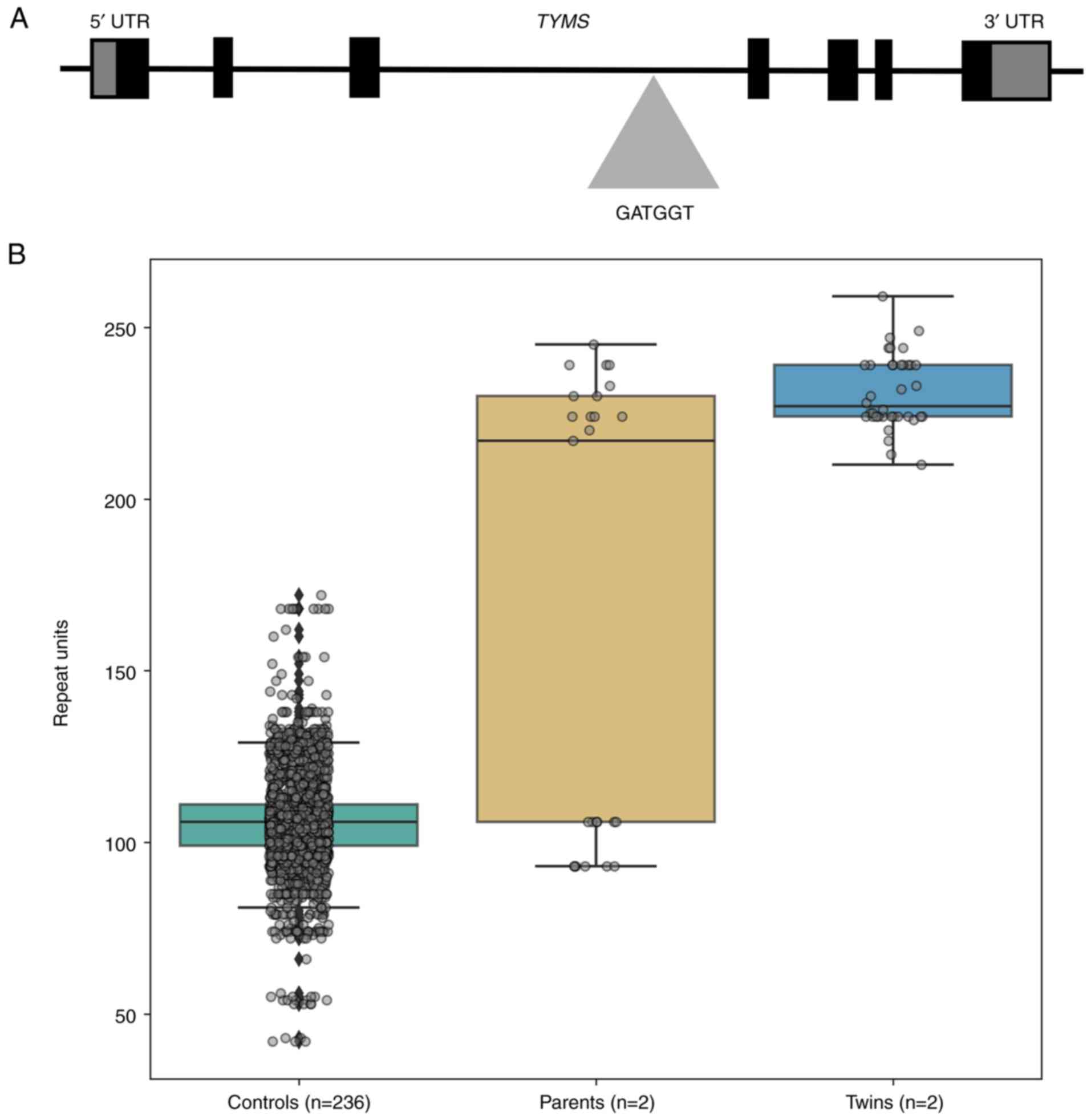
eight candidate patterns. Only one pattern (GATGGT) was consistent
with autosomal recessive inheritance, whereas other patterns were
not consistent between phenotype and mode of inheritance. There was
a biallelic GATGGT repeat expansion with a range of 210-259 repeats
in intron 3 of TYMS in both twins (Fig. 2A), whereas the parents exhibited a
heterozygous expansion. The father had 106 repeat units in one
allele and 230-245 repeat units expanded in the other allele. The
mother had 93 repeat units in one allele and 217-224 repeat units
expanded in the other allele compared with the reference genome
(data not shown). The 236 controls had ≤172 repeat units (Fig. 2B). The majority of controls (47%;
112/236) had 106 repeat units. All controls with 172 repeat units
carried the expansion on a single allele only. No homozygous
controls were identified, unlike in the twins.
Determination of the TYMS mRNA and
protein expression
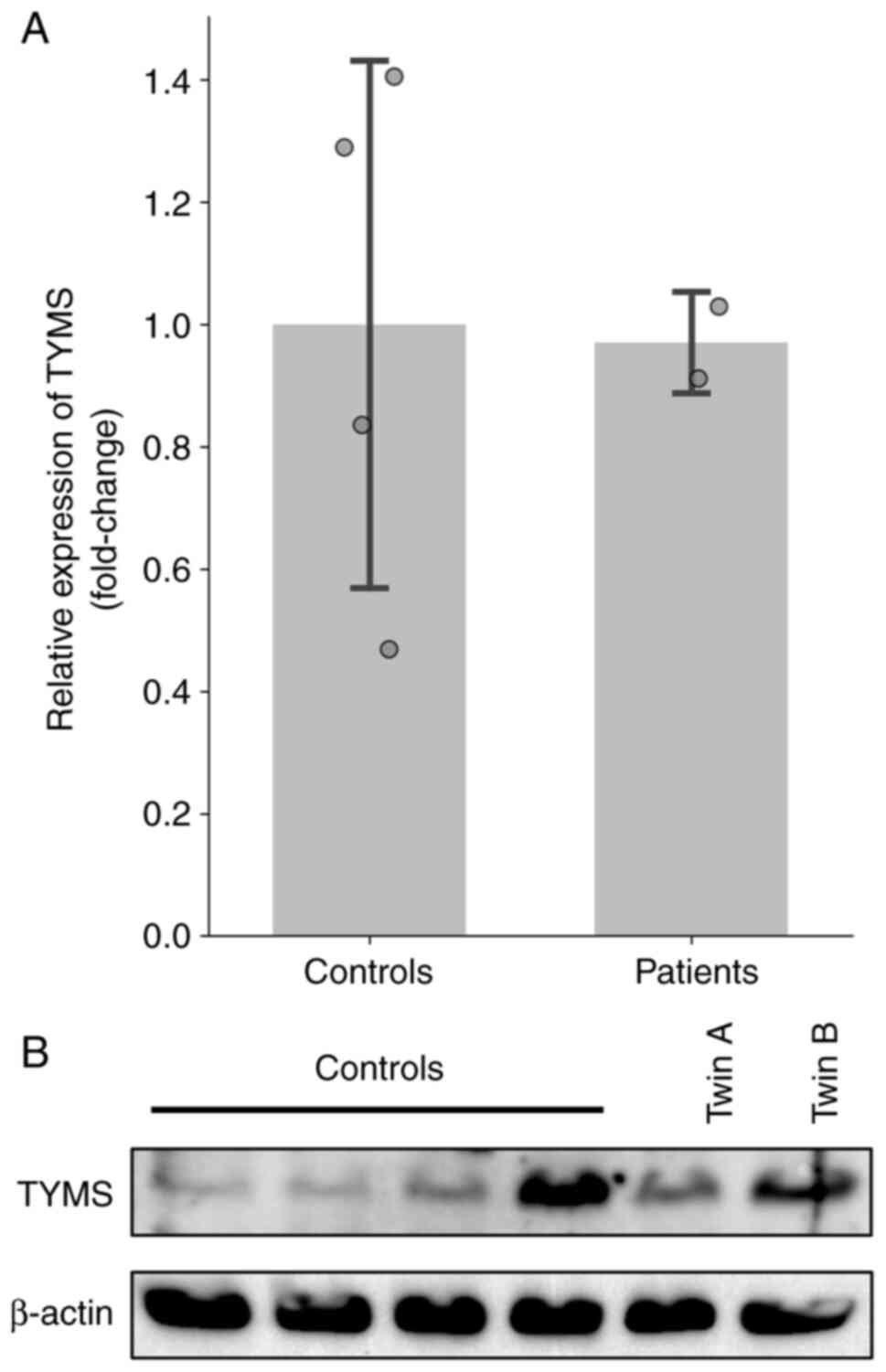
RT-qPCR was used to investigate the expression
levels of TYMS in identical twin patients. RNA was extracted
from cultured skin fibroblasts and PBMCs. The mean TYMS
expression levels in the patients (PBMCs, 0.74 and 0.76; skin
fibroblasts, 0.91 and 1.03) were within the range observed in
control samples (PBMCs, 0.30-1.00; skin fibroblasts, 0.47-1.41;
Figs. 3A and S1). No apparent differences were observed
in either cell type. Protein was extracted from cultured skin
fibroblasts. The western blot analysis showed that TYMS protein
levels varied between the two probands, with twin A showing lower
expression than twin B. Overall, the levels observed in both twins
were within the range seen in control samples (Fig. 3B, S2 and S3).
Discussion
The present study reported monozygotic twin girls
with distinct types of diffuse hyperpigmentation. Both had
hyperpigmentation on the trunk, although some areas showed
reticulated hypo- and hyperpigmentation, which differs from the
pigmentation of Addison's disease, which is most intense in the
flexures, at sites of pressure and friction and in the creases of
the palms and soles (9,24). Moreover, the investigation for
Addison's disease showed negative results. Unlike other diffuse
hyperpigmentation disorders, the present condition was
characterized by a congenital onset, progressive maturation and
absence of accompanying symptoms. By contrast with the acquired
onset in AUM (25), localized
ashy-gray patches in EDP (26),
pruritus and sun-exposed areas in LPP (27) and irregular patches and specific
area involvement in FPH (28), the
twins exhibited symptom-free, whole-body pigmentation. This
distinct presentation suggests a novel entity in the spectrum of
diffuse hyperpigmentation disorders.
Single gene disorders, such as X-linked recessive
hyperpigmentation, Dowling-Degos disease, dyschromatosis
universalis hereditarian and Kitamura disease, have reticulate
lesions and other signs and symptoms (29-32).
For example, a case report of hyperpigmentation in a Taiwanese
child demonstrated an inborn error of vitamin B12 metabolism with
identification of a homozygous mutation in ATP binding cassette
subfamily D member 4 (c.423C>G; p.Asn141Lys) (33). Due to these distinguished features,
the present condition was termed congenital progressive universal
melanosis (CPUM).
In the present case, the condition was similar to
AUM, also known as carbon baby syndrome (diffuse hyperpigmentation
subtype). To the best of our knowledge, only a few reports of AUM
have been published (25,34-46).
In patients with AUM, pigmentation of basal and suprabasal layers
is increased without melanocyte proliferation (44). Histopathological examination of the
present patients also showed these traits but hyperpigmentation was
congenital. The present patients presented with heavily pigmented
areas especially on the lips, tongue, knuckles and feet, with
generalized hypopigmented patches around the abdomen.
Electrocardiogram test, serum cortisol, ACTH stimulation and
physical examination results were within normal limits and no
retinitis pigmentosa was detected. Patients had no history of drug
intake and no family history of hyperpigmentation or associated
conditions. While melanin production regulation, excessive
production of β melanocyte-stimulating hormone, abnormal
sensitivity of melanocytes or neural stimuli may be involved in AUM
(46), the actual etiology of AUM
is yet to be elucidated.
The present study performed long-read genome
sequencing owing to its ability to provide a more comprehensive and
accurate representation of the genome than short-read sequencing.
SNV/Indel, SV, CNV, methylation profile and STR filtering were
performed, focusing on variants present in both affected
individuals and absent in the unaffected parents. A total of eight
variants from the SNV/Indel analysis and a novel unreported
biallelic repeat expansion in the third intron of the TYMS
gene from the STR analysis were found. No pathogenic or likely
pathogenic variants were identified in other variant classes.
The eight SNV/Indel pairs of variants were found in
eight genes. Mutations of the five genes (UGT1A1,
SERPINH1, CHRNE, TRIM10 and MYO7A) have
been linked to human disorders, but none of the disorders manifest
as abnormal skin pigmentation. Specifically, UGT1A1 mutation
causes Crigler-Najjar syndrome (47), whereas SERPINH1 mutations
result in osteogenesis imperfecta due to abnormal collagen
biosynthesis (48). CHRNE
mutations lead to congenital myasthenic syndrome (49) and TRIM10 mutation is
associated with nephronophthisis (50). Additionally, a mutation in
MYO7A causes non-syndromic hearing loss (51). To the best of our knowledge, for the
remaining three genes, MYH13, KRT36 and TECPR1, no
disease has yet been directly associated. However, MYH13 is
associated with muscle contraction (52). KRT36 is a member of the
keratin gene family involved in hair and nail formation (53) and TECPR1 serves a role in
autophagy (54). Given the
functions and associated conditions of these genes, no candidate
gene among the SNV/Indel variants appeared to be responsible for
causing CPUM.
An unreported biallelic GATGGT hexanucleotide repeat
expansion in the third intron of the TYMS gene was
identified from the STR analysis in the twins, with 210-159 repeat
units (124 repeat units of the hexanucleotide repeats are present
in the human reference genome hg38). The father and mother each had
one non-expanded allele, with the other allele measuring 230-245
units in the father and 217-224 units in the mother. A total of 206
controls carried the GATGGT repeat in a range of 42 to 172 and
almost half of the controls had 106 units. The repeat expansion was
present in both alleles of the affected twins and heterozygous
state in each parent, consistent with an autosomal recessive mode
of inheritance.
The TYMS gene catalyzes the methylation of
deoxyuridine monophosphate to form deoxythymidine monophosphate by
transferring a methyl group from 5,10-methylenetetrahydrofolate.
This conversion is key for maintaining the balance of nucleotides
necessary for DNA replication and repair (55). Dysregulation of TYMS
expression causes DNA damage, leading to defective DNA synthesis.
This triggers cell stress responses, including activation of the
tumor suppressor protein p53 and potentially increasing
microphthalmia-associated transcription factor (MITF)
expression (56). MITF
activation upregulates both tyrosinase and associated proteins,
resulting in increasing melanin synthesis (57,58).
Therefore, TYMS may indirectly influence skin pigmentation
via its involvement in DNA repair mechanisms. Hyperpigmentation can
arise from genes involved in DNA repair or early senescence
(58). A total of >150 DNA
damage repair genes, including TYMS, have been identified in
the hallmark gene set of the Molecular Signatures Database as
playing a role in DNA repair mechanisms (59).
Digenic germline variants in both TYMS and
enolase superfamily member 1 result in dyskeratosis congenita (DC)
(60). In the aforementioned study,
RT-qPCR analysis revealed a significantly reduced expression of
TYMS in lymphoblastoid cells derived from affected probands
compared with controls. TYMS deficiency due to loss-of-function
missense mutations disrupts the nucleotide metabolism resulting in
genome instability, which was measured by the level of protein
associated with DNA damage. Clinical manifestations of DC include
abnormal skin pigmentation, nail dystrophy and oral leucoplakia.
This suggests that the abnormal skin pigmentation in DC is a result
of genome instability. Certain mutations in TYMS causing a
dysmorphic syndrome may lead to non-syndromic anomaly. For example,
p63 mutations cause Mendelian clefting syndromes, including
ectrodactyly-ectodermal dysplasia-clefting syndrome [Online
Mendelian Inheritance in Man (OMIM no. #129900)],
ankyloblepharon-ectodermal dysplasiaclefting syndrome (OMIM
#106260) and RappHodgkin syndrome (OMIM #129400), while some
missense mutations can lead to non-syndromic cleft lip and palate
(61). Therefore, it was
hypothesized that since DC, a syndrome with hyperpigmentation, can
be caused by certain forms of genetic variants in TYMS,
another form of genetic variant in the same gene may lead to
non-syndromic hyperpigmentation.
The present study attempted to identify the
molecular mechanism underlying GATGGT repeats in non-coding regions
of the TYMS gene which lead to hyperpigmentation. Non-coding
repeat expansions can lead to disease through three major
mechanisms: Epigenetic silencing resulting in loss-of-function of
the expanded allele, sequestration of RNA-binding splicing factors
and repeat-associated non-AUG translation (62). For example, Friedreich ataxia
disorder exhibits GAA repeat expansion within the first intron of
frataxin (FXN) gene (63),
which leads to increases in DNA methylation in the upstream region
of the FXN repeats, resulting in changes of chromatin
structures. Benign adult familial myoclonic epilepsy type 1
(BAFME1) exhibits TTTTA repeats expansion with TTTCA repeats
insertion in the intron 4 of sterile alpha motif domain containing
12 (SAMD12) (64,65). BAFME3, BAFME4, BAFME6, BAFME7 and
BAFME8 also have the same pattern of repeat expansion in the intron
of membrane associated ring-ch-type finger 6 (MARCH6)
(66), yeats domain containing 2
(YEATS2) (67),
trinucleotide repeat containing adaptor 6A (TNRC6A)
(68), rap guanine nucleotide
exchange factor 2 (RAPGEF2) (68) and retinoic acid induced 1
(RAI1) (69). BFAME2 is
caused by ATTTC repeats expansion within the first intron of StAR
related lipid transfer domain containing 7(70). To the best of our knowledge,
however, the exact mechanisms underlying BAFME have not yet been
explained.
It was hypothesized that the intronic repeat
expansions in the present twins decreased TYMS RNA levels
and subsequently lowered TYMS protein levels, leading to genome
instability. This, in turn, activates p53, similarly to DNA damage
caused by UV exposure (71),
increasing MITF expression and resulting in increased melanin
synthesis and hyperpigmentation. RNA and protein levels in
fibroblasts were assessed because of the possible role of
fibroblasts in skin pigmentation. According to previous studies,
fibroblasts produce paracrine factors that bind receptors on
melanocytes, leading to melanogenesis (72,73).
In addition, fibroblast-derived growth factors including hepatocyte
growth factor (HGF), neuregulin 1 (NRG-1) and
corticotropin-releasing hormone (CRH) have been shown to increase
pigment contents in melanocytes (74). However, RT-qPCR to quantify
TYMS RNA levels and immunoblot analyses to determine TYMS
protein levels showed no differences between the twins and
controls. It is important to note that statistical analysis was not
performed due to the limited sample size (n=2).
Based on the present results, it is hypothesized
that the GATGGT repeats in non-coding regions of the TYMS
gene may reduce TYMS expression specifically in melanocytes
or keratinocytes as no decrease was observed in PBMCs or cultured
skin fibroblasts. This decreased expression could lead to the
disruption of nucleotide balance in DNA repair and skin
hyperpigmentation. Future studies should investigate melanocytes
and keratinocytes to verify the role of repeat expansion in
TYMS associated with CPUM. The present study did not
directly assess TYMS expression in melanocytes or
keratinocytes, the primary cells responsible for pigmentation.
Moreover, the present study was based on a single rare case of
monozygotic twins with CPUM, and no additional cases were available
for validation. Independent replication studies with additional
cases are needed to confirm the pathogenic relevance of TYMS
repeat expansion in CPUM. Despite these limitations, the present
findings provide novel insights into a potential genetic mechanism
underlying CPUM and offer a foundation for future functional
studies.
In summary, the present study suggested a
association link between intronic hexanucleotide repeat expansion
in TYMS and CPUM. However, further studies are necessary to
confirm this association and understand its pathogenesis.
Supplementary Material
Data S1
Mean expression of TYMS mRNA in
PBMCs. Reverse transcription-quantitative PCR was performed on
PBMCs from control (n=3) and patient samples (n=2). PBMC,
peripheral blood mononuclear cell; TYMS, thymidylate synthase.
Original blots of thymidylate synthase
detection.
Original blots of β-actin
detection.
Compound heterozygous single
nucleotide variant pairs identified.
Significantly enriched Gene Ontology
biological processes associated with differentially methylated
regions.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by Health Systems
Research Institute (grant no. 67-095) and Bioinformatics and
Computation Biology Program, Chulalongkorn University.
Availability of data and materials
The data generated in the present study are not
publicly available due to privacy constraints but may be requested
from the corresponding author.
Authors' contributions
VS conceptualized and supervised the study. VS, ST,
SP and MP designed the experiments and edited the manuscript. SC,
TK, CV and PW collected clinical data and biological samples. CS
performed the sequencing analysis. ST and KS performed RT-qPCR
analysis and western blotting. SK and MP analyzed the data. SK and
MP confirmed the authenticity of all the raw data. SK, SP, KS and
MP drafted the manuscript. All authors have read and approved the
final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Review Board of the Faculty of Medicine, Chulalongkorn University
(approval nos. 264/62 and 813/63) and was conducted in accordance
with the 1964 Helsinki Declaration and its later amendments, as
well as all relevant ethical guidelines and regulations. The rights
and interests of all participants were fully protected. Written
informed consent was obtained from the parents of the patients.
Patient consent for publication
The authors confirm that the parents of the patients
provided written informed consent for publication of potentially
identifying images.
Competing interests
The authors declare that they have no competing
interests.
Use of artificial intelligence tools
ChatGPT (OpenAI) was used to refine the clarity and
readability of English sentences in the manuscript. The AI-assisted
revisions were limited to grammar, syntax and phrasing, without
altering the scientific content, interpretation or originality of
the work. All final edits were reviewed and approved by the
authors.
References
|
1
|
Bolognia J, Jorizzo JL and Schaffer JV:
Disorders of hyperpigmentation. In: Dermatology. 3rd edition.
Elsevier, London, pp1049-1074, 2012.
|
|
2
|
Malviya N and Pandya A: Disorders of
hyperpigmentation. In: Dermatoanthropology of Ethnic Skin and Hair.
Vashi NA and Maibach HI (eds). Springer International Publishing,
Cham, pp197-214, 2017.
|
|
3
|
Ghosh A, Das A and Sarkar R: Diffuse
hyperpigmentation: A comprehensive approach. Pigment Int. 5:4–13.
2018.
|
|
4
|
Sandhu S, Neema S and Radhakrishnan S:
Dermoscopy of disorders of hyperpigmentation. Pigment Int. 8:14–24.
2021.
|
|
5
|
Alkhowailed MS, Otayf M, Albasseet A,
Almousa A, Alajlan Z and Altalhab S: Clinical approach to linear
hyperpigmentation: A review article. Clin Cosmet Investig Dermatol.
14:23–35. 2021.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Sinha S and Kulhari A: Reticulate
pigmentary disorders: A review. Pigment Int. 6:67–76.
2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Grimes P, Nordlund JJ, Pandya AG, Taylor
S, Rendon M and Ortonne JP: Increasing our understanding of
pigmentary disorders. J Am Acad Dermatol. 54 (Suppl 2):S255–S261.
2006.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Thawabteh AM, Jibreen A, Karaman D,
Thawabteh A and Karaman R: Skin pigmentation types, causes and
treatment-a review. Molecules. 28(4839)2023.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Ten S, New M and Maclaren N: Clinical
review 130: Addison's disease 2001. J Clin Endocrinol Metab.
86:2909–2922. 2001.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Betterle C and Morlin L: Autoimmune
Addison's disease. Endocr Dev. 20:161–172. 2011.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Liu JW, Habulieti X, Wang RR, Ma DL and
Zhang X: Two novel SASH1 mutations in Chinese families with
dyschromatosis universalis hereditaria. J Clin Lab Anal.
35(e23803)2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Murthy AB, Palaniappan V, Karthikeyan K
and Anbarasan V: Dyschromatosis universalis hereditaria. Int J
Dermatol. 62:1218–1227. 2023.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Orphanet: Prevalence and Incidence of Rare
Diseases: Bibliographic Data. No. 1. Available from: http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_diseases.pdf.
Accessed June 4, 2025.
|
|
14
|
Khattab S, Nasser H, Al-Janabi MH and
Hasan F: Dyskeratosis congenita: A rare case report. Oxf Med Case
Reports. 2024(omae049)2024.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Logsdon GA, Vollger MR and Eichler EE:
Long-read human genome sequencing and its applications. Nat Rev
Genet. 21:597–614. 2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Rhoads A and Au KF: PacBio Sequencing and
its applications. Genomics Proteomics Bioinformatics. 13:278–289.
2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Adewale BA: Will long-read sequencing
technologies replace short-read sequencing technologies in the next
10 years? Afr J Lab Med. 9(1340)2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Hamada M, Ono Y, Asai K and Frith MC:
Training alignment parameters for arbitrary sequencers with
LAST-TRAIN. Bioinformatics. 33:926–928. 2016.
|
|
19
|
Mitsuhashi S, Frith MC, Mizuguchi T,
Miyatake S, Toyota T, Adachi H, Oma Y, Kino Y, Mitsuhashi H and
Matsumoto N: Tandem-genotypes: Robust detection of tandem repeat
expansions from long DNA reads. Genome Biol. 20(58)2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Manichaikul A, Mychaleckyj JC, Rich SS,
Daly K, Sale M and Chen WM: Robust relationship inference in
genome-wide association studies. Bioinformatics. 26:2867–2873.
2010.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Kisiel MA and Klar AS: Isolation and
culture of human dermal fibroblasts. Methods Mol Biol. 1993:71–78.
2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2 (-Delta Delta C (T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Thorvaldsdóttir H, Robinson JT and Mesirov
JP: Integrative Genomics Viewer (IGV): High-performance genomics
data visualization and exploration. Brief Bioinform. 14:178–192.
2013.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Paller AS and Mancini AJ: Hurwitz Clinical
Pediatric Dermatology. Fifth edition. Elsevier, Amsterdam,
pp245-278, 2016.
|
|
25
|
Kaviarasan PK, Prasad PV, Joe JM, Nandana
N and Viswanathan P: Universal acquired melanosis (Carbon baby).
Indian J Dermatol Venereol Leprol. 74:38–40. 2008.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Convit J, Kerdel-Vegas F and Rodríguez G:
Erythema dyschromicum perstans: A hitherto undescribed skin
disease*. J Invest Dermatol. 36:457–462. 1961.
|
|
27
|
Bhutani LK, Bedi TR, Pandhi RK and Nayak
NC: Lichen planus pigmentosus. Dermatologica. 149:43–50.
1974.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Chernosky ME, Anderson DE, Chang JP, Shaw
MW and Romsdahl MM: Familial progressive hyperpigmentation. Arch
Dermatol. 103:581–591, passim. 1971.PubMed/NCBI
|
|
29
|
Betz RC, Planko L, Eigelshoven S, Hanneken
S, Pasternack SM, Büssow H, Van Den Bogaert K, Wenzel J,
Braun-Falco M, Rütten A, et al: Loss-of-function mutations in the
keratin 5 gene lead to dowling-degos diseasnue. Am J Hum Genet.
78:510–519. 2006.PubMed/NCBI View
Article : Google Scholar
|
|
30
|
Kono M, Sugiura K, Sugama M, Hayashi M,
Takama H, Suzuki T, Matsunaga K, Tomita Y and Akiyama M:
Whole-exome sequencing identifies ADAM10 mutations as a cause of
reticulate acropigmentation of Kitamura, a clinical entity distinct
from Dowling-Degos disease. Hum Mol Genet. 22:3524–3533.
2013.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Pezzani L, Brena M, Callea M, Colombi M
and Tadini G: X-linked reticulate pigmentary disorder with systemic
manifestations: A new family and review of the literature. Am J Med
Genet A. 161A:1414–1420. 2013.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Cao L, Zhang R, Yong L, Chen S, Zhang H,
Chen W, Xu Q, Ge H, Mao Y, Zhen Q, et al: Novel missense mutation
of SASH1 in a Chinese family with dyschromatosis universalis
hereditaria. BMC Med Genomics. 14(168)2021.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Takeichi T, Hsu CK, Yang HS, Chen HY, Wong
TW, Tsai WL, Chao SC, Lee JY, Akiyama M, Simpson MA and McGrath JA:
Progressive hyperpigmentation in a Taiwanese child due to an inborn
error of vitamin B12 metabolism (cblJ). Br J Dermatol.
172:1111–1115. 2015.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Baxter LL and Pavan WJ: The etiology and
molecular genetics of human pigmentation disorders. Wiley
Interdiscip Rev Dev Biol. 2:379–392. 2013.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Chakrabarti N and Chattopadhyay C: Ashy
dermatosis: A controversial entity. Indian J Dermatol. 57:61–62.
2012.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Furuya T and Mishima Y: Progressive
pigmentary disorder in Japanese child. Arch Dermatol. 86:412–418.
1962.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Ghosh SK, Ghoshal L, Bhunia D and Ghoshal
AM: Acquired universal melanosis (carbon baby syndrome). Pediatr
Dermatol. 31:620–622. 2014.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Kint A, Oomen C, Geerts ML and Breuillard
F: Congenital diffuse melanosis. Ann Dermatol Venereol. 114:11–16.
1987.PubMed/NCBI(In French).
|
|
39
|
Mahajan BB, Budhwar J, Chojer P and Singla
C: Carbon baby syndrome: A rare case report. JDA Indian J Clin
Dermatol. 1:61–63. 2018.
|
|
40
|
Malik P, Pathania M and Rathaur VK: A case
of a 4-year-old carbon baby: Acquired universal melanosis and
literature review. Int J Inn Res Med Sci. 5:92–94. 2020.
|
|
41
|
Mammen A, Deepthi B, Majhi C, Kumar M and
Kumar S: Carbon baby syndrome-A rare case. Kerala Med J. 9:138–140.
2015.
|
|
42
|
Naskar S, Kharkar V, Hershada M and
Maharshtra M: Carbon baby-A unique manifestation of cryptic
mastocytosis. Am J Dermatol Res Rev. 37(3)2020.
|
|
43
|
Niiyama S, Bando Y, Ishii M and Katsuoka
K: Universal acquired melanosis: Carbon baby. Dermatol Online J.
19(18961)2013.PubMed/NCBI
|
|
44
|
Parviz T, Sarah E and Ehsan A: Acquired
universal melanosis (Carbon baby syndrome) in a 4-year old girl.
Iran J Dermatol. 16:162–164. 2013.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Ruiz-Maldonado R, Tamayo L and
Fernández-Diez J: Universal Acquired Melanosis: The Carbon Baby.
Arch Dermatol. 114:775–778. 1978.PubMed/NCBI
|
|
46
|
Shome K, Seth J, Samanta AB, Halder S, Das
I and Sarkar P: Carbon baby syndrome: Two case reports. J Pak Assoc
Dermatol. 22:59–62. 2012.
|
|
47
|
Wanlapakorn N, Nilyanimit P,
Vorawandthanachai T, Deesudjit T, Dumrongpisutikul N and Poovorawan
Y: A novel stop codon mutation in exon 1 (558C>A) of the UGT1A1
gene in a Thai neonate with Crigler-Najjar syndrome type I. Genet
Mol Res. 14:419–425. 2015.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Song Y, Zhao D, Xu X, Lv F, Li L, Jiang Y,
Wang O, Xia W, Xing X and Li M: Novel compound heterozygous
mutations in SERPINH1 cause rare autosomal recessive osteogenesis
imperfecta type X. Osteoporos Int. 29:1389–1396. 2018.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Huang K, Luo YB, Bi FF and Yang H:
Pharmacological strategy for congenital myasthenic syndrome with
CHRNE mutations: A meta-analysis of case reports. Curr
Neuropharmacol. 19:718–729. 2021.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Alizadeh R, Jamshidi S, Keramatipour M,
Moeinian P, Hosseini R, Otukesh H and Talebi S: Whole exome
sequencing reveals a XPNPEP3 novel mutation causing
nephronophthisis in a pediatric patient. Iran Biomed J. 24:405–408.
2020.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Lu J, Chen P, Chen T, Li L, Fu X, Yang T
and Wu H: The p.R206C mutation in MYO7A leads to autosomal dominant
nonsyndromic hearing loss. ORL J Otorhinolaryngol Relat Spec.
82:181–187. 2020.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Schiaffino S, Hughes SM, Murgia M and
Reggiani C: MYH13, a superfast myosin expressed in extraocular,
laryngeal and syringeal muscles. J Physiol. 602:427–443.
2024.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Brychtova V, Coates PJ, Hrabal V, Boldrup
L, Fabian P, Vojtesek B, Sgaramella N and Nylander K: Keratin 36, a
specific marker of tongue filiform papillae, is downregulated in
squamous cell carcinoma of the mobile tongue. Mol Clin Oncol.
12:421–428. 2020.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Cao L and Lin F: TECPR1 induces apoptosis
in non-small cell lung carcinoma via ATG5 upregulation-induced
autophagy promotion. Ann Clin Lab Sci. 52:580–592. 2022.PubMed/NCBI
|
|
55
|
Ozer U, Barbour KW, Clinton SA and Berger
FG: Oxidative stress and response to thymidylate synthase-targeted
antimetabolites. Mol Pharmacol. 88:970–981. 2015.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Cui R, Widlund HR, Feige E, Lin JY,
Wilensky DL, Igras VE, D'Orazio J, Fung CY, Schanbacher CF, Granter
SR and Fisher DE: Central role of p53 in the suntan response and
pathologic hyperpigmentation. Cell. 128:853–864. 2007.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Niu C and Aisa HA: Upregulation of
melanogenesis and tyrosinase activity: Potential agents for
vitiligo. Molecules. 22(1303)2017.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Speeckaert R, Van Gele M, Speeckaert MM,
Lambert J and van Geel N: The biology of hyperpigmentation
syndromes. Pigment Cell Melanoma Res. 27:512–524. 2014.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Liberzon A, Birger C, Thorvaldsdóttir H,
Ghandi M, Mesirov JP and Tamayo P: The Molecular signatures
database (MSigDB) hallmark gene set collection. Cell Syst.
1:417–425. 2015.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Tummala H, Walne A, Buccafusca R, Alnajar
J, Szabo A, Robinson P, McConkie-Rosell A, Wilson M, Crowley S,
Kinsler V, et al: Germline thymidylate synthase deficiency impacts
nucleotide metabolism and causes dyskeratosis congenita. Am J Hum
Genet. 109:1472–1483. 2022.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Leoyklang P, Siriwan P and Shotelersuk V:
A mutation of the p63 gene in non-syndromic cleft lip. J Med Genet.
43(e28)2006.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Depienne C and Mandel JL: 30 years of
repeat expansion disorders: What have we learned and what are the
remaining challenges? Am J Hum Genet. 108:764–785. 2021.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Greene E, Mahishi L, Entezam A, Kumari D
and Usdin K: Repeat-induced epigenetic changes in intron 1 of the
frataxin gene and its consequences in Friedreich ataxia. Nucleic
Acids Res. 35:3383–3390. 2007.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Cen Z, Jiang Z, Chen Y, Zheng X, Xie F,
Yang X, Lu X, Ouyang Z, Wu H, Chen S, et al: Intronic
pentanucleotide TTTCA repeat insertion in the SAMD12 gene causes
familial cortical myoclonic tremor with epilepsy type 1. Brain.
141:2280–2288. 2018.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Yeetong P, Chunharas C, Pongpanich M,
Bennett MF, Srichomthong C, Pasutharnchat N, Suphapeetiporn K,
Bahlo M and Shotelersuk V: Founder effect of the TTTCA repeat
insertions in SAMD12 causing BAFME1. Eur J Hum Genet. 29:343–348.
2021.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Florian RT, Kraft F, Leitão E, Kaya S,
Klebe S, Magnin E, van Rootselaar AF, Buratti J, Kühnel T, Schröder
C, et al: Unstable TTTTA/TTTCA expansions in MARCH6 are associated
with Familial Adult Myoclonic Epilepsy type 3. Nat Commun.
10(4919)2019.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Yeetong P, Pongpanich M, Srichomthong C,
Assawapitaksakul A and Shotelersuk V, Tantirukdham N, Chunharas C,
Suphapeetiporn K and Shotelersuk V: TTTCA repeat insertions in an
intron of YEATS2 in benign adult familial myoclonic epilepsy type
4. Brain. 142:3360–3366. 2019.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Ishiura H, Doi K, Mitsui J, Yoshimura J,
Matsukawa MK, Fujiyama A, Toyoshima Y, Kakita A, Takahashi H,
Suzuki Y, et al: Expansions of intronic TTTCA and TTTTA repeats in
benign adult familial myoclonic epilepsy. Nat Genet. 50:581–590.
2018.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Yeetong P, Dembélé ME, Pongpanich M, Cissé
L, Srichomthong C, Maiga AB, Dembélé K, Assawapitaksakul A, Bamba
S, Yalcouyé A, et al: Pentanucleotide repeat insertions in RAI1
cause benign adult familial myoclonic epilepsy type 8. Mov Disord.
39:164–172. 2024.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Corbett MA, Kroes T, Veneziano L, Bennett
MF, Florian R, Schneider AL, Coppola A, Licchetta L, Franceschetti
S, Suppa A, et al: Intronic ATTTC repeat expansions in STARD7 in
familial adult myoclonic epilepsy linked to chromosome 2. Nat
Commun. 10(4920)2019.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Hida T, Kamiya T, Kawakami A, Ogino J,
Sohma H, Uhara H and Jimbow K: Elucidation of melanogenesis cascade
for identifying pathophysiology and therapeutic approach of
pigmentary disorders and melanoma. Int J Mol Sci.
21(6129)2020.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Duval C, Cohen C, Chagnoleau C, Flouret V,
Bourreau E and Bernerd F: Key regulatory role of dermal fibroblasts
in pigmentation as demonstrated using a reconstructed skin model:
Impact of photo-aging. PLoS One. 9(e114182)2014.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Wang Y, Viennet C, Robin S, Berthon JY, He
L and Humbert P: Precise role of dermal fibroblasts on melanocyte
pigmentation. J Dermatol Sci. 88:159–166. 2017.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Li PH, Liu LH, Chang CC, Gao R, Leung CH,
Ma DL and David Wang HM: Silencing stem cell factor gene in
fibroblasts to regulate paracrine factor productions and enhance
c-Kit expression in melanocytes on melanogenesis. Int J Mol Sci.
19(1475)2018.PubMed/NCBI View Article : Google Scholar
|