Introduction
Comprehensive genomic profiling (CGP) is a molecular
diagnostic tool, based on next-generation sequencing (NGS)
technology, used in the field of cancer research and treatment. CGP
identifies various genetic alterations and mutations within the
tumor (1). Results from CGP guide
oncologists to make decisions about treatment options and select
targeted therapies tailored to the specific genetic alterations
present within the tumor. CGP examines a broad panel of genes,
detecting a wide range of genomic alterations. This includes point
mutations (such as PIK3CA p.E453Q and c.1357G>C),
insertions/deletions (indels; such as BRCA2 p.E1571Gfs*3 and
c.4712_4713delAG), copy number variations (CNVs; such as
ERBB2 amplification, copy number:4) and gene fusions (such
as TBL1XR1/PIK3CA fusion). It can detect multiple genetic
alterations in a single test, reducing the need for multiple
individual tests. CGP is an application of NGS that specifically
focuses on a detailed analysis of the genetic profile of patients,
and can provide a broad overview of the genomic alterations in a
tumor. Consequently, precision, efficiency and therapeutic guidance
are potential advantages of CGP, which has been advocated for
advanced-stage cancers (2-5).
A large-sized panel can provide more opportunities for matching
patients to targeted therapies or for increased mutation detection
in clinical trials, while the use of CGP is limited by its high
cost, data complexity and increased turnaround time.
Triple-negative breast cancer (TNBC) is a specific
subtype of breast cancer characterized by the absence of three
deterministic receptors commonly found in other breast cancers:
Estrogen receptor (ER), progesterone receptor (PR) and human
epidermal growth factor receptor 2 (HER2). TNBC cells lack the
expression of these three receptors and do not rely on them for
growth. TNBC accounts for 10-20% of all breast cancer cases world
(6), and is more commonly diagnosed
in younger (#x003C;40 years old) women, African-American women and
those with a family history of breast cancer (7). TNBC is known for its aggressive
behavior and tends to grow and spread quickly, while it is also
associated with a higher risk of recurrence and metastasis
(8), and ~40% of people with stage
I to stage III TNBC will exhibit tumor recurrence after standard
treatment (9). Treatment options
for TNBC often involve chemotherapy, as endocrine therapies and
anti-HER2 therapies that target ER, PR or HER2 are not effective
due to the absence of these receptors (8). The prognosis for TNBC varies depending
on factors such as the diagnosed stage and response to treatment
(10). Due to its aggressiveness,
there is an unmet need to identify actionable targets such as
AKT1, BRCA1/2, PALB2, ERBB2,
PIK3CA and PTEN, for patients with TNBC (11,12).
Research focusing on developing targeted therapies
for TNBC continues to improve treatment outcomes and broaden
therapeutic options (13,14). For example, biomarker-driven
therapies are being applied with the advent of immune checkpoint
inhibitors, poly-adenosine-diphosphate-ribose polymerase (PARP)
inhibitors, selective PI3K and AKT inhibitors, as well as
antibody-drug conjugates, which target immune-enriched, DNA
repair-deficient, PI3K/AKT/mTOR-activated and surface
antigen-overexpressed TNBC, respectively. However, most umbrella
trials for advanced breast cancer are not TNBC-specific, while a
recent review highlighted that large-sized panels could identify
novel markers, emphasizing the potential of CGP to uncover
actionable targets in TNBC (3).
Commercialized CGP assays may be beneficial;
however, the size of the panel to be employed to deliver the most
optimal coverage of actionable genes for TNBC remains to be
explored. Some studies comparing distinct CGP platforms and
resulting biomarkers have been conducted for gastric, head and neck
squamous cell carcinoma, non-small cell lung cancer, ovarian and
prostate cancer, but rarely for TNBC (15,16).
In the present study, a subgroup of patients with TNBC from the
Veterans General Hospital TAipei-Yung-Ling foundation sinO-canceR
(VGH-TAYLOR) study was used (17).
TNBC samples were initially assayed using a medium-sized CGP panel,
and the remaining specimens of nucleic acid were re-sequenced with
a large-sized GCP panel. The aim of the present study was to
investigate whether a larger CGP panel offers clinically
significant advantages in detecting actionable variants for TNBC
management compared with a medium-sized panel.
Materials and methods
Study overview
The present study was conducted in two phases: The
first phase involved prospective and retrospective tissue
collection as part of the VGH-TAYLOR study (medium-sized panel).
The second phase involved the reuse of biobanked samples from the
VGH-TAYLOR study (large-sized panel). The study was approved by the
Institutional Review Board of Taipei Veterans General Hospital
(approval nos. 2021-01-007B and 2023-08-002B; Taipei, Taiwan).
Participants provided written informed consent for both phases of
the study.
The VGH-TAYLOR study was designed to examine the
genetic profiling of different subtypes of breast cancer in Taiwan
(17). The prospective study
comprised diverse clinical scenarios of breast cancer: i) Group 1,
planned to receive first-line surgery followed by adjuvant therapy
(1A) or exhibited early relapse within 3 years of diagnosis (1B);
ii) group 2, planned to receive first-line neoadjuvant therapy
followed by surgery; and iii) group 3, exhibited de novo
stage IV (3-I) or recurrence beyond 3 years of diagnosis
(3-II).
In addition, a retrospective cohort with biobanked
samples from recurrent/metastatic breast cancers or patients with
non-pathological complete response following neoadjuvant therapy
was also included (18-21).
Inclusion criteria were a diagnosis of TNBC from the
VGH-TAYLOR study, availability of sufficient biobanked nucleic acid
and willingness to sign informed consent. Patients without residual
samples and those unwilling to participate in the retesting program
were excluded. A total of 120 female patients were screened, with
108 cases included for comprehensive genomic profiling. The age
range was 27 to 84 years, with a mean ± SD age of 56.1±12.8 years.
All enrolled subjects were treated according to contemporary
guidelines and underwent regular follow-up (22,23).
Regarding immunohistochemistry (IHC) testing, ER and PR were scored
by percentage of nuclear labeling (0-100%). HER2 expression was
scored using a 0 to 3+ membrane staining intensity score (24). Hormone receptor positivity was
defined by either ER or PR with ≥1% of tumor cells exhibiting
nuclear staining while HR-negative breast cancer was defined by the
absence of ER and PR on the cancer cells. HER2 overexpression was
indicated by either a 3+ (positive) or 2+ (equivocal) IHC score
with fluorescence in situ hybridization (FISH) amplification
(25). For patients with equivocal
IHC scoring of HER2, a negative FISH result was a prerequisite. All
patients of the VGH-TAYLOR study were enrolled between November
2018 and December 2021 from the Comprehensive Breast Health Center,
Taipei Veterans General Hospital, a tertiary referral medical
center at Taipei, Taiwan.
Samples and nucleic acid
preparation
Formalin-fixed paraffin-embedded (FFPE) samples were
collected after obtaining informed consent. For consistent results,
a fixation time of 6-48 h in 10% neutral buffer formalin at room
temperature for breast cancer biomarker testing was performed. At
least seven unstained tumor sections with 10 µm in thickness were
retrieved, with one used for hematoxylin and eosin (H&E)
staining and six used for nucleic acid extraction. Hematoxylin (3.5
min at room temperature) was used for nuclear staining, and eosin
(60 sec at room temperature) was used for cytoplasmic staining,
allowing for differentiation of cellular components.
H&E-stained slides were reviewed to ascertain the presence of
adequate breast cancer cells (>70% of cancer composition).
Paraffin was removed by xylene and ethanol serial
washes. Nucleic acid was extracted from 5-µm sections with the
QIAmp DNA FFPE Tissue Kit (cat. no. 56404; Qiagen, Inc.) or AllPrep
DNA/RNA FFPE Kit (cat. no. 80234; Qiagen, Inc.), while quality
control and concentration were checked and determined using the
Qubit fluorimeter (Invitrogen; Thermo Fisher Scientific, Inc.),
Qubit dsDNA HS and Qubit dsDNA BR Assay Kits (cat. nos. Q32851 and
Q32850; Thermo Fisher Scientific, Inc.). In the current study,
treatment-naïve cancerous tissue was used for NGS. Targeted
sequencing for the TruSight Oncology 500 (TSO500) panel was
conducted on residual specimens from the VGH-TAYLOR study.
The storage conditions for the remaining nucleic
acids after CGP aimed at maintaining their integrity for potential
future use. Therefore, nuclear acid was stored at -80˚C in
Tris-EDTA buffer or nuclease-free water. Repeated freeze-thaw
cycles were avoided. A total of 120 pairs of DNA/RNA were tested
for integrity and 108 passed quality control (QC) matrices for NGS
experiments: QC parameters for TSO500 included DNA integrity number
>7 and RNA integrity number >7, size distribution of 200-500
bp for library construction and >90% on-target enrichment with
uniform coverage. Under optimal storage conditions with minimal
nucleic acid degradation, a ~90% success rate (108 out of 120
paired DNA/RNA samples) in sequencing with quality metrics was
reported.
Oncomine comprehensive panel
The Ion Torrent Oncomine Comprehensive Assay Panel
v3 (OCP; cat. no. A35805; Thermo Fisher Scientific, Inc.) was used
as the default CGP for the VGH-TAYLOR study, which enabled the
detection of 161 cancer-related genes and the identification of
single nucleotide variants (SNVs), CNVs, gene fusions and indels. A
total of 10 ng of DNA and RNA sample input was required. Libraries
were generated according to the standard protocols, which was
constructed with the Ion AmpliSeq Library Kit Plus (cat. no.
4488990; Thermo Fisher Scientific Inc.), and were multiplexed for
templating on the Ion OneTouch 2 System and subsequently sequenced
on the Ion GeneStudio S5 Prime System (Thermo Fisher Scientific,
Inc.) using the Ion 318 Chip Kit (cat. no. 488146; Thermo Fisher
Scientific Inc.) with single-end semiconductor-based sequencing and
a read length of 200 bp, according to the manufacturer's
instructions (guide MAN0015885 Revision C of Thermo Fisher
Scientific, Inc.). Sequencing data were analyzed, aligned and
annotated through Torrent Suite v5.10.0 (Thermo Fisher Scientific,
Inc.) and Ion Reporter v5.10 (Thermo Fisher Scientific, Inc.)
software with the default Coverage Analysis (v5.10.0.3), Sample ID
(v5.10.0.1) and Variant Caller (v5.10.0.18) plugin. Variants were
further analyzed and interpreted for clinical actionability using
the Oncomine Knowledgebase Reporter (Thermo Fisher Scientific,
Inc.) database. The coverage metrics indicated that the number of
mapped reads ranged from 4 to 6 million, with a mean depth of
1,100-1,600 times.
TSO500
TSO500 (cat. no. 20032626; Illumina, Inc.) was
designed to identify known and emerging tumor biomarkers, using
both DNA and RNA from tumor samples. These pan-cancer biomarkers
aligned with key guidelines and clinical trials (22,23),
including 523 genes for assessment of DNA and RNA variant types,
plus microsatellite instability, tumor mutational burden and
homologous recombination deficiency (optional). Libraries were
prepared according to the manufacturer's guidelines from up to 80
ng DNA and 40 ng of RNA, using the TSO 500 library prep kit (cat.
no. 20028216; Illumina, Inc.). Adapter ligation with unique
molecular identifiers (UMIs) was performed with target fragments
amplified and indexed. NGS was performed with a NextSeq 2000
sequencing system optimized for a minimum read length of 2x101 bp,
operated by the Department of Pathology and Laboratory Medicine of
the Taipei Veterans General Hospital using the NextSeq 1000/2000 P2
Reagents (300 cycles) v3 (Illumina, Inc.). Data were analyzed with
the TSO500 Local App v2.2 (Illumina, Inc.), and variant call format
files were further processed and annotated with the PierianDx
software version CGW_v6.20 (PierianDx), which offered an integrated
interpretation. The read collapsing analysis step executed an
algorithm that collapses sets of reads (known as families) with
similar genomic locations into representative sequences using UMI
tags. Median exon fragment coverage across all exon bases was
≥150.
Benchmark comparisons
The difference in sequencing technology between
amplicon-based (OCP) and hybrid capture-based (TSO500) methods has
implications for detecting genetic variants, especially in
homopolymer regions, consisting of a series of consecutive
identical bases (26).
Hybridization capture-based approaches show better uniformity,
which can be relevant for detecting variants in homopolymer
regions. Amplicon-based methods are faster and cost-effective but
prone to errors in homopolymer regions. Hybrid capture methods are
more reliable for these regions due to uniform coverage and reduced
PCR bias, but they are more resource-intensive. Final reports from
the medium- (OCP) and large-sized (TSO500) panels and accompanied
tab-separated values files were collected. Variants reported from
actionable genes were the primary endpoints in the current study.
Clinical actionability was defined by the joint consensus of the
Association for Molecular Pathology, American Society of Clinical
Oncology and College of American Pathologists, published in
2017(27). Additional annotations
for actionability and OncoPrinter (28,29)
visualization were conducted using the OncoKB database (30) and European Society for Medical
Oncology (ESMO) Scale for Clinical Actionability of molecular
Targets (ESCAT) criteria (11,12).
Clinical actionability was categorized as follows: i) Tier I,
actionability indicated an alteration-drug match associated with
improved outcome in clinical trials; ii) tier II, anti-tumor
activity was associated with the matched alteration-drug but lacked
prospective outcome data; and iii) tier III, the matched
drug-alteration led to clinical benefit in another tumor type other
than the tumor of interest. Polymorphisms were identified through
population databases, such as the Single Nucleotide Polymorphism
Database (31), the Genome
Aggregation Database (32), 1000
Genomes Project and Exome Aggregation Consortium (33). Variants with minor allele frequency
>1% were excluded. Variants outside the targeted regions of the
sequencing panel were masked. Considering the sequencing depth, a
10% variant allele frequency (VAF) threshold assumed standard
sequencing depths (500-1,000 times). The limit of detection was set
to 5% for SNVs/indels and VAF #x003C;10% was considered a low-VAF
status. An average copy number ≥4 was interpreted as a gain
(amplification) and #x003C;1 as a loss (deletion). The concordance
of filtered actionable genes (AKT1, BRCA1/2,
PALB2, ERBB2, PIK3CA and PTEN) and
TP53 based on the original reports between the two panels
was calculated (concordant variants divided by the sum of both
concordant and discordant variants) and reported.
Data description and clinical
usage
ESCAT-defined actionable genes, including fusions,
amplifications, copy number gains, point mutations and indels, were
extracted for downstream analysis. Categorical variables are
presented as numbers and percentages. Official reports from the Ion
Reporter and the Oncomine Knowledgebase Reporter, as well as those
from Pierian Dx software were released to all participants when
requested. In addition, primary care physicians received the same
reports once they were available, augmenting clinical decision
making.
Results
Study population and targeted
actionable genes
A total of 108 patients with breast cancer from the
VGH-TAYLOR study with adequate remaining nucleic acid (both DNA and
RNA) were recalled. After explaining the purpose of this
re-sequencing study, all participants signed informed consent forms
and their specimens were assayed with the large-sized CGP, TSO500
panel. There were 54 patients in group 1A, 6 in group 1B, 25 in
group 2, 5 in group 3-I, 7 in group 3-II and 11
biobank/retrospective cohort breast cancer samples. Early-stage
breast cancer (groups IA and 2) constituted the majority of samples
(73.1%, n=79). Four patients initially classified as TNBC at the
time of enrollment were re-tested by FISH and found to be
HER2-positive. Due to discrepancy in the interrogated genes (523
vs. 161 genes), only actionable genes listed by the ESCAT criteria
for breast cancer were analyzed, as follows: i) Tier IA:
ERBB2 amplification, BRCA1/2 germline mutation and
PIK3CA mutation; ii) tier IC: NTRK translocation;
iii) tier IIA: PTEN loss and ESR1 mutation; iv) tier
IIB: AKT1 mutation and ERBB2 mutation; v) tier IIIA:
BRCA1/2 somatic mutation and MDM2 amplification; and
vi) Tier IIIB: ERBB3 mutation (11,12).
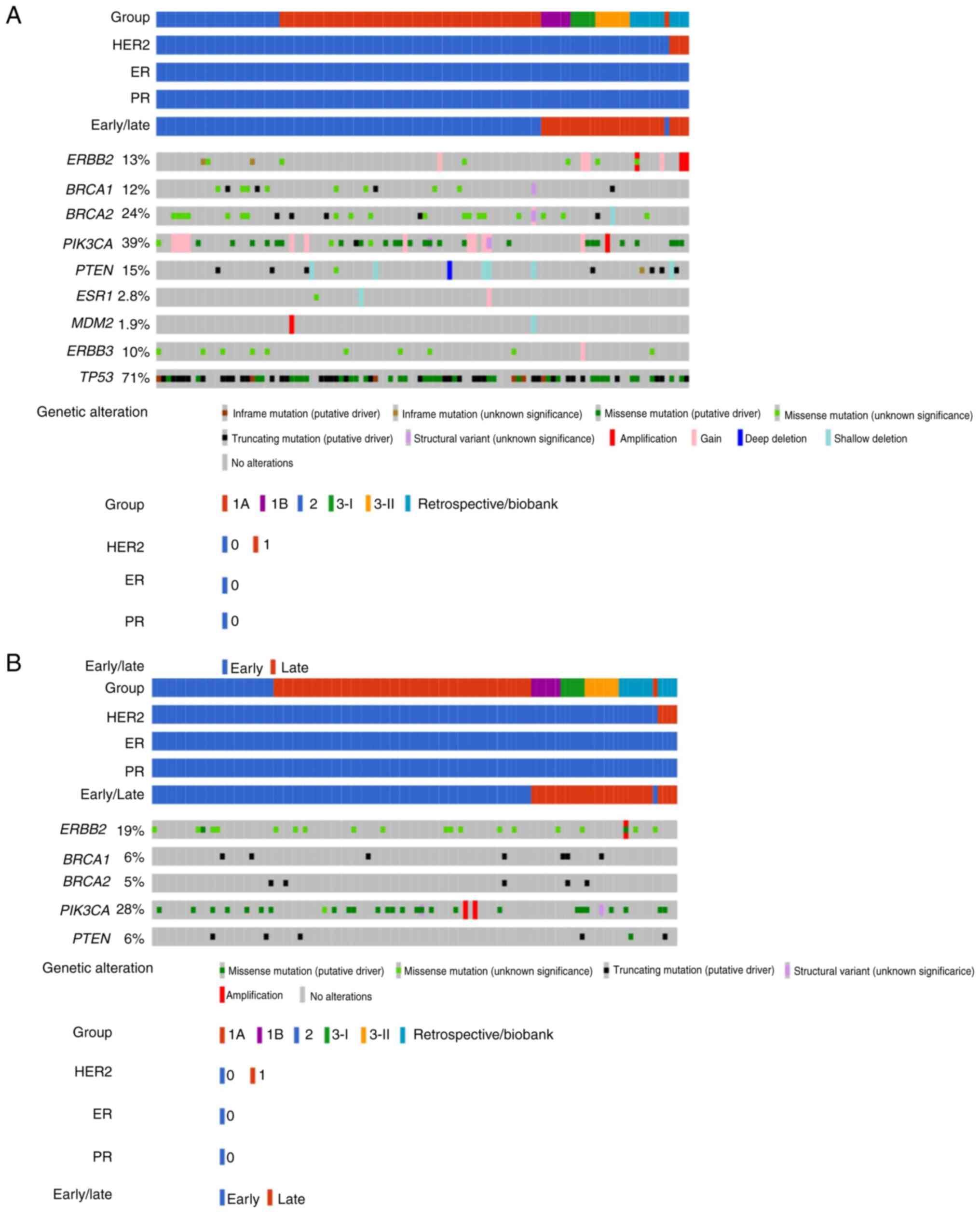
Mutational landscape of actionable
genes with TSO500
Fig. 1A shows the
mutational landscape of ESCAT-defined actionable genes among
Taiwanese patients with TNBC examined with TSO500. Among 108
Taiwanese patients with breast cancer (all samples were TNBC except
for four samples that were HER2-positive), PIK3CA was the
most common actionable gene (39%, n=42), including fusion,
amplification, copy number gain and point mutation, followed by
BRCA2, including fusion, copy number gain, heterozygous
loss, truncating and point mutation (24%, n=26). BRCA1
variants, including fusions, truncating and point mutations were
detected in 12% (n=13) of samples. ERBB2 amplification, copy
number gain, in-frame and point mutations were identified in 13%
(n=14) of the study population. Importantly, among three breast
cancer samples with HER2 amplification reported by TSO500, two
coincided with the four clinically HER2-positive (overexpression)
cases.
PTEN mutations including
heterozygous/homozygous loss and truncating/in-frame/point
mutations were reported in 15% (n=16) of breast cancer samples.
Copy number gain and point mutations in ERBB3 were
identified in 10% (n=11) of the samples, while ESR1 (2.8%,
n=3; copy number gain/heterozygous loss and point mutation) and
MDM2 variants (1.9%, n=2; amplification and heterozygous
loss) were infrequently mutated in the Taiwanese population. No
NTRK translocations were reported. Table SI presents the actionable mutations
assayed with TSO500 CGP.
Mutational landscape of actionable
genes with OCP
Fig. 1B shows the
mutational landscape of actionable genes reported by OCP. Compared
with TSO500, OCP reported a higher number of ERBB2 variants
(19 vs. 13%), most of which came from higher proportion of missense
mutations with unknown significance. Of the four clinical
HER2-positive breast cancer samples, none reported a HER2
alteration, except for another HER2-equivocal case associated with
an ERBB2 I665V missense mutation, which was a variant of
uncertain significance (VUS). Conversely, the frequencies of
BRCA1 and BRCA2 variants were lower than those
reported from TSO500 (BRCA1, 6 vs. 12%; and BRCA2, 5
vs. 24%). PIK3CA variants and PTEN mutations were
also less frequent than those with TSO500 (28 vs. 39% and 6 vs.
15%, respectively). No alterations were identified by OCP in
ESR1, MDM2 and ERBB3. Table SII details the actionable mutations
identified with OCP.
Comparisons of reported actionable
variants between OCP and TSO500
For benchmark comparisons, the four HER2-positive
samples were excluded, and the analysis was focused on tumor DNA
sequence variants from actionable genes. Amino acid change (protein
coordinate), genomic and transcript-dependent cDNA coordinates were
used to identify variants reported as least once from either
TSO500, OCP or both (Table I).
Among 272 variants, 94 (34.6%) were identified by both platforms;
therefore, the concordance rate was slightly higher than one-third
based on the original reports. To understand the mechanisms
underpinning the high discordance between TSO500 and OCP, a manual
review of conflicting variants was conducted with all binary
alignment map (BAM) files visualized through integrative genomic
viewer by an author who is expert in precision oncology and
bioinformatics (34). Fig. S1 provides such an example.
 | Table IVariants of actionable genes and
TP53 (n=202) reported from both the TSO500 and OCP. |
Table I
Variants of actionable genes and
TP53 (n=202) reported from both the TSO500 and OCP.
| Gene | Variant | ESCAT | Both platforms | TSO500 only |
|---|
| AKT1 | E17K | II-B | 3 | |
| BRCA1 | S405* | III-A | 1 | |
| | R1203* | III-A | 1 | |
| | S1286fs | III-A | 1 | |
| | S632fs | III-A | 1 | |
| | c.5470-1G>A | III-A | 1 | |
| | G1350C | III-A | | 1 |
| | M1783L | III-A | | 2 |
| | N909S | III-A | | 1 |
| | R1583K | III-A | | 1 |
| | R762S | III-A | | 1 |
| | S1389N | III-A | | 1 |
| | V191I | III-A | | 1 |
| | c.1A>G | III-A | | 1 |
| BRCA2 | S521* | III-A | 1 | |
| | E1571fs | III-A | 1 | |
| | N2135fs | III-A | 1 | |
| | C315S | III-A | | 1 |
| | F2254Yfs*6 | III-A | | 1 |
| | G2508S | III-A | | 1 |
| | G2901D | III-A | | 1 |
| | H523R | III-A | | 1 |
| | I1929V | III-A | | 4 |
| | N72S | III-A | | 2 |
| | P3292L, V2109I | III-A | | 1 |
| | R2108C | III-A | | 5 |
| | R2842H | III-A | | 1 |
| | V2109I | III-A | | 1 |
| | V2151Ffs*17 | III-A | | 1 |
| | V783A | III-A | | 1 |
| ERBB2 | L811V, L839R | II-B | 1 | |
| | L725S | | 1 | |
| | P1140A | | | 1 |
| | V743_ | | | 1 |
| | M744insHV | | | 1 |
| | Y742_ A745dup | | | 1 |
| NTRK1 | K167R | | | 1 |
| | M530T | | | 1 |
| | R190Q | | | 1 |
| | R190W | | | 1 |
| NTRk3 | D611E | | | 2 |
| PALB2 | K353fs | | 1 | |
| | A38G | | | 1 |
| | D498Y | | | 2 |
| | R825T | | | 2 |
| PIK3CA | E542K | I-A | 3 | |
| | E542K, E726K | I-A | 1 | |
| | E542Q, H1047R | I-A | 1 | |
| | E545K | I-A | 3 | |
| | E545Q, H1047Y | I-A | 1 | |
| | E726K | | 1 | |
| | G1049R | | 1 | |
| | H1047L | I-A | 2 | |
| | H1047R | I-A | 9 | |
| | N345I | | 1 | |
| | Q546R | | 1 | |
| | D350N | | | 1a |
| | D725N | | | 1b |
| PTEN | E150* | II-A | 1 | |
| | E43fs | II-A | 1 | |
| | P38fs | II-A | 1 | |
| | R130* | II-A | 1 | |
| | Q245* | II-A | 1 | |
| | c.1026+1G>A | II-A | 1 | |
| | V290Sfs*8 | II-A | | 1 |
| | G127_ G129del,
G129E | II-A | | 1 |
| TP53 | C176R | IV-A | 1 | |
| | C242Afs | IV-A | 1 | |
| | C275Y | IV-A | 1 | |
| | E271* | IV-A | 1 | |
| | E56Kfs | IV-A | 2 | |
| | F109Sfs | IV-A | 1 | |
| | G108Vfs | IV-A | 4 | |
| | G245S | IV-A | 2 | |
| | H179R | IV-A | 2 | |
| | H179Y | IV-A | 1 | |
| | H193P | IV-A | 1 | |
| | H193R | IV-A | 1 | |
| | H193Y | IV-A | 1 | |
| | H214Qfs | IV-A | 1 | |
| | K132N | IV-A | 1 | |
| | L111Dfs, R196 | IV-A | 1 | |
| | L111Ffs | IV-A | 1 | |
| | L194H | IV-A | 1 | |
| | L252Hfs | IV-A | 1 | |
| | P151S | IV-A | 1 | |
| | Q192* | IV-A | 1 | |
| | R158Lfs | IV-A | 1 | |
| | R175H | IV-A | 2 | |
| | R196* | IV-A | 2 | |
| | R248Q | IV-A | 4 | |
| | R248W | IV-A | 1 | |
| | R273H | IV-A | 4 | |
| | R282W | IV-A | 1 | |
| | R333Vfs | IV-A | 1 | |
| | R342* | IV-A | 1 | |
| | S149Pfs | IV-A | 1 | |
| | S166* | IV-A | 1 | |
| | S241F | IV-A | 1 | |
| | T253Pfs | IV-A | 1 | |
| | V147* | IV-A | 1 | |
| | W146* | IV-A | 1 | |
| | W53* | IV-A | 1 | |
| | W91* | IV-A | 1 | |
| | Y103Afs | IV-A | 1 | |
| | Y107* | IV-A | 1 | |
| | Y205D | IV-A | 1 | |
| | c.560-2A>T | IV-A | 1 | |
| | c.993+1G>A | IV-A | 1 | |
| | c.994-2A>C | IV-A | 1 | |
| | c.993+1G>A,
526T>A | IV-A | 1 | |
| | c.920-2A>G | IV-A | 1 | |
| | c.993+1G>T | IV-A | 1 | |
| | c.560-1G>A | IV-A | 1 | |
| | c.919+1G>T | IV-A | 1 | |
| | c.993+2T>G | IV-A | 1 | |
| | c.993+1G>A | IV-A | 1 | |
| | D281_ K292del | IV-A | | 1 |
| | E271Q | IV-A | | 1 |
| | F113V | IV-A | | 1 |
| | I251L | IV-A | | 1 |
| | L265P | IV-A | | 1 |
| | N131del | IV-A | | 1 |
| | P190L | IV-A | | 1 |
| | Q333E, R213* | IV-A | | 1 |
| | R213* | IV-A | | 4 |
| | T140_ C141
delinsS | IV-A | | 1 |
| | V157_ R158insL | IV-A | | 1 |
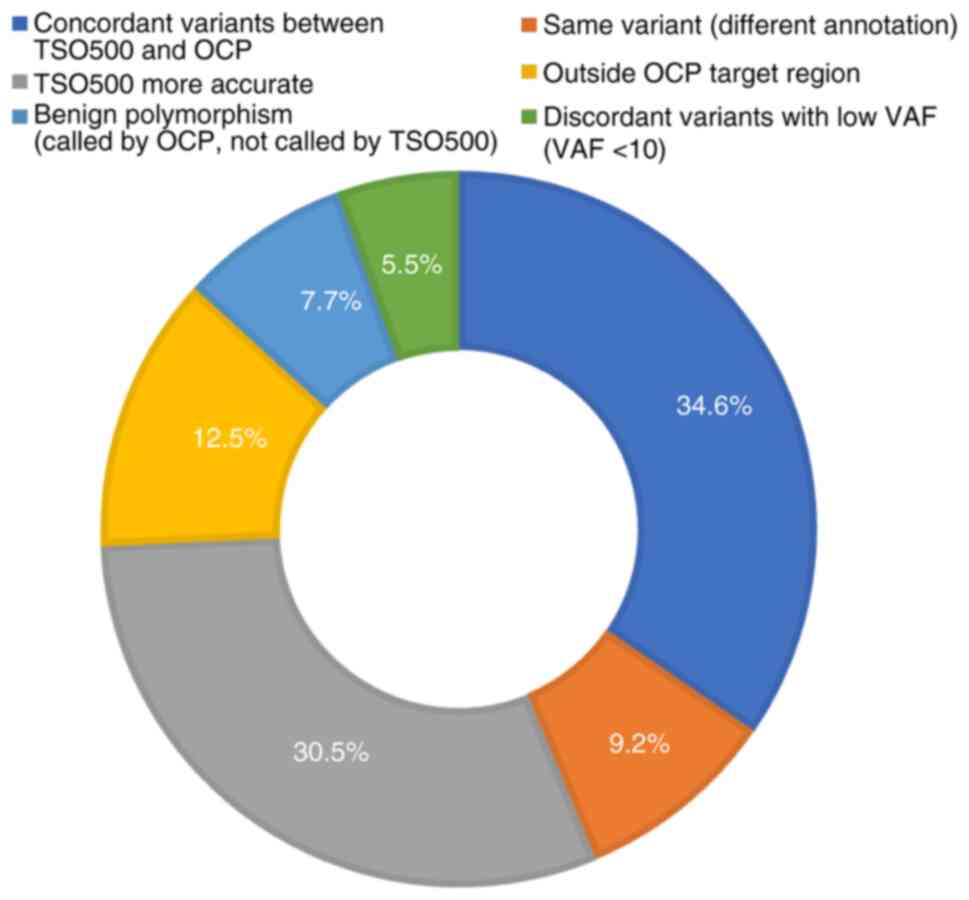
Fig. 2 shows the
interpretation categories of the comparison: A total of 25 (9.2%)
variants were the same with different annotations, 34 (12.5%) were
beyond the OCP targeted regions, 21 (7.7%) were benign
polymorphisms called by OCP but filtered out by TSO500, 15 (5.5%)
were discordant variants with low variant allele frequency
(VAF#x003C;10%) while TSO500 were more accurate in 83 cases
(30.5%). After discarding out-of-targeted region variants, benign
and low-VAF variants, the concordance rate approached 60% (119 of
202 variants, 58.9%) and the large-sized panel (TSO500) detected
more variants even for the same set of actionable genes and
TP53. It deserves notice that certain variants were called
by OCP but ignored by TSO500, which were proven to be homopolymer
regions, including variants in BRCA1 (n=2), BRCA2
(n=2), PALB2 (n=1) and TP53 (n=3), as well as a
misalignment with PTEN in one case and with TP53 in
two cases (data not shown).
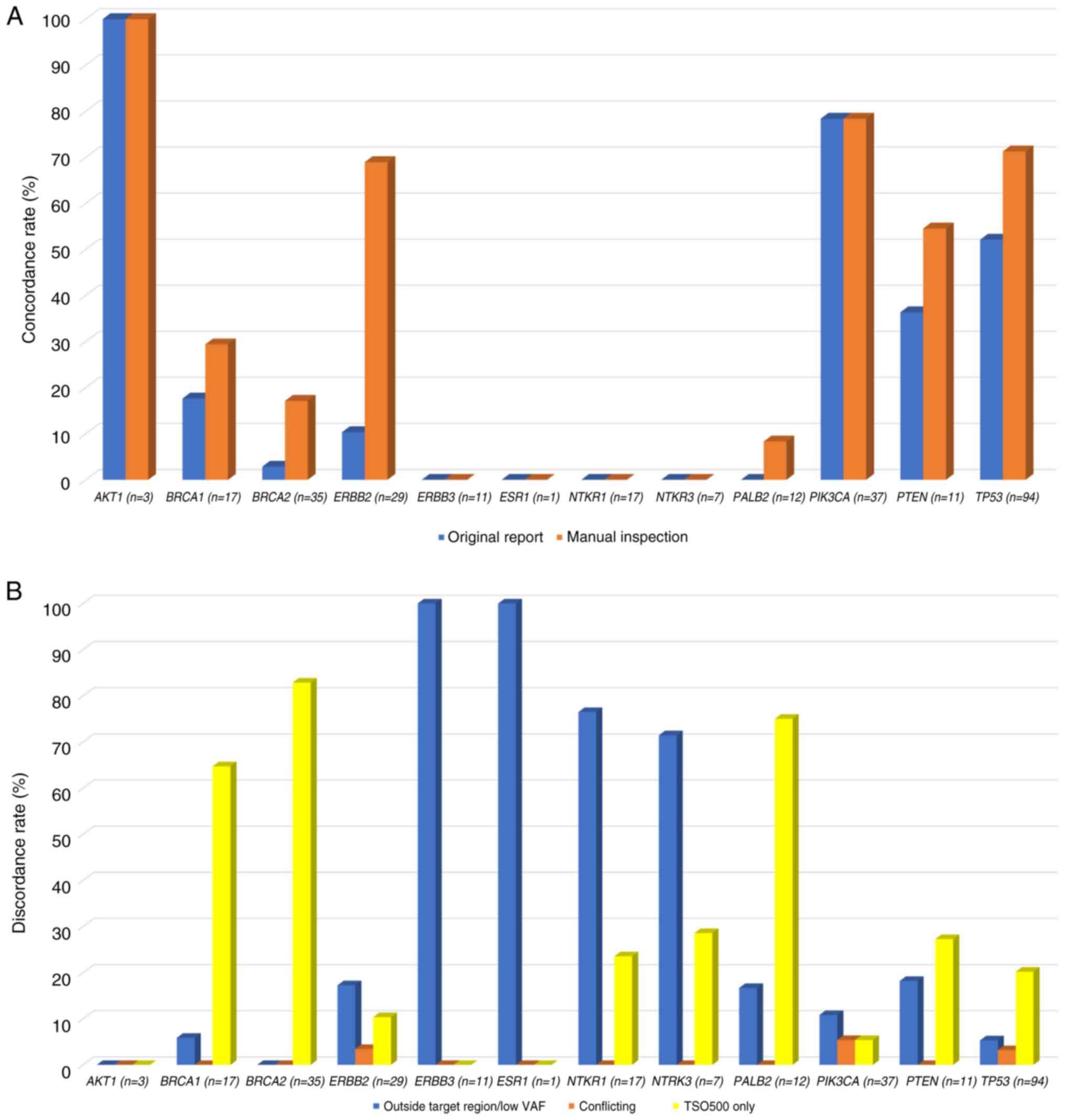
Fig. 3A and B and Table
II present concordant and discordant variants among actionable
genes between two CGP panels. AKT1 E17K exhibited a perfect
concordance (100%, n=3), followed by PIK3CA (78%, n=29) and
TP53 (52%, n=49). With manual corrections of polymorphism
and aliasing variants, the concordance of TP53 (71%, n=67),
ERBB2 (69%, n=20), PTEN (55%, n=6), BRCA1,
BRCA2 and PALB2 (29, 17 and 8%, respectively) was
enhanced substantially. Most of the discordance came from those
detected by TSO500 only, including variants in BRCA1 (65%,
n=11), BRCA2 (83%, n=29) and PALB2 (75%, n=9). Some
variants were undetectable by OCP due to out-of-target regions or
low VAF, which occurred in all ERBB3 and ESR1
variants, as well as NTRK1 and NTRK3 (76 and 71%,
respectively) variants. True conflicting calling results were rare,
including 3 TP53 (3%), 2 PIK3CA (5%) and 1
ERBB2 (3%) variants.
| Table IIDeconvolution of variants among
actionable genes and TP53 with concordant (original report
and manual inspection) and discordant (outside targeted region/low
VAF), conflicting results and TSO500-detected only results. |
Table II
Deconvolution of variants among
actionable genes and TP53 with concordant (original report
and manual inspection) and discordant (outside targeted region/low
VAF), conflicting results and TSO500-detected only results.
| | AKT1
(n=3) | BRCA1
(n=17) | BRCA2
(n=35) | ERBB2
(n=29) | ERBB3
(n=11) | ESR1
(n=1) | NTKR1
(n=17) | NTRK3
(n=7) | PALB2
(n=12) | PIK3CA
(n=37) | PTEN
(n=11) | TP53
(n=94) |
|---|
| Original
report | 100 | 18 | 3 | 10 | 0 | 0 | 0 | 0 | 0 | 78 | 36 | 52 |
| Enhanced with
manual inspection | 100 | 29 | 17 | 69 | 0 | 0 | 0 | 0 | 8 | 78 | 55 | 71 |
| Outside targeted
region/low VAF | 0 | 6 | 0 | 17 | 100 | 100 | 76 | 71 | 17 | 11 | 18 | 5 |
| Conflicting
result | 0 | 0 | 0 | 3 | 0 | 0 | 0 | 0 | 0 | 5 | 0 | 3 |
| TSO500-detected
only | 0 | 65 | 83 | 10 | 0 | 0 | 24 | 29 | 75 | 5 | 27 | 20 |
Discussion
To achieve the goal of personalized and precision
medicine, CGP has been recognized as a key tool that can
potentially transform cancer risk prediction, detection, diagnosis,
treatment and monitoring. CGP can augment clinical decision-making
in the form of either in vitro diagnostics (IVD) or
companion diagnostics. Most importantly, the continuous price
reduction across small- to large-sized CGP panels has improved
patient access (35). Consequently,
therapy assignments for newly diagnosed breast cancers and
monitoring of patients who are in treatment are expected to benefit
from CGP.
In the context of CGP, actionable genes refer to
specific genetic alterations or mutations in a patient's (tumor)
DNA that can guide therapeutic decisions or clinical interventions.
These genetic changes are considered ‘actionable’ because they
suggest potential treatments or clinical strategies that can
directly impact the patient's care. This may include targeting
specific mutations with precision medicine therapies, selecting
clinical trials based on the genomic profile, such as basket or
umbrella trials, or monitoring the patient for disease progression.
Actionable genes typically include those that are associated with
known therapies (such as targeted therapies and immunotherapies,
including PARP inhibition for pathogenic BRCA1/2 mutations),
have established clinical guidelines for intervention based on
their presence, or may offer prognostic or predictive value for
disease outcomes, guiding treatment options (1-5).
In the present study, the performance of medium- to large-sized
gene panels in a TNBC patient cohort was compared. Both OCP and
TSO500 are used worldwide, but there are few direct comparison
studies for breast cancer actionable genes in the literature
(18,19,36,37).
The four HER2-positive breast cancer cases provided
an opportunity to evaluate the association between clinical
HER2-status and NGS-based ERBB2 CNVs. None of these cases
were reported as HER2-amplified by OCP while two of three
HER2-amplified cases reported by TSO500 coincided with the four
clinically HER2-positive breast cancers. A higher number of
HER2-gained breast cancers were identified by TSO500, indicating
the potential for anti-HER2 targeting therapy. As NTRK
translocation was not listed as the targeted alteration of OCP, the
detectability of structure variants (CNV and fusion) was not
compared further.
In the present study, the genes and variants
reported from either OCP v3 or TSO500 when genomic, cDNA and
protein coordinates were used for variant sorting were detailed.
This subset represented the most actionable variants revealed by
medium- and large-sized CGP. AKT1 is an intra-cellular
kinase and is predominantly altered in breast and endometrial
cancer. The AKT1 E17K is the most common alteration across
various tumor types, including breast and gynecological cancers
(38,39). The CAPItello-291 phase III trial
demonstrated that capivasertib (AZD5363) and fulvestrant
combination therapy resulted in markedly longer progression-free
survival compared with the placebo and fulvestrant groups (40). The three AKT1 E17K-mutant
cases in the present study were identified by both panels.
There were two BRCA1 (S405* and R1203*) and
one BRCA2 (S521*) truncating mutations, and reflex germline
testing should precede if PARP inhibitor was considered for these
patients, as only germline BRCA1/2 mutations are actionable
in breast cancer (41). The
BRCA1 c.5470-1G>A SNP has been recognized as pathogenic
by both the Breast Cancer Information Core and Consortium of
Investigators of Modifiers of BRCA1/2 (42,43).
The BRCA2 S521* truncating mutation impairs nuclear
localization of BRCA2, which is essential for normal BRCA2 function
(44). Despite common truncating
and frameshift mutations (five for BRCA1 and three for
BRCA2) called by both panels, only TSO500 revealed more
suspicious variants (eight variants among 9 cases for BRCA1
and 13 variants among 21 subjects), while OCP was flawed by
spurious mutations (BRCA1: S1180fs and T1376fs; and
BRCA2: S538fs, T598fs, S973fs, E33*, T912fs, S3041fs and
S3147fs).
PALB2 K353fs is a truncating mutation in a
tumor suppressor gene and therefore is likely oncogenic. The phase
II TBCRC 048 study also indicated the predictive power of the
germline PALB2 mutation for metastatic breast cancer, as a
high response rate (82%) was observed when olaparib, a PARP
inhibitor, was used (45). In the
present study, TSO500 identified three additional variants in 5
cases while OCP reported 1 false positive case with up to four
variants (D1125fs, N368fs, S357fs and N342fs). Tumor-only
sequencing made differentiating germline from somatic
BRCA1/2 and PALB2 mutations challenging. However,
LOH-germline inference calculator and the somatic-germline-zygosity
algorithms helped distinguish these mutations, with one somatic and
eight germline BRCA1/2 mutations identified (46).
One patient harbored two mutations in ERBB2
(L811V and L839R) and another with the L725S mutation was
recognized by both platforms, none of which were among the
oncogenic mutations approved by U.S. Food and Drug Administration
for the use of neratinib, a pan-HER kinase inhibitor that binds
irreversibly to the ATP-kinase domain of HER2 inhibiting the
downstream phosphorylation of AKT and MAPK (47). TSO500 identified three additional
ERBB2 mutations (P1140A, V743_M744insHV and Y742_A745dup).
It is important to note that TSO500 detected more complex mutations
(such as Y742_A745dup and V743_M744insHV, both classified as VUS)
than did OCP.
Certain PIK3CA hotspot mutations have been
identified from the SOLAR-1 trial for the usage of alpelisib, a
selective PI3K-α inhibitor, namely C420R, E542K, E545A, E545D,
E545G, E545K, Q546E, Q546R, H1047L, H1047R and H1047Y (48). Our previous study also observed
double mutations of PIK3CA among the Taiwanese population
(21). Only four cases with E726K,
G1049R, N345I and Q546R mutations were outside the hotspot region,
while two cases with D350N or D725N were co-mutant with other
hotspots, which were only reported by TSO500. Consequently, both
platforms identified the same number of PIK3CA-altered
breast cancer cases.
Conversely, selective PI3K-β inhibitors, GSK2636771
and AZD8186, are ATP competitors and have shown preclinical
antitumor response and durability for PTEN-deficient solid
tumors including TNBC (49). The
PTEN c.1026+1G>A SNP has been reported as clinically
pathogenic at least twice (Invitae Clinical Genomics Group and
ClinGen PTEN Variant Curation Expert Panel), with germline origin
(50,51). The remaining five consensual
variants (E150*, E43fs, P38fs, R130* and Q245*) were truncating
mutations; TSO500 identified two additional cases with PTEN
mutations (G127_G129del and V290Sfs*8) and OCP called one
inappropriate variant due to misalignment (c.386G>A).
Quite a few TP53 alterations were identified
by both OCP and TSO500. Despite not being currently druggable,
TP53 may act as an independent prognostic factor for early
and advanced-stage cancer with a wide range of mutational frequency
(52). It has been argued that all
TP53 mutations from tumor-only sequencing are somatic, and
rarely representative of the germline Li-Fraumeni syndrome
(53). TSO500 identified more
variants than OCP did, while the latter reported three cases with
V73fs in homopolymer region and one case with misaligned V73fs
mutation, both of which were spurious.
A total of 272 variants were identified among
actionable genes and TP53 from at least one panel.
Interestingly, the concordance rate from original reports was low
at slightly more than one-third (34.6%). With extensive
bioinformatics analyses and manual curation, three-fifths of
concordance (58.9%) were achieved after exclusion of unfiltered
polymorphisms, low-VAF variants and those beyond the targeted scope
of OCP. Among inconsistent variants, some were identical after
manual inspections, further indicating the necessity of analytical
abilities and domain knowledge in genomic nomenclature (54). Furthermore, an additional 62
variants were revealed by TSO500, indicating missed opportunities
by OCP, which may be explained by fundamental discrepancies in
sequencing technology. The accuracy of the amplicon-based OCP vs.
the hybrid capture-based TSO500 panel can be influenced by their
underlying methodologies. Amplicon-based methods utilize PCR
amplification with sequence-specific primers to enrich target
regions. By contrast, hybrid capture sequencing employs
hybridization-based capture to enrich for desired genomic regions.
Hybrid capture may offer a more robust and accurate approach,
especially for clinical applications (55).
In the context of TNBC research, a VAF cut-off of
#x003C;10% is used to filter out low-confidence or likely spurious
SNVs identified through NGS of tumor samples (56,57).
The threshold may be beneficial to eliminate technical, background
noise and subclonal mutations, as tumor samples can be contaminated
with normal cells, and low-level DNA damage or degradation can
introduce background noise that interferes with accurate SNV
calling. In addition, tumors are often composed of multiple
subclones with distinct genetic profiles. SNVs present in minor
subclones may have VAFs #x003C;10%, making them difficult to
distinguish from background noise (58). A VAF cut-off of 5-10% is commonly
used in various cancer genomics studies, including those focused on
TNBC. This range is considered to achieve a reasonable balance
between sensitivity (detecting true SNVs) and specificity
(excluding false positives) (59).
Despite being one of the pioneers to directly
compare two commercialized targeted panels, there were some
limitations to the present study. First, sequencing was not
conducted concurrently, and the 1-2 year delay of the TSO500
analysis following OCP may have introduced some bias from nucleic
acid degradation, despite all samples being stored under
temperature-controlled conditions. Second, variants called by each
panel were considered based on the standard or formal algorithm of
each platform (Ion Reporter/Oncomine Knowledgebase Reporter for OCP
and PierianDx for TSO500), which limited the comparability between
distinct platforms. For unbiased comparison, the same aligner,
caller and annotator should be applied. However, browser extensible
data files were unavailable from manufacturers to confirm the
jointly interrogated regions. On the other hand, all commercial CGP
solutions are under regulation as either laboratory developed tests
or IVD, and practically these pre-set algorithms should not be
modified arbitrarily to enhance reproducibility. Despite this, the
present study conducted an exhaustive bioinformatic analysis from
BAM files to dissect the conflicting results from the same samples.
Third, NTRK is not within the targeted fusion genes of OCP;
therefore, a comparison with TSO500 was not possible. Moreover,
NTRK sequence variants, amplifications or fusions are not
actionable for breast cancer. As larotrectinib and entrectinib are
approved in a number of countries, NTRK fusion as a
tumor-agnostic marker is important, and its detection should be
incorporated into CGP for breast cancer (60). Fourth, as most cases are triple
negative phenotypically, a cohort of ~100 patients with TNBC is
suitable for a comparative sequencing panel study. However, if the
goal is to perform more detailed subgroup analyses or identify rare
mutations, a larger cohort may be more appropriate. In the future,
conducting a prospective and parallel study across distinct CGP
platforms could reveal whether larger sequencing panels justify
their higher costs by improving the detection of actionable
mutations, refining novel therapeutic strategies, and
characterizing the complex genomic landscape of TNBC. Furthermore,
it would provide critical insights to optimize genomic profiling
for other breast cancer subtypes, advancing both research and
clinical care.
As an observational study, the present study could
not directly assess the impact of CGP on treatment outcomes, as no
intervention was performed. During the study period, the
compassionate use of alpelisib (a selective PI3K inhibitor)
benefited nine patients, and the out-of-pocket use of olaparib (a
PARP inhibitor) benefited two patients, demonstrating the
actionability of the testing. Since May 2024, NGS, specifically
whole-exome sequencing for BRCA1/2, has been reimbursed in
Taiwan for patients with stage II or higher TNBC. The Regulations
of Special Medical Techniques of Taiwan require that all NGS
testing results (including both whole-exome sequencing and targeted
panels) be submitted to the National Health Research Institutes
Biobank. It is anticipated that cost-effective implementation of
larger NGS panels in routine clinical practice will be possible in
the future, once sufficient data from NGS results and accompanying
clinical outcomes are available.
In conclusion, the present study comparing medium-
and large-sized sequencing panels in a cohort of patients with TNBC
may provide critical insights into the balance between actionable
gene findings, cost and complexity. TSO500, the larger panel,
detected more variants than OCP did, even from the same set of
ESCAT-defined actionable genes and TP53. Conversely, a
proportion of inconsistent variants could be manually curated and
were identical with aliasing coordinates or starting positions.
Finally, there were variants detectable only by TSO500, indicating
potential and fundamental differences in sequencing technology,
bioinformatic algorithms and variant filtering. The value of a
large-sized panel in clinical usage is ascertained, considering the
beneficiaries of PARP inhibition for the higher number of
BRCA1/2 and PALB2 mutations detected, as indicated by
the National Comprehensive Cancer Network and ESMO guidelines
(61,62). Given the experience from the present
study, the updated Oncomine Comprehensive Assay Plus with >500
genes profiled is anticipated in future studies (63). The results could guide panel
selection for precision oncology, ensuring optimal clinical
outcomes while maximizing resource efficiency. This approach would
be particularly impactful in refining strategies for TNBC
management.
Supplementary Material
Examples of discordant variants
between TSO500 and OCP visualized through integrative genomic
viewer. The upper half of each panel represents aligned reads of
TSO500, and the lower half represents aligned reads of OCP. (A)
Variants beyond the OCP targeted regions. There was a ERBB3
c.1463G>A (p.R488Q) variant detected by TSO500. In OCP, no read
was present in this region and therefore no variant was reported by
OCP. (B) Variants in homopolymer region. There was a PTEN
c.V290Sfs*8 (p.867dupA) variant clearly observed in the aligned
reads of TSO500 and OCP. TSO500 accurately reported this variant,
whereas OCP missed this variant and therefore did not report it.
(C) Likely artifactual variants in homopolymer region. In the
aligned reads of OCP, some reads showed an insertion of a single A
nucleotide, and some showed an insertion of double A nucleotides.
Due to these alterations, OCP reported that there was a BRCA2
c.2916_2917insA (p.S973fs) variant in this region. However, in the
aligned reads of TSO500, no variant can be observed. As false
indels in homopolymer repeats are a well-known weakness of the Ion
Torrent sequencing platform, which is used in OCP, BRCA2
c.2916_2917insA (p.S973fs) is likely to be an artifactual variant
in homopolymer region introduced by errors of sequencing. (D)
Inappropriately annotated variant due to misalignment. In the
aligned reads of TSO500, a three-nucleotide deletion of TP53
c.419_421delCCT (p.T140_C141delinsS) can be clearly observed. The
alignment of OCP in this region is more disorganized. Although the
three-nucleotide deletion of c.419_421delCCT still can be seen, OCP
erroneously reported this variant as TP53 c.421T>A (p.C141S).
TSO500, TruSight Oncology 500; OCP, Oncomine Comprehensive Assay
Panel v3.
Summary of actionable mutations among
108 Taiwanese patients with breast cancer assayed with the TruSight
Oncology 500.
Summary of actionable mutations among
108 Taiwanese patients with breast cancer assayed with the Oncomine
Comprehensive Assay Plus v3.
Acknowledgements
The authors would like to thank Dr Morris Chang
(retired from the Taiwan Semiconductor Manufacturing Company) for
professional consultation about the technical feasibility of the
present study. The authors also appreciate the kind support with
NGS experiments from the Illumina team (Illumina, Inc.; San Diego,
CA), including Dr Mark M. Wang, Dr Vincent Hsieh, Mr. Thomas Tang,
Mrs. Jora Lin, Mr. Dr Wang and Mr. Jerry Cheng throughout the
execution of this study.
Funding
Funding: This work was supported in part by Taipei Veterans
General Hospital research fund grant nos. V110E-005-3, V111E-006-3,
V112E-004-3 and V112C-013, Melissa Lee Cancer Foundation (grant no.
MLCF_V114_B11404) and National Science and Technology Council
(grant no. NSTC 111-2314-B-075-063-MY3).
Availability of data and materials
The data generated in the present study may be
found in the Sequence Read Archive database under accession number
PRJNA1256731 or at the following URL: https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1256731.
Authors' contributions
CCH and LMT conceived the study. CCH drafted the
manuscript. YCY conducted bioinformatics analyses. YFT, YSL, TCC
and CYL collected samples and certified the NGS reports. HLH
conducted NGS experiments. CCH and YCY confirm the authenticity of
all the raw data. LMT approved the final submission. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
The study protocol was reviewed and approved by the
Institutional Review Board of Taipei Veterans General Hospital
(approval nos. 2021-01-007B and 2023-08-002B; Taipei, Taiwan).
Participants provided written informed consent for both the
prospective/retrospective VGH-TAYLOR study (approval no.
2021-01-007B) and for the retrospective re-sequencing study
(approval no. 2023-08-002B).
Patient consent for publication
All participants agreed to the publication of the
present study, with all identifying information removed.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Huang CC, Yeh YC, Ho HL, Liu CY, Tsai YF
and Tseng LM: Comprehensive genomic profiling of Taiwanese patients
with breast cancer using a novel targeted panel: Preliminary
analyses from a prospective triple-negative cohort. J Clin Oncol.
41 (Suppl 16)(e12553)2023.
|
|
2
|
Wang L, Zhai Q, Lu Q, Lee K, Zheng Q, Hong
R and Wang S: Clinical genomic profiling to identify actionable
alterations for very early relapsed triple-negative breast cancer
patients in the Chinese population. Ann Med. 53:1358–1369.
2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Tierno D, Grassi G, Scomersi S, Bortul M,
Generali D, Zanconati F and Scaggiante B: Next-generation
sequencing and triple-negative breast cancer: Insights and
applications. Int J Mol Sci. 24(9688)2023.PubMed/NCBI View Article : Google Scholar
|
|
4
|
O'Haire S, Degeling K, Franchini F, Tran
B, Luen SJ, Gaff C, Smith K, Fox S, Desai J and IJzerman M:
Comparing survival outcomes for advanced cancer patients who
received complex genomic profiling using a synthetic control arm.
Target Oncol. 17:539–548. 2022.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Cifuentes C, Lombana M, Vargas H, Laguado
P, Ruiz-Patiño A, Rojas L, Navarro U, Vargas C, Ricaurte L, Arrieta
O, et al: Application of comprehensive genomic profiling-based
next-generation sequencing assay to improve cancer care in a
developing country. Cancer Control.
30(10732748231175256)2023.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Almansour NM: Triple-negative breast
cancer: A brief review about epidemiology, risk factors, signaling
pathways, treatment and role of artificial intelligence. Front Mol
Biosci. 9(836417)2022.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Dent R, Trudeau M, Pritchard KI, Hanna WM,
Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P and Narod SA:
Triple-negative breast cancer: Clinical features and patterns of
recurrence. Clin Cancer Res. 13:4429–4434. 2007.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Foulkes WD, Smith IE and Reis-Filho JS:
Triple-negative breast cancer. N Engl J Med. 363:1938–1948.
2010.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Stewart RL, Updike KL, Factor RE, Henry
NL, Boucher KM, Bernard PS and Varley KE: A multigene assay
determines risk of recurrence in patients with triple-negative
breast cancer. Cancer Res. 79:3466–3478. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Jie H, Ma W and Huang C: Diagnosis,
prognosis, and treatment of triple-negative breast cancer: A
review. Breast Cancer (Dove Med Press). 17:265–274. 2025.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Mateo J, Chakravarty D, Dienstmann R,
Jezdic S, Gonzalez-Perez A, Lopez-Bigas N, Ng CKY, Bedard PL,
Tortora G, Douillard JY, et al: A framework to rank genomic
alterations as targets for cancer precision medicine: The ESMO
scale for clinical actionability of molecular targets (ESCAT). Ann
Oncol. 29:1895–1902. 2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Condorelli R, Mosele F, Verret B, Bachelot
T, Bedard PL, Cortes J, Hyman DM, Juric D, Krop I, Bieche I, et al:
Genomic alterations in breast cancer: Level of evidence for
actionability according to ESMO scale for clinical actionability of
molecular targets (ESCAT). Ann Oncol. 30:365–373. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Bianchini G, Balko JM, Mayer IA, Sanders
ME and Gianni L: Triple-negative breast cancer: Challenges and
opportunities of a heterogeneous disease. Nat Rev Clin Oncol.
13:674–690. 2016.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Masci D, Naro C, Puxeddu M, Urbani A,
Sette C, La Regina G and Silvestri R: Recent advances in drug
discovery for triple-negative breast cancer treatment. Molecules.
28(7513)2023.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Baden J, Zhao C, Pratt J, Kirov S, Pant A,
Seminara A, Green G, Bilke S, Deras I, Fabrizio DA and Pawlowski T:
90PD-Comparison of platforms for determining tumour mutational
burden (TMB) in patients with non-small cell lung cancer (NSCLC).
Ann Oncol. 30 (Suppl 5)(v25)2019.
|
|
16
|
Wei B, Kang J, Kibukawa M, Arreaza G,
Maguire M, Chen L, Qiu P, Lang L, Aurora-Garg D, Cristescu R and
Levitan D: Evaluation of the TruSight oncology 500 assay for
routine clinical testing of tumor mutational burden and clinical
utility for predicting response to pembrolizumab. J Mol Diagn.
24:600–608. 2022.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Liu CY, Huang CC, Tsai YF, Chao TC, Lien
PJ, Lin YS, Feng CJ, Chen JL, Chen YJ, Chiu JH, et al: VGH-TAYLOR:
Comprehensive precision medicine study protocol on the
heterogeneity of Taiwanese breast cancer patients. Future Oncol:
Oct 19, 2021 (Epub ahead of print).
|
|
18
|
Huang CC, Tsai YF, Liu CY, Chao TC, Lien
PJ, Lin YS, Feng CJ, Chiu JH, Hsu CY and Tseng LM: Comprehensive
molecular profiling of Taiwanese breast cancers revealed potential
therapeutic targets: prevalence of actionable mutations among 380
targeted sequencing analyses. BMC Cancer. 21(199)2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Huang CC, Tsai YF, Liu CY, Lien PJ, Lin
YS, Chao TC, Feng CJ, Chen YJ, Lai JI, Phan NN, et al: Prevalence
of tumor genomic alterations in homologous recombination repair
genes among taiwanese breast cancers. Ann Surg Oncol. 29:3578–3590.
2022.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Huang CC, Tsai YF, Liu CY, Lien PJ, Lin
YS, Chao TC, Feng CJ, Chen YJ, Lai JI, Cheng HF, et al: Concordance
of targeted sequencing from circulating tumor DNA and paired tumor
tissue for early breast cancer. Cancers (Basel).
15(4475)2023.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Chao TC, Tsai YF, Liu CY, Lien PJ, Lin YS,
Feng CJ, Chen YJ, Lai JI, Hsu CY, Lynn JJ, et al: Prevalence of
PIK3CA mutations in Taiwanese patients with breast cancer: A
retrospective next-generation sequencing database analysis. Front
Oncol. 13(1192946)2023.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Gradishar WJ, Moran MS, Abraham J,
Abramson V, Aft R, Agnese D, Allison KH, Anderson B, Burstein HJ,
Chew H, et al: NCCN guidelines® insights: Breast cancer,
version 4.2023. J Natl Compr Canc Netw. 21:594–608. 2023.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Gennari A, André F, Barrios CH, Cortés J,
de Azambuja E, DeMichele A, Dent R, Fenlon D, Gligorov J, Hurvitz
SA, et al: ESMO clinical practice guideline for the diagnosis,
staging and treatment of patients with metastatic breast cancer.
Ann Oncol. 32:1475–1495. 2021.PubMed/NCBI View Article : Google Scholar
|
|
24
|
No authors listed. Pathologists' guideline
recommendations for immunohistochemical testing of estrogen and
progesterone receptors in breast cancer. Breast Care (Basel).
5:185–187. 2010.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Wolff AC, Somerfield MR, Dowsett M,
Hammond MEH, Hayes DF, McShane LM, Saphner TJ, Spears PA and
Allison KH: Human epidermal growth factor receptor 2 testing in
breast cancer: ASCO-college of american pathologists guideline
update. J Clin Oncol. 41:3867–3872. 2023.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Schirmer M, Ijaz UZ, D'Amore R, Hall N,
Sloan WT and Quince C: Insight into biases and sequencing errors
for amplicon sequencing with the Illumina MiSeq platform. Nucleic
Acids Res. 43(e37)2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Li MM, Datto M, Duncavage EJ, Kulkarni S,
Lindeman NI, Roy S, Tsimberidou AM, Vnencak-Jones CL, Wolff DJ,
Younes A and Nikiforova MN: Standards and guidelines for the
interpretation and reporting of sequence variants in cancer: A
joint consensus recommendation of the association for molecular
pathology, American society of clinical oncology, and college of
American pathologists. J Mol Diagn. 19:4–23. 2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Gao J, Aksoy BA, Dogrusoz U, Dresdner G,
Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al:
Integrative analysis of complex cancer genomics and clinical
profiles using the cBioPortal. Sci Signal. 6(pl1)2013.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Cerami E, Gao J, Dogrusoz U, Gross BE,
Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et
al: The cBio cancer genomics portal: An open platform for exploring
multidimensional cancer genomics data. Cancer Discov. 2:401–404.
2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Chakravarty D, Gao J, Phillips SM, Kundra
R, Zhang H, Wang J, Rudolph JE, Yaeger R, Soumerai T, Nissan MH, et
al: OncoKB: A precision oncology knowledge base. JCO Precis Oncol.
2017(PO.17.00011)2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Phan L, Zhang H, Wang Q, Villamarin R,
Hefferon T, Ramanathan A and Kattman B: The evolution of dbSNP: 25
Years of impact in genomic research. Nucleic Acids Res.
53:D925–D931. 2025.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Chen S, Francioli LC, Goodrich JK, Collins
RL, Kanai M, Wang Q, Alföldi J, Watts NA, Vittal C, Gauthier LD, et
al: A genomic mutational constraint map using variation in 76,156
human genomes. Nature. 625:92–100. 2024.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Karczewski KJ, Weisburd B, Thomas B,
Solomonson M, Ruderfer DM, Kavanagh D, Hamamsy T, Lek M, Samocha
KE, Cummings BB, et al: The ExAC browser: Displaying reference data
information from over 60 000 exomes. Nucleic Acids Res.
45:D840–D845. 2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Robinson JT, Thorvaldsdottir H, Turner D
and Mesirov JP: igv.js: An embeddable JavaScript implementation of
the Integrative Genomics Viewer (IGV). Bioinformatics.
39(btac830)2023.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Wolff L and Kiesewetter B: Applicability
of ESMO-MCBS and ESCAT for molecular tumor boards. Memo.
15:190–195. 2022.
|
|
36
|
Huang CC, Yeh YC, Cheng HF, Chen BF, Liu
CY, Tsai YF, Ho HL and Tseng LM: Comprehensive genomic profiling of
Taiwanese triple-negative breast cancer with a large targeted
sequencing panel. J Chin Med Assoc: Jun 20, 2025 (Epub ahead of
print).
|
|
37
|
Loderer D, Hornáková A, Tobiášová K,
Lešková K, Halašová E, Danková Z, Biringer K, Kúdela E, Rokos T,
Dzian A, et al: Comparison of next-generation sequencing quality
metrics and concordance in the detection of cancer-specific
molecular alterations between formalin-fixed paraffin-embedded and
fresh-frozen samples in comprehensive genomic profiling with the
Illumina® TruSight oncology 500 assay. Exp Ther Med.
29(64)2025.
|
|
38
|
Carpten JD, Faber AL, Horn C, Donoho GP,
Briggs SL, Robbins CM, Hostetter G, Boguslawski S, Moses TY, Savage
S, et al: A transforming mutation in the pleckstrin homology domain
of AKT1 in cancer. Nature. 448:439–444. 2007.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Kalinsky K, Hong F, McCourt CK, Sachdev
JC, Mitchell EP, Zwiebel JA, Doyle LA, McShane LM, Li S, Gray RJ,
et al: Effect of capivasertib in patients with an AKT1 E17K-mutated
tumor: NCI-MATCH subprotocol EAY131-Y nonrandomized trial. JAMA
Oncol. 7:271–278. 2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Turner NC, Oliveira M, Howell SJ, Dalenc
F, Cortes J, Gomez Moreno HL, Hu X, Jhaveri K, Krivorotko P, Loibl
S, et al: Capivasertib in hormone receptor-positive advanced breast
cancer. N Engl J Med. 388:2058–2070. 2023.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Daly GR, AlRawashdeh MM, McGrath J,
Dowling GP, Cox L, Naidoo S, Vareslija D, Hill ADK and Young L:
PARP inhibitors in breast cancer: A short communication. Curr Oncol
Rep. 26:103–113. 2024.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Szabo C, Masiello A, Ryan JF and Brody LC:
The breast cancer information core: Database design, structure, and
scope. Hum Mutat. 16:123–131. 2000.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Chenevix-Trench G, Milne RL, Antoniou AC,
Couch FJ, Easton DF and Goldgar DE: CIMBA. An international
initiative to identify genetic modifiers of cancer risk in BRCA1
and BRCA2 mutation carriers: The consortium of investigators of
modifiers of BRCA1 and BRCA2 (CIMBA). Breast Cancer Res.
9(104)2007.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Spain BH, Larson CJ, Shihabuddin LS, Gage
FH and Verma IM: Truncated BRCA2 is cytoplasmic: Implications for
cancer-linked mutations. Proc Natl Acad Sci USA. 96:13920–13925.
1999.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Tung NM, Robson ME, Ventz S, Santa-Maria
CA, Nanda R, Marcom PK, Shah PD, Ballinger TJ, Yang ES, Vinayak S,
et al: TBCRC 048: Phase II study of olaparib for metastatic breast
cancer and mutations in homologous recombination-related genes. J
Clin Oncol. 38:4274–4282. 2020.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Cheng HF, Tsai YF, Liu CY, Hsu CY, Lien
PJ, Lin YS, Chao TC, Lai JI, Feng CJ, Chen YJ, et al: Prevalence of
BRCA1, BRCA2, and PALB2 genomic alterations among 924 Taiwanese
breast cancer assays with tumor-only targeted sequencing: Extended
data analysis from the VGH-TAYLOR study. Breast Cancer Res.
25(152)2023.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Hyman DM, Piha-Paul SA, Won H, Rodon J,
Saura C, Shapiro GI, Juric D, Quinn DI, Moreno V, Doger B, et al:
HER kinase inhibition in patients with HER2- and HER3-mutant
cancers. Nature. 554:189–194. 2018.PubMed/NCBI View Article : Google Scholar
|
|
48
|
André F, Ciruelos EM, Juric D, Loibl S,
Campone M, Mayer IA, Rubovszky G, Yamashita T, Kaufman B, Lu YS, et
al: Alpelisib plus fulvestrant for PIK3CA-mutated, hormone
receptor-positive, human epidermal growth factor
receptor-2-negative advanced breast cancer: Final overall survival
results from SOLAR-1. Ann Oncol. 32:208–217. 2021.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Choudhury AD, Higano CS, de Bono JS, Cook
N, Rathkopf DE, Wisinski KB, Martin-Liberal J, Linch M, Heath EI,
Baird RD, et al: A phase I study investigating AZD8186, a potent
and selective inhibitor of PI3Kβ/δ, in patients with advanced solid
tumors. Clin Cancer Res. 28:2257–2269. 2022.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Chen HJ, Romigh T, Sesock K and Eng C:
Characterization of cryptic splicing in germline PTEN intronic
variants in Cowden syndrome. Hum Mutat. 38:1372–1377.
2017.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Mester JL, Ghosh R, Pesaran T, Huether R,
Karam R, Hruska KS, Costa HA, Lachlan K, Ngeow J, Barnholtz-Sloan
J, et al: Gene-specific criteria for PTEN variant curation:
Recommendations from the ClinGen PTEN expert panel. Hum Mutat.
39:1581–1592. 2018.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Nykamp K, Anderson M, Powers M, Garcia J,
Herrera B, Ho YY, Kobayashi Y, Patil N, Thusberg J, Westbrook M, et
al: Sherloc: A comprehensive refinement of the ACMG-AMP variant
classification criteria. Genet Med. 19:1105–1117. 2017.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Garrido-Navas MC, García-Díaz A,
Molina-Vallejo MP, González-Martínez C, Alcaide Lucena M,
Cañas-García I, Bayarri C, Delgado JR, González E, Lorente JA and
Serrano MJ: The polemic diagnostic role of TP53 mutations in liquid
biopsies from breast, colon and lung cancers. Cancers (Basel).
12(3343)2020.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Zhou W, Chen T, Chong Z, Rohrdanz MA,
Melott JM, Wakefield C, Zeng J, Weinstein JN, Meric-Bernstam F,
Mills GB and Chen K: TransVar: A multilevel variant annotator for
precision genomics. Nat Methods. 12:1002–1003. 2015.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Mosteiro M, Azuara D, Villatoro S, Alay A,
Gausachs M, Varela M, Baixeras N, Pijuan L, Ajenjo-Bauza M,
Lopez-Doriga A, et al: Molecular profiling and feasibility using a
comprehensive hybrid capture panel on a consecutive series of
non-small-cell lung cancer patients from a single centre. ESMO
Open. 8(102197)2023.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Fox EJ, Reid-Bayliss KS, Emond MJ and Loeb
LA: Accuracy of Next generation sequencing platforms. Next Gener
Seq Appl. 1(1000106)2014.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Song P, Chen SX, Yan YH, Pinto A, Cheng
LY, Dai P, Patel AA and Zhang DY: Selective multiplexed enrichment
for the detection and quantitation of low-fraction DNA variants via
low-depth sequencing. Nat Biomed Eng. 5:690–701. 2021.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Apostoli AJ and Ailles L: Clonal evolution
and tumor-initiating cells: New dimensions in cancer patient
treatment. Crit Rev Clin Lab Sci. 53:40–51. 2016.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Edsjö A, Gisselsson D, Staaf J, Holmquist
L, Fioretos T, Cavelier L and Rosenquist R: Current and emerging
sequencing-based tools for precision cancer medicine. Mol Aspects
Med. 96(101250)2024.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Cocco E, Scaltriti M and Drilon A: NTRK
fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin
Oncol. 15:731–747. 2018.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Mosele MF, Westphalen CB, Stenzinger A,
Barlesi F, Bayle A, Bièche I, Bonastre J, Castro E, Dienstmann R,
Krämer A, et al: Recommendations for the use of next-generation
sequencing (NGS) for patients with advanced cancer in 2024: A
report from the ESMO precision medicine working group. Ann Oncol.
35:588–606. 2024.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Gradishar WJ, Moran MS, Abraham J,
Abramson V, Aft R, Agnese D, Allison KH, Anderson B, Bailey J,
Burstein HJ, et al: Breast cancer, version 3.2024, NCCN clinical
practice guidelines in oncology. J Natl Compr Canc Netw.
22:331–357. 2024.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Kang J, Na K, Kang H, Cho U, Kwon SY,
Hwang S and Lee A: Prediction of homologous recombination
deficiency from oncomine comprehensive assay plus correlating with
SOPHiA DDM HRD solution. PLoS One. 19(e0298128)2024.PubMed/NCBI View Article : Google Scholar
|