Introduction
The diagnosis of central nervous system (CNS)
tumours has evolved significantly over the past decade, moving
beyond traditional clinical and histopathological assessments to an
integrated, multi-dimensional approach that incorporates molecular
diagnostics (1). The 2021 WHO
classification of CNS tumours introduced a paradigm shift by
emphasizing the need for a molecularly informed diagnosis that
combines histopathological findings with specific genetic,
epigenetic, and protein expression profiles (2,3). This
integrated diagnosis framework allows for more accurate tumour
classification, as numerous CNS tumours exhibit overlapping
histological features but distinct molecular signatures that
directly impact prognosis and therapeutic decisions.
Immunohistochemistry (IHC) remains a cornerstone for
initial tumour characterization, providing insights into cellular
lineage and differentiation. However, it often falls short in cases
with ambiguous morphology or treatment-induced changes.
Next-generation sequencing (NGS) technologies, such as targeted
gene panels, whole-exome sequencing, and methylation profiling, are
increasingly essential to identify diagnostic, prognostic, and
therapeutic markers. For example, the detection of specific
mutations (such as IDH1/2 in gliomas, BRAF V600E in gangliogliomas
or pleomorphic xanthoastrocytomas), chromosomal alterations (such
as 1p/19q co-deletion in oligodendrogliomas), or methylation
patterns [such as O6-methylguanine-DNA
methyltransferase (MGMT) promoter methylation or genome-wide
methylation classifiers] is now pivotal in classifying CNS tumours
(2). This molecular classification
not only refines diagnostic accuracy but also provides critical
information on tumour behaviour, treatment response, and patient
outcomes, underscoring the necessity for a comprehensive,
integrated diagnostic approach in modern neuropathology (4).
The accurate diagnosis of CNS tumours is the most
challenging for cases that have been previously treated. Tumours
subjected to pre-treatment modalities such as radiation or
chemotherapy frequently undergo morphological and molecular changes
that can obscure their histopathological features. This phenomenon,
known as treatment-related distortion, complicates subsequent
diagnostic efforts, making it difficult to discern between tumour
recurrence, radiation-induced changes, or an entirely new entity
(5). For instance, gliomas, such as
high-grade gliomas, often exhibit post-radiation changes including
nuclear and cytoplasmic enlargement, which may also mimic
pseudo-malignant radiation-associated changes (6). Similarly, meningiomas post-irradiation
may exhibit increased cellular atypia, loss of classic whorl
patterns, and elevated Ki-67 indices reflecting either
radiation-induced proliferation or cell death (7). These changes can mimic features of
higher-grade malignancies, leading to diagnostic uncertainty when
relying solely on histopathology and IHC.
Post-treatment CNS tumours, especially those
subjected to high-dose radiation, pose a significant diagnostic
dilemma due to treatment-related changes. Recurrent CNS tumours
exhibit histopathological features altered by prior therapies,
complicating differentiation between tumour recurrence,
treatment-induced necrosis, or secondary malignancies (8-10)
For instance, a 2023 study by Wood et al (5) highlighted that paediatric high-grade
astrocytoma post-treatment showed increased tumour mutation burden
and morphological ambiguity in a subset of recurrences,
underscoring the prevalence of diagnostic challenges (5). Similarly, radiation-induced changes,
such as fibrosis, vascular alterations, and cellular atypia, can
mimic aggressive tumour features, leading to misclassification
rates as high as 20% when relying solely on histopathology and IHC
(11). IHC, while valuable for
initial tumour characterization, often yields inconclusive or
conflicting results in these cases due to altered protein
expression profiles induced by therapy.
Recent advances in epigenomic profiling, such as
whole genome methylation profiling, have emerged as potent tools
for resolving such diagnostic dilemmas (12,13).
Unlike protein expression or histopathological features, which are
highly susceptible to treatment-induced alterations, DNA
methylation patterns exhibit high stability, even in the presence
of morphological distortions. This stability arises because DNA
methylation, an epigenetic modification, is regulated by DNA
methyltransferases and remains relatively unaffected by the
cellular stress or protein turnover induced by therapies such as
radiation, whereas downstream protein expression is disrupted by
changes in transcription, translation, or protein degradation
pathways (14). For example,
radiation may upregulate stress-response genes or silence tumour
suppressor genes via histone modifications, altering IHC-detectable
proteins such as p53 or mucin 1, cell surface associated (EMA),
while the underlying CpG methylation patterns at promoter regions
remain conserved (15). This
phenomenon has been reported in brain malignancies; a study on
recurrent glioblastomas demonstrated that methylation profiles of
MGMT were preserved despite chemoradiation (16). This was likely because promoter
methylation is relatively stable, and does not require active
maintenance of methylation status by DNA methyltransferases (DNMTs)
(17).
Methylation profiling leverages this conserved
epigenetic signature to provide robust molecular classification,
offering a distinct advantage over existing methodologies.
Methylation profiling can provide a rigorous molecular
classification that remains largely unaffected by histopathological
distortions. In the present study, a case of a patient with a
heavily irradiated CNS tumour that exhibited perplexing
histological features, making definitive diagnosis difficult, is
reported. Despite extensive immunohistochemical analysis, the
tumour remained unclassifiable. To address this diagnostic impasse,
low-coverage whole genome methylation profiling, a technique that
has shown promise in accurately predicting the molecular subtype of
various cancers, even in the presence of histological ambiguity,
was employed.
Case report
Patient information
A clinical sample was obtained from a patient
48-year-old female who initially presented with a parasellar mass,
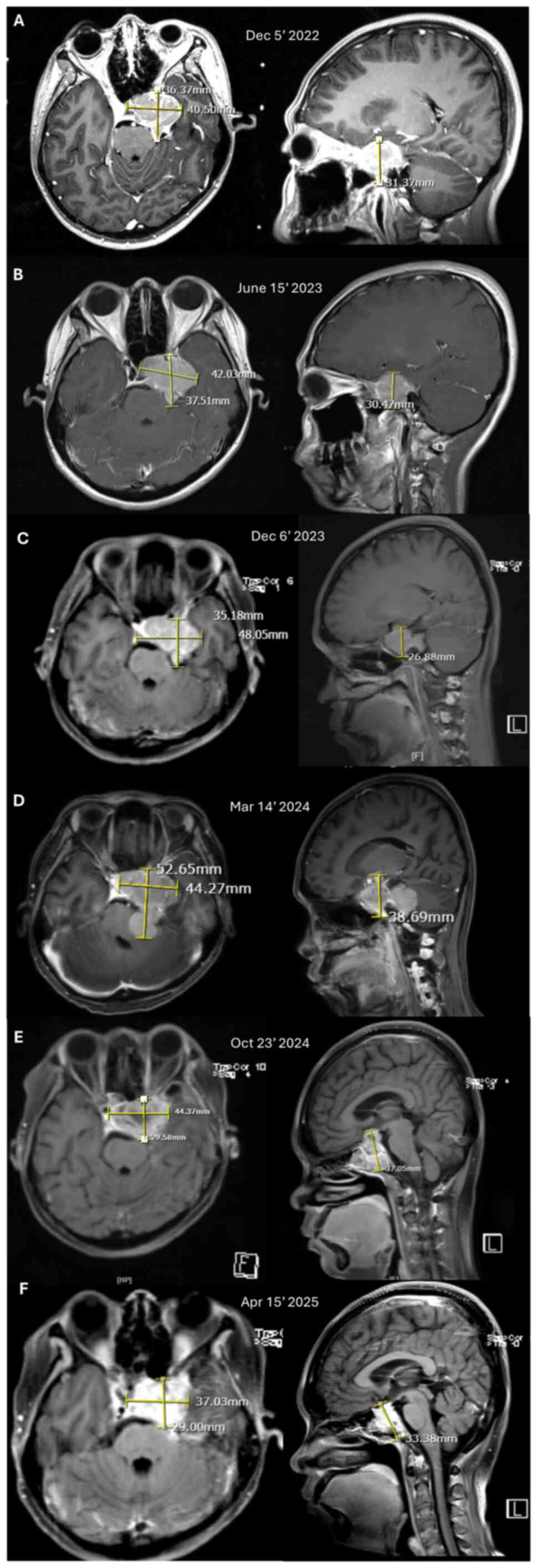
clinically resembling an extra-axial tumour. An initial MRI
performed in December 2022 revealed a 3.6x4.3x3.1 cm tumour in left
parasellar region, raising suspicion for meningioma or
craniopharyngioma (Fig. 1A). The
patient initially declined surgical intervention and was instead
treated with Gamma Knife stereotactic radiosurgery, receiving a
dose of 14 Gy in December 2022. Subsequent MRI follow-ups were
conducted every six months. The tumour remained stable in size for
up to one year post-radiation, as shown in MRI scans from June 2023
and December 2023 (Fig. 1B and
C, respectively). After ~1 year and
3 months post-radiation treatment, a brain MRI performed in March
2024 revealed a progressive tumour at the same location extending
to the left cerebellopontine angle and abutting the brainstem
(Fig. 1D). Surgery was offered and
the patient agreed. A subtotal resection was performed in March
2024; however the surgeon was unable to achieve gross total
resection due to the tumour encasing the cavernous sinus. At the
follow-up 6 months after the surgery, the patient did not exhibit
any significant symptoms, and an MRI scan performed in October
2024, revealed stable disease (Fig.
1E). An MRI was performed again 6 months later in April 2025
and revealed a slight reduction of tumour size (Fig. 1F). Since there was no progression of
disease, no further intervention was undertaken.
A tissue sample was obtained during craniotomy
performed in in March 2024 from the left retroclival region.
Histopathological analysis and further immunohistochemical
assessment performed in April 2024 were both inconclusive;
therefore, further molecular examination was performed. Ethical
clearance was obtained from the Ethics Committee of the Faculty of
Medicine, University of Indonesia (Jakarta, Indonesia) in
accordance with the Declaration of Helsinki, for the collection and
molecular testing of biological specimens from rare and recurrent
tumours at any site. Written informed consent was obtained from the
patient prior to molecular testing, ensuring full disclosure of the
purpose, procedures and potential use of biological specimens for
research in the present study. The specimen was retrieved from the
Department of Pathology of Cipto Mangunkusumo National General
Hospital (Jakarta, Indonesia) in the form of formalin-fixed
paraffin-embedded (FFPE) tissue. DNA extraction was performed on
the FFPE tissue sample, followed by NGS sequencing using a third
generation sequencing platform, and the methylation profile was
analysed. The DNA extraction and NGS were performed in August 2024.
The molecular information was then matched with available clinical,
imaging, histopathology and immunohistochemcial results.
Tissue processing and IHC
The tumour tissue sample was initially fixed in 10%
neutral buffered formalin for 24 at 4˚C to preserve cellular
structure and protein antigenicity, followed by dehydration and
embedding in paraffin wax. Once embedded, the tissue was sectioned
into thin slices with a thickness of 4-5 µm using a microtome, and
was then mounted onto positively charged glass slides to ensure
optimal adherence. The slides were baked at 60˚C to solidify tissue
attachment before further processing. The next step was
deparaffinization and rehydration. Tissue sections were
deparaffinized by immersing them in xylene, followed by rehydration
through a graded series of ethanol solutions of decreasing
concentrations (100, 95 and 70%) and finally in distilled water. To
expose the antigenic sites, antigen retrieval was performed using a
heat-induced epitope retrieval (HIER) technique in an EDTA buffer
(pH 9.0). This step resulted in unmasking the target epitopes and
enhancing antibody binding.
Following antigen retrieval, non-specific binding
was minimized by incubating the slides with a blocking solution,
10% bovine serum albumin (cat. no. 37520; Thermo Fisher Scientific,
Inc.) for 30 min at room temperature. After blocking, primary
antibodies (Table I) specific to
each target protein marker were applied at their optimized
dilutions following each antibody kit manufacturer guideline. The
slides were then incubated in a humid chamber for 2 h at room
temperature. This step enabled the primary antibodies to bind
specifically to their corresponding antigens in the tissue
sections. The detection of bound primary antibodies was carried out
using an appropriate secondary antibody conjugated to a biotin
enzyme, horseradish peroxidase (HRP) (mouse anti-human IgG4 Fc,
secondary antibody, HRP; 1:1,000; cat. no. A-10654; Thermo Fisher
Scientific, Inc.) for 1 h at room temperature. A chromogenic
substrate, 3,3'-diaminobenzidine (DAB), was used to develop a brown
colour, indicating the presence of the target antigen.
Counterstaining with hematoxylin was performed for 5 min at room
temperature to provide contrast by staining cell nuclei blue,
aiding in the morphological assessment of the tissue.
 | Table IAntibodies for immunohistochemical
staining. |
Table I
Antibodies for immunohistochemical
staining.
| Immunohistochemical
marker | Supplier | Cat. no. | Dilution |
|---|
| Vimentin | Leica
Biosystems | PA0640 | RTU |
| AE1/3 | Leica
Biosystems | PA0909 | RTU |
| EMA | Leica
Biosystems | NCL-L-EMA | 1:400 |
| p63 | Biocare Medical,
LLC | CM163C | 1:50 |
| S100 | Biocare Medical,
LLC | ACR3237C | 1:200 |
| Synaptophysin | Biocare Medical,
LLC | CM371C | 1:200 |
| Chromogranin | Cell Marque; Merck
KGaA | 238M-96 | 1:500 |
| PR | Roche
Diagnostics | 5277990001 | RTU |
| CD34 | BioSB, Inc. | BSB5230 | 1:100 |
| Ki-67 | Roche
Diagnostics | 5278384001 | RTU |
| p40 | BioSB, Inc. | BSB2075 | 1:200 |
| CK7 | BioSB, Inc. | BSB5412 | 1:600 |
| CK8 | BioSB, Inc. | BSB6665 | 1:100 |
| CK19 | Biocare Medical,
LLC | CM242C | 1:100 |
| HMWCK | BioSB, Inc. | BSB5398 | 1:50 |
| Cam5.2 | Cell Marque; Merck
KGaA | 452M-96 | 1:100 |
| CK20 | Biocare Medical,
LLC | CM062C | 1:200 |
| ER | Roche
Diagnostics | 5278406001 | RTU |
| STAT6 | Biocare Medical,
LLC | ACI3244C | 1:100 |
| TTF-1 | Leica
Biosystems | PA0364 | RTU |
| BRAF V600E | BioSB, Inc. | BSB2824 | 1:250 |
| Beta catenin | Biocare Medical,
LLC | CM406C | 1:200 |
| TLE-1 | Cell Marque; Merck
KGaA | 401M-16 | 1:100 |
| BCL2 | Leica
Biosystems | PA0117 | RTU |
| CD99 | Cell Marque; Merck
KGaA | 199R-16 | 1:100 |
| CD56 | Leica
Biosystems | NCL-L-CD56-504 | 1:100 |
After staining, the slides underwent a series of
dehydration steps using graded alcohols, followed by clearing in
xylene, and were finally mounted with a permanent mounting medium.
The prepared slides were then examined under a light microscope by
a senior neuropathologist to evaluate the expression patterns of
the specific markers. The staining intensity and proportion of
positive cells were recorded for each marker. The
immunohistochemical markers examined were Vimentin, AE1/3, EMA,
p63, S100, PR, CD34, Ki-67, p40, CK7, CK8, HMWCK, Cam5.2, CK20, ER,
STAT6, TTF-1, BRAF V600E, β-catenin, TLE-1, BCL2, CD99, and
CD56.
DNA extraction
The DNA extraction of the FFPE tissue was carried
out using the QIAamp DNA FFPE Tissue Kit (cat. no. 56404; QIAGEN)
which involved several key steps. Initially, the FFPE tissue sample
was deparaffinized using a xylene and ethanol wash to remove any
residual paraffin. Then the tissue was prepared for next process,
lysis. This lysis step involved breakdown of the cross-linked
proteins in the FFPE samples. The sample underwent incubation with
a lysis buffer and proteinase K (both included in the
aforementioned kit) to digest the tissue and release the DNA into
the solution.
Once lysis was completed, the sample was treated
with a heat incubation step up to 90˚C to reverse formalin-induced
crosslinking, which helped to ensure optimal DNA recovery and
purity. The lysate was then mixed with ethanol and passed through a
QIAamp MinElute column, which selectively bonded the DNA. Following
several washes to remove contaminants, the purified DNA was eluted
in a low-salt buffer, ready for downstream sequencing applications.
The authors adhered strictly to the manufacturer-recommended
protocol for extracting FFPE tissue. The complete and detailed
protocol was available publicly in the corresponding kit
documentation online. The extracted DNA was measured for purity
using Nanodrop Spectrophotometer and the quantity was measured
using Qubit Fluorometers (both from Thermo Fisher Scientific,
Inc.).
NGS sequencing
The extracted DNA was sequenced using a third
generation sequencer from (Oxford Nanopore Technologies plc). A
ligation-based protocol was used to prepare the library DNA
[Ligation sequencing DNA V14 (SQK-LSK114); Oxford Nanopore
Technologies plc]. There was no PCR step involved to preserve the
native methylation information within the DNA. The type of
sequencing was direct DNA sequencing with modified bases for
methylation detection. The DNA was sequenced using PromethION flow
cell (cat. no. FLO-PRO114M; Oxford Nanopore Technologies plc) with
chemistry version R 10.4.1. The library loading concentration was
20 fmol and the software used for analysis (basecalling and
alignment) was MinKNOW Version 25.02 (Oxford Nanopore Technologies
plc). The detailed library preparation and sequencing steps were
carried out according to the publicly available Nanopore protocol
on the Nanopore Community webpage. The present study aimed to
obtain low coverage whole genome sequencing, and thus the
sequencing was carried out for 3 h until the raw data in POD5
format reached ~50 GB. The sequencing was then interrupted and
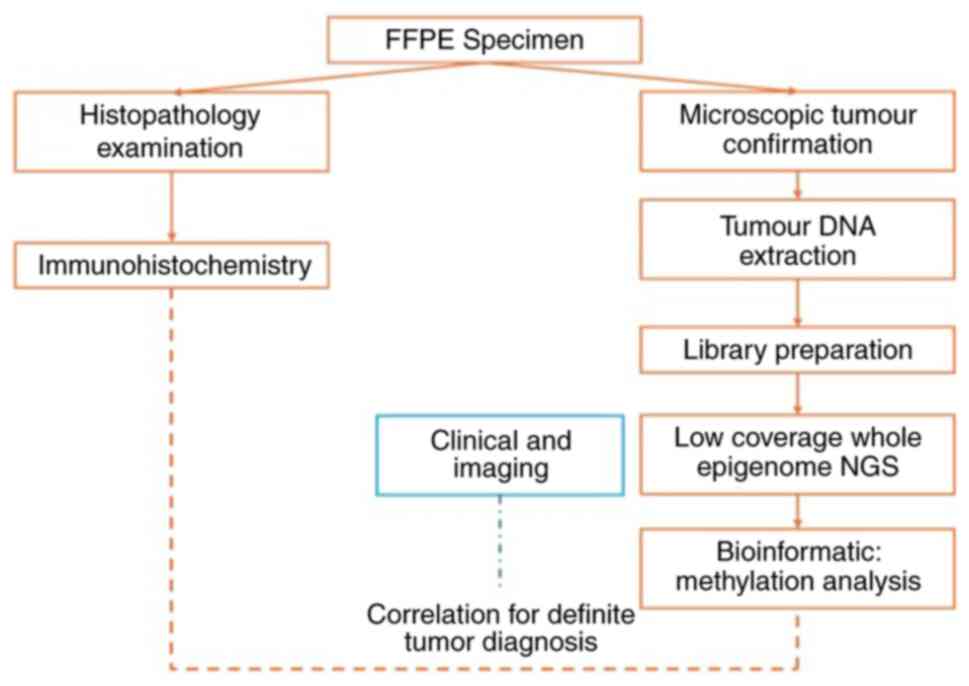
stopped. The overview process for molecular testing, from IHC and
methylation testing to correlation with clinical and imaging data,
is illustrated in Fig. 2.
Methylation analysis
After sequencing was completed, post-run basecalling
was performed using the dna_r10.4.1_e8.2_400bps_sup@v5.0.0
model, which included 5mCG and 5hmCG base modification calling with
Dorado (18). The methylation
analysis was then carried out using NanoDx pipeline, which is
publicly available (12,19). Briefly, the basecalled sequencing
reads were aligned to the hg19 human reference genome using
Minimap2(20). Methylation
information was then extracted using Modkit version 0.4.0
(https://github.com/nanoporetech/modkit), which
provides single-base resolution of methylation data. The
methylation matrix was then constructed representing all CpG sites.
Furthermore, dimensionality reduction using t-SNE was applied to
visualise and analyse the high-dimensional methylation data. The
methylation data was classified using the public Heidelberg brain
tumour classifier v11b4 reference set (21).
Clinical findings
The histopathology results revealed a tumour mass
that under microscopic examination, was arranged in irregular short
lines, forming sheets, lobules and a patternless architecture
(Fig. 3A). The tumour cells
exhibited round to oval nuclei with spindle shapes, mild to
moderate pleomorphism, slightly coarse chromatin, and eosinophilic
cytoplasm. The mitotic count was 6 per 10 low-power fields. Blood
vessels appeared partially congested. There was dense infiltration
of macrophages or foamy cells around the tumour. This
histopathological feature was inconclusive, with the histology
suggesting the possibility of meningioma, solitary fibrous tumour,
pituitary adenoma or carcinoma.
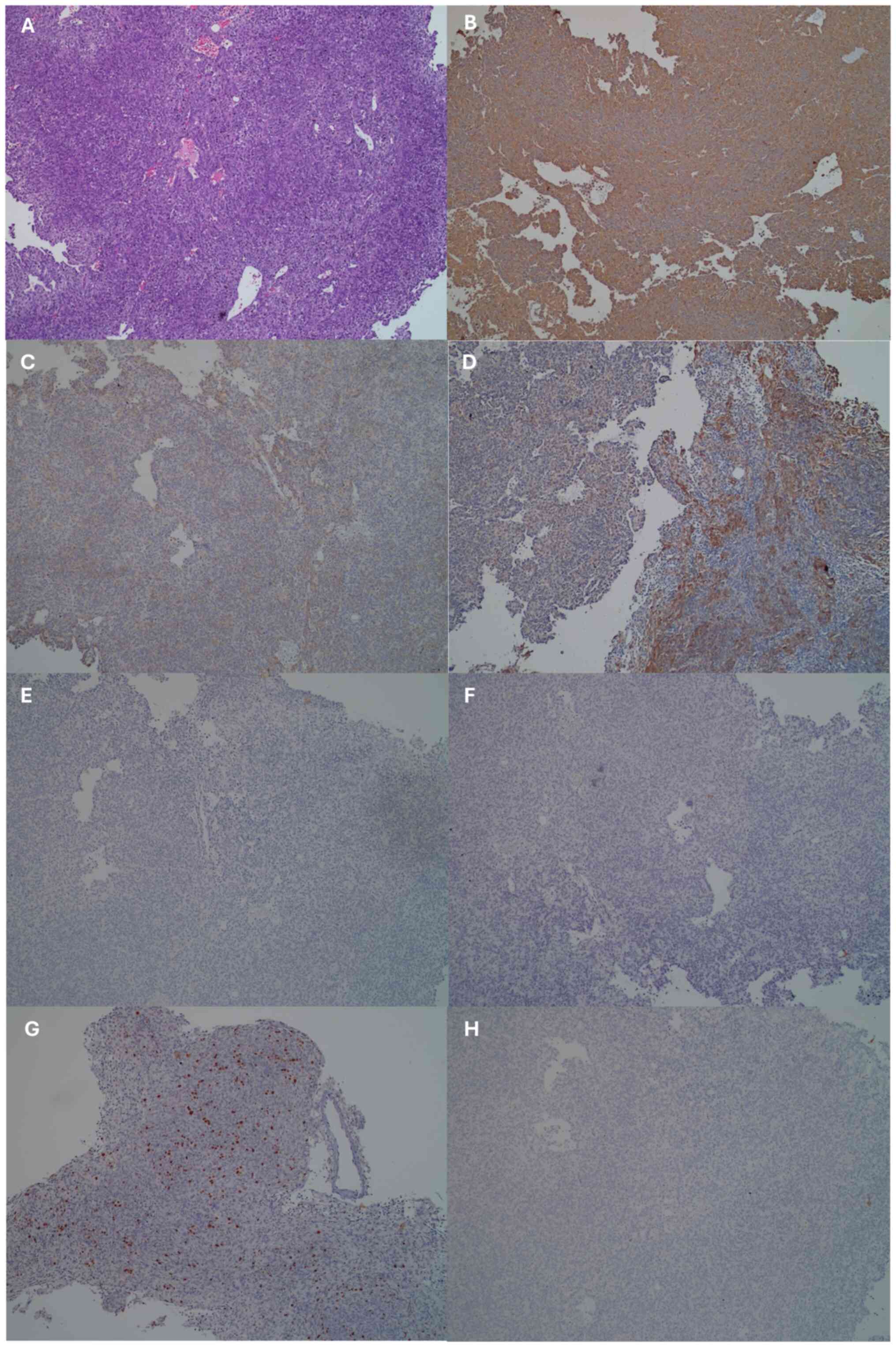 | Figure 3Histopathology and
immunohistochemical findings. (A) Haematoxylin and eosin staining.
(B-H) Immunohistochemical staining of the following markers: (B)
Vimentin, (C) EMA, (D) AE1/3, (E) S100, (F) PR, (G) Ki-67 and (H)
synaptophysin. All images were obtained at a magnification of x100.
EMA, mucin 1, cell surface associated. EMA, mucin 1, cell surface
associated; AE1/3, pan-cytokeratin antibody cocktail; S100,
calcium-binding proteins; PR, progesterone receptor. |
IHC was performed and the results obtained from all
the tested markers are summarized in Table II, Fig.
3B-H, and Fig. S1. The Ki-67
index of up to 20% was indicative of moderately high proliferative
activity, suggesting a potentially aggressive tumour. Positive BRAF
V600E expression suggested the presence of this mutation in the
tumour, which was also indicative of a more aggressive tumour and
poorer prognosis. The findings of IHC indicated partial epithelial
and mesenchymal marker expression (Vimentin, AE1/3 and EMA) and
BRAF V600E positivity, along with negative markers for neural and
neuroendocrine differentiation, suggesting that the tumour may be a
type of mixed epithelial-mesenchymal tumour or a variant of glioma
or meningioma with complex differentiation. The neuropathologist
concluded that this was more likely a craniopharyngioma. The
ambiguous morphological findings on the histopathology and atypical
expression were likely due to prior high-dose stereotactic
radiotherapy to the tumour. Based on the histopathology and
immunohistochemical results alone, a definitive classification
could not be made; however, the most likely diagnosis was
craniopharyngioma.
| Table IIResults of immunohistochemical
analysis. |
Table II
Results of immunohistochemical
analysis.
| Immunohistochemical
marker | Result |
|---|
| Vimentin | Expressed in most
tumour cells |
| AE1/3 | Positive in some
tumour cells |
| EMA | Positive in some
tumour cells |
| p63 | Positive in some
tumour cells |
| S100 | Negative |
| Synaptophysin | Negative |
| Chromogranin | Negative |
| PR | Negative |
| CD34 | Only expressed in
vessel walls |
| Ki-67 | Increased,
variable, up to 20% in some areas |
| p40 | Positive in some
tumour cells |
| CK7 | Positive in some
tumour cells |
| CK8 | Positive in some
tumour cells |
| CK19 | Positive in some
tumour cells |
| HMWCK | Positive in some
tumour cells |
| Cam5.2 | Positive in some
tumour cells |
| CK20 | Negative |
| ER | Negative |
| STAT6 | Expressed in tumour
cells (cytoplasmic reactivity, rather than nucleus) |
| TTF-1 | Negative |
| BRAF V600E | Positive in most
tumour cells |
| Beta catenin | Cytoplasmic and
membranous expression in some tumour cells |
| TLE-1 | Positive in some
tumour cells with weak intensity |
| BCL2 | Expressed in some
(few) tumour cells |
| CD99 | Weak cytoplasmic
and membranous expression in some tumour cells |
| CD56 | Positive in a small
number of tumour cells |
Further molecular analysis was performed using NGS
to conduct direct sequencing in order to detect random whole genome
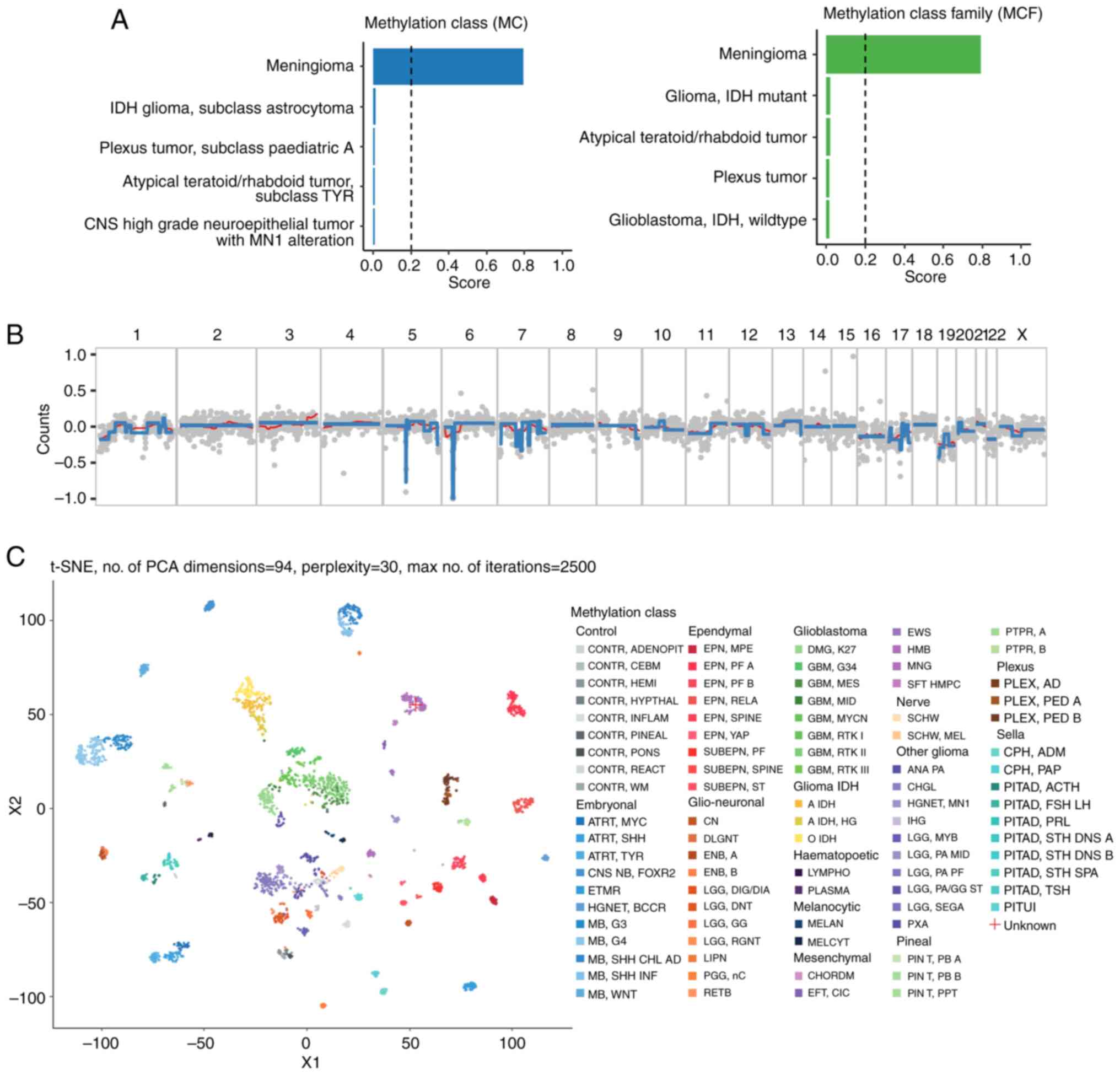
methylation from the tumour DNA. The methylation classification
analysis of the CNS tumour sample identified the tumour as a
meningioma at both the methylation class and methylation class
family levels. The confidence score for this classification was
0.791, which was above the commonly used threshold of 0.2,
indicating a reliable prediction. The Heidelberg classifier,
validated in large cohorts, demonstrates a sensitivity of 92-95%
and specificity of 90-93% for meningioma classification in
untreated cases (22-24),
although its performance in irradiated tumours remains less well
studied. In this context, the 0.791 score suggests reliable
discrimination despite post-treatment complexity. This results
suggested that the epigenetic profile of the tumour was most
consistent with meningioma when compared to the reference profiles
of CNS tumours. Additionally, the classification was supported by
the absence of strong signals for other CNS tumour entities among
the top five categories, which included high-grade neuroepithelial
tumours, atypical teratoid/rhabdoid tumours, and various gliomas
(Fig. 4A).
The copy number variation (CNV) profile, shown in
Fig. 4B, was generated based on
low-coverage whole genome sequencing data. Due to the low coverage,
the resolution of the CNV analysis may be limited, and fine-scale
alterations could be missed. Nonetheless, it provided an overview
of the broader chromosomal changes present in the tumour specimen.
Overall, the CNV plot revealed regions with potential copy number
gains (positive log2 values represented by red bars) and losses
(negative log2 values represented by blue bars) across multiple
chromosomes. The copy number profile showed relative stability
across most chromosomes, with the majority of regions falling
within the-0.5 to 0.5 range on the y-axis, indicating no major
large-scale chromosomal gains or losses.
Chromosome 5 and 6 exhibited large segments of copy
number loss, indicating possible deletions. A slight gain was
visible on the q arm of chromosome 1, extending approximately from
the middle to the end of the chromosome. The 1q gain observed in
the copy number profile was consistent with the classification of a
meningioma, as gain of 1q is a known recurrent alteration in
certain meningioma subtypes (25).
Chromosome 22 had a noticeable deletion, which was consistent with
known chromosomal patterns in meningiomas (26,27).
Despite the lower resolution of this analysis, these broad CNV
changes provide valuable insights into the overall genomic
landscape of the tumour and could serve as a preliminary indicator
for further validation using higher-coverage sequencing
methods.
The dimensionality reduction plot further
corroborated this classification by visually clustering the sample
close to the meningioma reference group, distinct from other tumour
classes such as glioblastomas or embryonal tumours (Fig. 4C). This visualization tool, while
only qualitative, reinforces the classification outcome by showing
that the methylation pattern of the sample overlaps predominantly
with meningioma profiles. Given the relatively high number of CpG
sites used in the classification (11,653 sites), the prediction was
considered robust, as it comprehensively covered relevant loci for
distinguishing CNS tumour types. A final diagnosis was obtained
following multidisciplinary assessment of all available data, and
considered this entity to be a meningioma. Compared with IHC, which
required 5-10 days and yielded inconclusive results, methylation
profiling provided a definitive diagnosis in 4-7 days (from DNA
extraction to classification), with an estimated accuracy of
>90% based on classifier benchmarks (21,23,24).
This quantitative advantage highlights the superiority of
methylation profiling in both speed and diagnostic precision,
particularly in complex cases.
Discussion
The present case report highlights the complexities
encountered in diagnosing CNS tumours post-radiation therapy. The
patient's initial clinical presentation and radiological features
were consistent with a meningioma. However, following Gamma Knife
treatment and subsequent tumour recurrence, the histopathological
and immunohistochemical profiles suggested craniopharyngioma, a
diagnosis with a vastly different therapeutic approach. The
discordance between clinical and pathological findings was likely
attributable to radiation-induced histological alterations
(28).
The immunohistochemical result of the proliferation
marker Ki-67 showed increased expression in some areas, reaching up
to 20%, indicative of a moderate proliferative index, which
suggests an intermediate to high level of tumour aggressiveness.
The positive BRAF V600E expressed in a substantial portion of
tumour cells was extremely rare in meningioma, even in grade 3
meningioma (29). It is commonly
mutated in certain gliomas, gangliogliomas and melanomas (30). The presence of the BRAF V600E
mutation is associated with more aggressive behavior in certain
types of cancer (31). In this
case, the mutation was likely acquired secondararily following
radiation therapy. Based on the immunohistochemical profile, it was
unlikely to be a meningioma.
The discordance between clinical, imaging,
histopathology and immunohistochemical findings hindered appropiate
management of the patient. Such cases underscore the limitations of
conventional histopathology and IHC, in the context of previously
treated tumours. Radiation can induce both morphological and
molecular changes, including altered expression of key diagnostic
markers. This phenomenon poses a significant challenge in
distinguishing true recurrence from treatment-related effects.
Methylation profiling emerged as a critical
diagnostic tool in this scenario, providing a definitive
classification based on the intrinsic epigenetic landscape of the
tumour. The methylation profile has been shown to be conserved
between primary and metastatic samples (32), although the driver mutations may
differ. Evidence from ovarian cancer, which is often heavily
treated with chemotherapeutic agents and prone to recurrance, has
shown that the methylation signature is preseerved between primary
and recurrent tumours (33).
Meningioma methylation profiles typically exhibit
hypermethylation at HOX gene clusters and hypomethylation at cell
cycle regulatory genes, distinguishing them from other CNS tumours
(34,35). For comparison, glioblastomas exhibit
widespread hypomethylation of oncogenes such as EGFR and
hypermethylation of tumour suppressor genes such as MGMT, patterns
that correlate with aggressive proliferation and resistance to
therapy (16,36,37).
Craniopharyngiomas, initially suspected in this case, display
distinct WNT/β-catenin pathway activation, linked to their
epithelial differentiation and cystic growth patterns (38-40).
In this case, the absence of WNT pathway activation, evidenced by
negative β-catenin staining, aided in ruling out
craniopharyngioma.
While meningioma methylation signatures are often
correlated with the risk of recurrence (such as hypermethylation at
NF2 in aggressive subtypes) (41),
physiological associations in irradiated cases remain underexplored
due to limited post-treatment data. A definitive link between the
observed methylation patterns and specific physiological traits
such as proliferation or invasiveness could not be established, as
this would require functional studies which were beyond the scope
of the present work. However, the conserved methylation profile
observed despite prior radiation, aligns with evidence from other
cancers, such as ovarian cancer, where methylation signatures
remain stable between primary and recurrent tumours despite
treatment (33).
Moreover, the copy number profile did not show any
substantial deviations that would suggest chromosomal aberrations
typical of more aggressive gliomas or embryonal tumours, which
often present with distinct copy number alterations (36). This finding aligns with the
relatively stable genomic profile typically observed in
meningiomas, especially in non-anaplastic forms. Collectively,
these findings provide a strong molecular basis for classifying the
sample as a meningioma, with implications for the prognosis of the
patient and potential therapeutic strategies.
The identification of a meningioma-specific
methylation signature was pivotal in resolving the present case.
With a conclusive diagnosis of meningioma, it is now understood
that gross total resection should be pursued in the event of
recurrence, an approach not typically required in cases of
craniopharyngioma. This demonstrated the utility of methylation
profiling as a complementary diagnostic approach for challenging
CNS tumours. Incorporating such advanced molecular techniques into
routine diagnostics can enhance the accuracy of CNS tumour
classification, particularly for complex cases where conventional
methods fall short (42). Adopting
methylation profiling as a standard in neuro-oncology could
transform diagnostics, particularly for recurrent CNS tumours with
treatment-altered histopathology. Unlike targeted gene panels,
which focus on specific mutations (such as IDH1/2) and may miss
broader epigenetic context, or RNA sequencing, which is sensitive
to RNA degradation in FFPE samples (43), methylation profiling offers a
stable, genome-wide view of tumor biology (44). Therefore, methylation profiling can
outperform gene panels in classifying ambiguous cases and is more
robust than RNA sequencing in post-treatment settings.
Cost-effectiveness is a key consideration. While the
initial setup cost for NGS platforms such as Oxford Nanopore
exceeds IHC, the improved diagnostic accuracy can reduce downstream
costs from misdiagnosis or unnecessary treatments, potentially
saving thousands of dollars per patient. Barriers to implementation
include limited access to equipment, the need for specialized
technical expertise, poor DNA quality of FFPE samples for NGS, and
challenges integrating these methods into clinical workflows,
particularly in resource-limited settings. However, declining
sequencing costs, improved NGS technology, and scalable protocols
(such as low-coverage approaches) could mitigate these challenges.
Overall, the analysis of the present study highlighted the utility
of methylation profiling in providing a precise, molecular-based
classification of CNS tumours, particularly in cases where
histopathological evaluation may be challenging. The integration of
methylation data with genomic information, as demonstrated in the
present study, is critical in the context of CNS tumours, where the
accurate classification can directly influence clinical
decision-making and patient management strategies. In conclusion,
methylation signatures can provide a robust, treatment-resistant
classification method, offering clarity in diagnostically
challenging cases and guiding appropriate therapeutic strategies.
Future research should focus on expanding the CNS methylation
classifier to include post-treatment profiles, validating its
utility across diverse tumour types, and assessing long-term
outcomes of methylation-guided management. Standardizing protocols
and addressing cost barriers will further enhance its integration
into routine diagnostics, improving patient outcomes in complex
neuro-oncology cases.
Supplementary Material
Immunohistochemical findings of
various markers. (A) p63, (B) chromogranin, (C) CD34, (D) p40, (E)
CK7, (F) CK8, (G) CK19, (H) HMWCK, (I) Cam5.2, (J) CK20, (K) ER,
(L) STAT6, (M) TTF-1, (N) BRAF V600E, (O) β-catenin, (P) TLE-1, (Q)
BCL2, (R) CD99, and (S) CD56. All images were obtained at a
magnification of x100. CD34, cluster of differentiation 34; p40,
ΔNp63 isoform of the p63 protein; CK7, cytokeratin 7; CK19,
cytokeratin 19; HMWCK, high molecular weight cytokeratin; Cam5.2,
anti-cytokeratin antibody; CK20, cytokeratin 20; ER, estrogen
receptor; STAT6, signal transducer and activator of transcription
6; TTF-1, thyroid transcription factor-1; BRAF V600E, B-Raf
proto-oncogene, serine/threonine kinase, with V600E mutation
(Val600Glu); TLE-1, transducin-like enhancer of split 1; BCL2,
B-cell lymphoma 2; CD99, cluster of differentiation 99; CD56,
cluster of differentiation 56.
Acknowledgements
Not applicable.
Funding
Funding: The present case report was supported by the PUTI grant
from the University of Indonesia (grant no.
NKB-412/UN2.RST/HKP.05.00/2023).
Availability of data and materials
The data generated in the present study may be found
in the SRA database under accession no. PRJNA1240044 (SRA accession
no. SRR32801446) or at the following URL: https://dataview.ncbi.nlm.nih.gov/object/PRJNA1240044?reviewer=hgbuness2eea3b45vlvo9bb134.
Authors' contributions
H contributed to the conceptualization, methodology,
investigation, formal analysis, and writing of the original draft.
ES contributed to acquisition of data, visualization and the
writing, review and editing of the manuscript. RA contributed to
conceptualization, resources, investigation, as well as the
writing, review and editing of the manuscript. TA contributed to
the conceptualization, investigation, as well as the writing,
review and editing of the manuscript. HW contributed to the
interpretation of data, obtaining resources, project
administration, as well as the writing, review and editing of the
manuscript. TBMP contributed to the conceptualization and funding
acquisition, as well as the writing, review and editing of the
manuscript. SAG contributed to the conceptualization, supervision,
obtaining resources, and funding acquisition, as well as the
writing, review and editing of the manuscript. H and ES confirm the
authenticity of all the raw data. All authors contributed equally
to this study, and have read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was performed in line with the
principles of the Declaration of Helsinki. Approval (ethics
approval no. KET-289/UN2.F1/ETIK/PPM.00.02/2024) was granted by the
Ethics Committee of the University of Indonesia (Jakarta,
Indonesia). Written informed consent was secured from the patient
prior to molecular testing, ensuring full disclosure of the study's
purpose, procedures, and potential use of biological specimens for
research.
Patient consent for publication
Written informed consent was secured from the
patient prior to molecular testing for the use of any patient data
or associated images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kristensen BW, Priesterbach-Ackley LP,
Petersen JK and Wesseling P: Molecular pathology of tumors of the
central nervous system. Ann Oncol. 30:1265–1278. 2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Louis DN, Perry A, Wesseling P, Brat DJ,
Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM,
Reifenberger G, et al: The 2021 WHO classification of tumors of the
central nervous system: A summary. Neuro Oncol. 23:1231–1251.
2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Smith HL, Wadhwani N and Horbinski C:
Major features of the 2021 WHO classification of CNS tumors.
Neurotherapeutics. 19:1691–1704. 2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Singer LS, Feldman AZ, Buerki RA,
Horbinski CM, Lukas RV and Stupp R: The impact of the molecular
classification of glioblastoma on the interpretation of therapeutic
clinical trial results. Chin Clin Oncol. 10(38)2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Wood MD, Beadling C, Neff T, Moore S,
Harrington CA, Baird L and Corless C: Molecular profiling of pre-
and post-treatment pediatric high-grade astrocytomas reveals
acquired increased tumor mutation burden in a subset of
recurrences. Acta Neuropathol Commun. 11(143)2023.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Ng WK: Radiation-associated changes in
tissues and tumours. Curr Diagn Pathol. 9:124–136. 2003.
|
|
7
|
Umekawa M, Shinya Y, Hasegawa H, Morshed
RA, Katano A, Shinozaki-Ushiku A and Saito N: Ki-67 labeling index
predicts tumor progression patterns and survival in patients with
atypical meningiomas following stereotactic radiosurgery. J
Neurooncol. 167:51–61. 2024.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Verma N, Cowperthwaite MC, Burnett MG and
Markey MK: Differentiating tumor recurrence from treatment
necrosis: A review of neuro-oncologic imaging strategies. Neuro
Oncol. 15:515–534. 2013.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Smith EJ, Naik A, Shaffer A, Goel M, Krist
DT, Liang E, Furey CG, Miller WK, Lawton MT, Barnett DH, et al:
Differentiating radiation necrosis from tumor recurrence: A
systematic review and diagnostic meta-analysis comparing imaging
modalities. J Neurooncol. 162:15–23. 2023.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Cacciotti C, Lenzen A, Self C and
Pillay-Smiley N: Recurrence patterns and surveillance imaging in
pediatric brain tumor survivors. J Pediatr Hematol Oncol.
46:e227–e232. 2024.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Lebrun L, Gilis N, Dausort M, Gillard C,
Rusu S, Slimani K, De Witte O, Escande F, Lefranc F, D'Haene N, et
al: Diagnostic impact of DNA methylation classification in adult
and pediatric CNS tumors. Sci Rep. 15(2857)2025.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Kuschel LP, Hench J, Frank S, Hench IB,
Girard E, Blanluet M, Masliah-Planchon J, Misch M, Onken J,
Czabanka M, et al: Robust methylation-based classification of brain
tumours using nanopore sequencing. Neuropathol Appl Neurobiol.
49(e12856)2023.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Park JW, Lee K, Kim EE, Kim SI and Park
SH: Brain tumor classification by methylation profile. J Korean Med
Sci. 38(e356)2023.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Lyko F: The DNA methyltransferase family:
A versatile toolkit for epigenetic regulation. Nat Rev Genet.
19:81–92. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Lee TG, Kim SY, Kim HR, Kim H and Kim CH:
Radiation induces autophagy via histone H4 lysine 20 trimethylation
in non-small cell lung cancer cells. Anticancer Res. 40:2537–2548.
2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Brandes AA, Franceschi E, Paccapelo A,
Tallini G, De Biase D, Ghimenton C, Danieli D, Zunarelli E, Lanza
G, Silini EM, et al: Role of MGMT methylation status at time of
diagnosis and recurrence for patients with glioblastoma: Clinical
implications. Oncologist. 22:432–437. 2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Jin B and Robertson KD: DNA
methyltransferases, DNA damage repair, and cancer. Adv Exp Med
Biol. 754:3–29. 2013.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Ahsan MU, Gouru A, Chan J, Zhou W and Wang
K: A signal processing and deep learning framework for methylation
detection using Oxford Nanopore sequencing. Nat Commun.
15(1448)2024.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Euskirchen P, Bielle F, Labreche K,
Kloosterman WP, Rosenberg S, Daniau M, Schmitt C, Masliah-Planchon
J, Bourdeaut F, Dehais C, et al: Same-day genomic and epigenomic
diagnosis of brain tumors using real-time nanopore sequencing. Acta
Neuropathol. 134:691–703. 2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Li H: Minimap2: Pairwise alignment for
nucleotide sequences. Bioinformatics. 34:3094–3100. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Capper D, Jones DTW, Sill M, Hovestadt V,
Schrimpf D, Sturm D, Koelsche C, Sahm F, Chavez L, Reuss DE, et al:
DNA methylation-based classification of central nervous system
tumours. Nature. 555:469–474. 2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Maas SLN, Stichel D, Hielscher T, Sievers
P, Berghoff AS, Schrimpf D, Sill M, Euskirchen P, Blume C, Patel A,
et al: Integrated molecular-morphologic meningioma classification:
A multicenter retrospective analysis, retrospectively and
prospectively validated. J Clin Oncol. 39:3839–3852.
2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Santana-Santos L, Kam KL, Dittmann D, De
Vito S, McCord M, Jamshidi P, Fowler H, Wang X, Aalsburg AM, Brat
DJ, et al: Validation of whole genome methylation profiling
classifier for central nervous system tumors. J Mol Diagn.
24:924–934. 2022.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Priesterbach-Ackley LP, Boldt HB, Petersen
JK, Bervoets N, Scheie D, Ulhøi BP, Gardberg M, Brännström T, Torp
SH, Aronica E, et al: Brain tumour diagnostics using a DNA
methylation-based classifier as a diagnostic support tool.
Neuropathol Appl Neurobiol. 46:478–492. 2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Ma J, Hong Y, Chen W, Li D, Tian K, Wang
K, Yang Y, Zhang Y, Chen Y, Song L, et al: High copy-number
variation burdens in cranial meningiomas from patients with diverse
clinical phenotypes characterized by hot genomic structure changes.
Front Oncol. 10(1382)2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Wang JZ, Nassiri F, Landry AP, Patil V,
Liu J, Aldape K, Gao A and Zadeh G: The multiomic landscape of
meningiomas: A review and update. J Neurooncol. 161:405–414.
2023.PubMed/NCBI View Article : Google Scholar
|
|
27
|
da Silveira MA, Ferreira WAS, Amorim CKN,
Brito JRN, Kayath AS, Sagica FDES and de Oliveira EHC: Meningiomas:
An overview of the landscape of copy number alterations in samples
from an admixed population. J Oncol. 2020(3821695)2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Whitehouse JP, Howlett M, Federico A, Kool
M, Endersby R and Gottardo NG: Defining the molecular features of
radiation-induced glioma: A systematic review and meta-analysis.
Neurooncol Adv. 3(vdab109)2021.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Behling F, Barrantes-Freer A, Skardelly M,
Nieser M, Christians A, Stockhammer F, Rohde V, Tatagiba M,
Hartmann C, Stadelmann C and Schittenhelm J: Frequency of BRAF
V600E mutations in 969 central nervous system neoplasms. Diagn
Pathol. 11(55)2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Kowalewski A, Durślewicz J, Zdrenka M,
Grzanka D and Szylberg Ł: Clinical relevance of BRAF V600E mutation
status in brain tumors with a focus on a novel management
algorithm. Target Oncol. 15:531–540. 2020.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Attia AS, Hussein M, Issa PP, Elnahla A,
Farhoud A, Magazine BM, Youssef MR, Aboueisha M, Shama M, Toraih E
and Kandil E: Association of BRAFV600E mutation with the
aggressive behavior of papillary thyroid microcarcinoma: A
meta-analysis of 33 studies. Int J Mol Sci.
23(15626)2022.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Zhang S, He S, Zhu X, Wang Y, Xie Q, Song
X, Xu C, Wang W, Xing L, Xia C, et al: DNA methylation profiling to
determine the primary sites of metastatic cancers using
formalin-fixed paraffin-embedded tissues. Nat Commun.
14(5686)2023.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Gull N, Jones MR, Peng PC, Coetzee SG,
Silva TC, Plummer JT, Reyes ALP, Davis BD, Chen SS, Lawrenson K, et
al: DNA methylation and transcriptomic features are preserved
throughout disease recurrence and chemoresistance in high grade
serous ovarian cancers. J Exp Clin Cancer Res.
41(232)2022.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Huntoon K, Toland AMS and Dahiya S:
Meningioma: A review of clinicopathological and molecular aspects.
Front Oncol. 10(579599)2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Hergalant S, Saurel C, Divoux M, Rech F,
Pouget C, Godfraind C, Rouyer P, Lacomme S, Battaglia-Hsu SF and
Gauchotte G: Correlation between DNA methylation and cell
proliferation identifies new candidate predictive markers in
meningioma. Cancers (Basel. 14(6227)2022.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Chen CH, Lin YJ, Lin YY, Lin CH, Feng LY,
Chang IY, Wei KC and Huang CY: Glioblastoma primary cells retain
the most copy number alterations that predict poor survival in
glioma patients. Front Oncol. 11(621432)2021.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Yin A, Etcheverry A, He Y, Aubry M,
Barnholtz-Sloan J, Zhang L, Mao X, Chen W, Liu B, Zhang W, et al:
Integrative analysis of novel hypomethylation and gene expression
signatures in glioblastomas. Oncotarget. 8:89607–89619.
2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Matsuda T, Kono T, Taki Y, Sakuma I,
Fujimoto M, Hashimoto N, Kawakami E, Fukuhara N, Nishioka H,
Inoshita N, et al: Deciphering craniopharyngioma subtypes:
Single-cell analysis of tumor microenvironment and immune networks.
iScience. 27(111068)2024.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Zhao C, Wang Y, Liu H, Qi X, Zhou Z, Wang
X and Lin Z: Molecular biological features of cyst wall of
adamantinomatous craniopharyngioma. Sci Rep.
13(3049)2023.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Jucá CEB, Colli LM, Martins CS, Campanini
ML, Paixão B, Jucá RV, Saggioro FP, de Oliveira RS, Moreira AC,
Machado HR, et al: Impact of the canonical wnt pathway activation
on the pathogenesis and prognosis of adamantinomatous
craniopharyngiomas. Horm Metab Res. 50:575–581. 2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Choudhury A, Magill ST, Eaton CD, Prager
BC, Chen WC, Cady MA, Seo K, Lucas CG, Casey-Clyde TJ, Vasudevan
HN, et al: Meningioma DNA methylation groups identify biological
drivers and therapeutic vulnerabilities. Nat Genet. 54:649–659.
2022.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Bertero L, Mangherini L, Ricci AA, Cassoni
P and Sahm F: Molecular neuropathology: An essential and evolving
toolbox for the diagnosis and clinical management of central
nervous system tumors. Virchows Arch. 484:181–194. 2024.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Lin Y, Dong ZH, Ye TY, Yang JM, Xie M, Luo
JC, Gao J and Guo AY: Optimization of FFPE preparation and
identification of gene attributes associated with RNA degradation.
NAR Genom Bioinform. 6(lqae008)2024.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Zhang P, Lehmann BD, Shyr Y and Guo Y: The
utilization of formalin fixed-paraffin-embedded specimens in high
throughput genomic studies. Int J Genomics.
2017(1926304)2017.PubMed/NCBI View Article : Google Scholar
|