Introduction
Sitosterolemia is a rare metabolic disease that
results in the buildup of phytosterols in the body. Phytosterols
are dietary plant sterols found in high-fat plant-derived foods
such as nuts and oils. While healthy individuals absorb ~5% of
these sterols, patients with sitosterolemia absorb 15-60%,
resulting in phytosterol accumulation in the blood and arteries
(1). This lipid buildup can lead to
heart attack, stroke, and death, making its early diagnosis
essential. Sitosterolemia is estimated to affect 1 in every 200,000
individuals globally and is often detected as early as childhood
(2). However, this rate may be
underestimated, as sitosterolemia is often misdiagnosed as other
more common hyperlipidemia disorders, such as familial
hypercholesterolemia (FH), which affects an estimated 1 in every
250 individuals globally, due to their similar signs of
hypercholesterolemia as well as the lack of plant sterol-specific
testing (2,3).
Sitosterolemia has been associated with variants in
either ABCG5 or ABCG8 on chromosome 2, which code for
sterol efflux transporters in the intestines (1). Sitosterolemia is an autosomal
recessive disease and biallelic missense, nonsense, frameshift, and
splice site variants have all been previously cited as causes of
sitosterolemia (4). Similar to
other autosomal recessive disorders, this mode of inheritance
requires a variant to be inherited from each parent.
In the present study, the first reported case of
sitosterolemia caused by uniparental disomy (UPD) is identified.
Specifically, a homozygous variant in ABCG5 was found as a
result of paternal segmental uniparental isodisomy (UPiD) of the p
arm of chromosome 2. The molecular diagnosis enabled effective
treatment after 13 years of therapeutic resistance.
Case report
A 2-year-old female patient from non-consanguineous
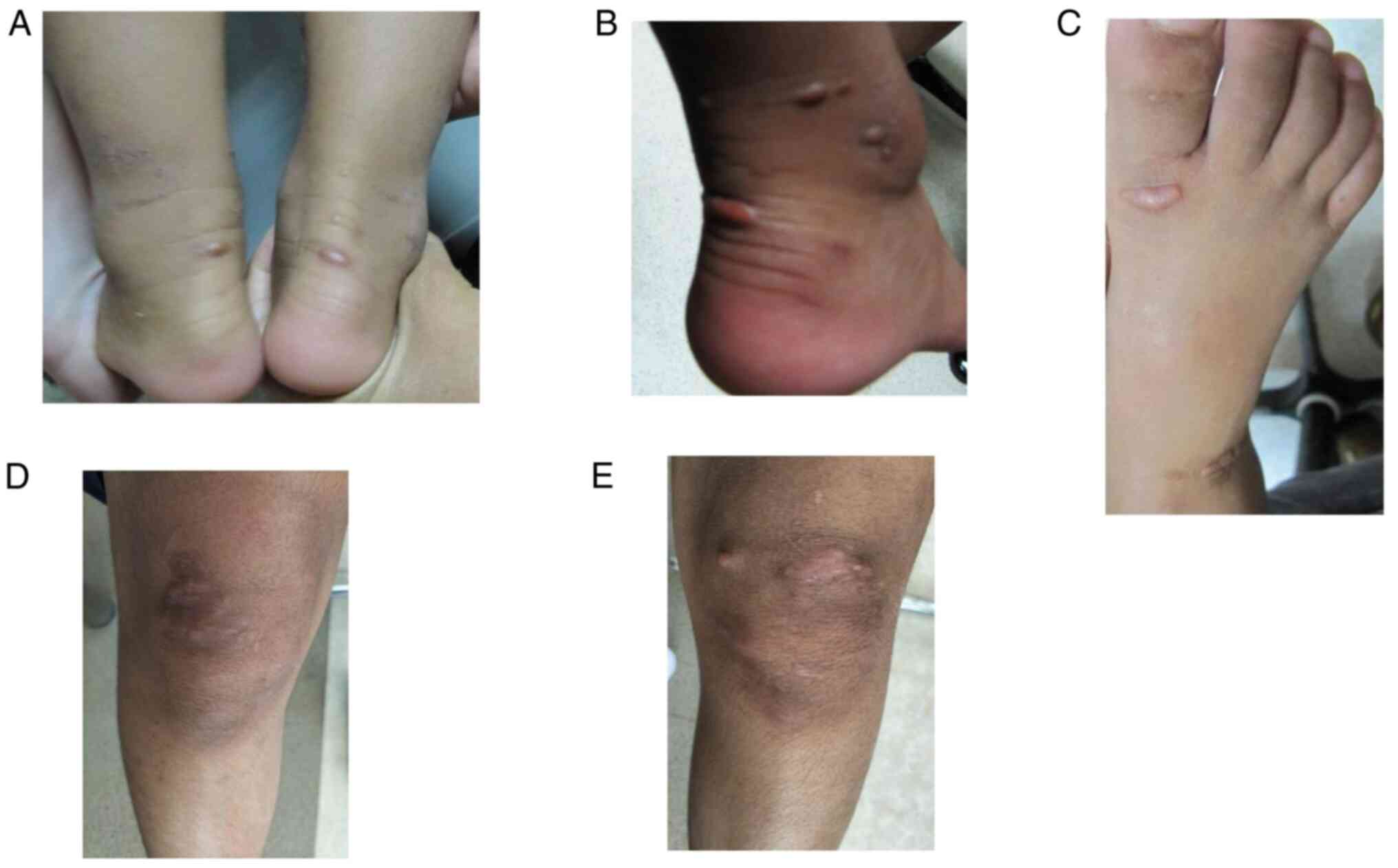
parents presented with xanthomas on both achilles tendons, knees
and toe knuckles (Fig. 1A-C) at
King Chulalongkorn Memorial Hospital (Bangkok, Thailand). The
patient had high total cholesterol (542 mg/dl; normal range,
<200 mg/dl) and high LDL cholesterol (419 mg/dl; normal range,
<130 mg/dl) (5). The father of
the patient had high cholesterol levels (230 mg/dl) in his 30s, and
the mother of the patient developed hypercholesterolemia (220
mg/dl) in her 40s. The patient was initially clinically diagnosed
with FH. The patient was advised to follow the Cardiovascular
Health Integrated Lifestyle Diet (CHILD) 2 diet including
consumption of skim milk, cholesterol restriction <200 mg/day,
avoidance of saturated fat and trans-fat, shifting to
monounsaturated fat and plant sterols, in addition to regular
physical activity (5). The patient
was encouraged to participate in school athletics and reported
running as much as 4-5 km on multiple days per week at the age of
7. Despite these lifestyle changes and strict dietary control (a
more plant-based, pescatarian diet) the cholesterol levels of the
patient did not improve over time, as observed in Table I. Around the age of 10, the patient
was prescribed simvastatin. The compliance of the patient with the
CHILD 2 diet worsened during adolescence, and the medication was
changed to atorvastatin with modest improvement. Around the age of
13, the patient experienced the first episode of acute arthritis in
both knee joints (Fig. 1D and
E). The investigations showed acute
inflammation, but none indicated other autoimmune diseases.
Therefore, it was likely ‘periarthritis’ resulted from
hypercholesterolemia. The arthritis recurred and subsided
periodically once or twice a year, requiring school breaks and
short-term NSAIDs. Atorvastatin was then titrated upward together
with stricter cholesterol restriction. With the lack of improvement
and this ‘periarthritis’ manifestation, genetic testing was
requested and revealed a sitosterolemia diagnosis at the age of 15.
Thus, in addition to the current treatment, the patient was
instructed to avoid phytosterols such as canola oil, olive oil,
nuts, whole grains, legumes, and avocados. The patient was also
prescribed ezetimibe which can lower cholesterol and phytosterol
absorption. Notably, 4 months later, on the first visit of the
patient to the hospital following diagnosis, the total and LDL
cholesterol levels of the patient returned to normal levels
(Table I).
 | Table ILipid profiles of the patient before
and after diagnosis of sitosterolemia. |
Table I
Lipid profiles of the patient before
and after diagnosis of sitosterolemia.
| Lipid profile | Reference range | First diagnosis (2
years old) | After dietary
counseling (Cardiovascular Health Integrated Lifestyle Diet 2) | After simvastatin
treatment (starting at 10 years old) | After atorvastatin
titration from 10-80 mg/day 12 years old) | During the diagnostic
visit for sitosterolemia, (starting at before initiating new
treatment (15 years old) | After ezetimibe
treatment and avoidance of phytosterols (15 years old, 4
months) |
|---|
| Total cholesterol
(mg/dl) | <200 | 542 | 187-457 | 217-384 | 155-244 | 242 | 117 |
| LDL (mg/dl) | <130 | 419 | 138-384 | 168-345 | 101-195 | 193 | 59 |
| HDL (mg/dl) | >40 | 52 | 26-45 | 30-44 | 39-47 | 46 | 48 |
| Triglycerides
(mg/dl) | <100-130 | 72 | 56-127 | 71-106 | 57-103 | 57 | 59 |
Genetic testing
After written informed consent was obtained from the
patient and both parents of the patient, peripheral blood from the
patient and the parents was collected and short-read genome
sequencing was conducted on the extracted genomic DNA. The present
study was approved (approval no. 264/62) by the Institutional
Review Board of the Faculty of Medicine, Chulalongkorn University
(Bangkok, Thailand). Genome sequencing was performed on the
DNBSEQ-T7 platform (BGI), achieving an average coverage depth of
30x. Variant calling was conducted using GATK 4.0 against the human
genome reference GRCh38(6).
Variants were annotated with Variant Effect Predictor version 110,
dbNSFP version 4.4a and our in-house Thai genome databases
(7-9).
The in-house Thai databases included the Thai Exome Database (ThEx)
and Thai Long Read Genome Database (Th-LR), both developed by the
Center of Excellence for Medical Genomics, Chulalongkorn University
(Bangkok, Thailand). genmod (version 3.1; https://github.com/Clinical-Genomics/genmod), a
variant-inheritance annotation tool developed by Måns Magnusson,
available at GitHub, Inc. was used for Trio analysis. All SNVs and
indels were filtered by the following filtering criteria: i)
Located in genes related to the phenotype of the patient; ii)
located in exons or flanking introns of the phenotype-associated
genes; iii) not synonymous; iv) total read depth ≥10x; v) present
at >1% in the Genome Aggregation Database (gnomAD) v4.1 and/or
our in-house Thai population database; and vi) predicted to be
damaging (≥0.644) by REVEL score (for missense variants) or
predicted to be splice-altering (≥0.2) by SpliceAI (for splice
variants) (10-12).
UPD detection was performed with TrioMix v.0.0.1(13).
Short-read genome sequencing identified a homozygous
splice variant c.904+1G>A in the ABCG5 gene
(NM_022436.3). The father of the patient is heterozygous for this
variant. This variant is present in gnomAD in 8 alleles of
1,613,770 with no homozygotes and is absent from our in-house Thai
population database. SpliceAI predicts this variant to strongly
disrupt the exon 7 donor splice site with a score of 0.99. This
variant has been reported to occur in trans with other
ABCG5 variants in two other unrelated individuals affected
with sitosterolemia (14). As a
result, the variant was classified as pathogenic in accordance with
ACMG/AMP guidelines (PVS1 + PM3_Strong + PM2_Supporting) (15). Examination of the BAM file showed
paternal UPiD of the p arm of chromosome 2 in the patient
[seq(GRCh38) 2p25.3p11.2 (1-85,128,508)x2upat]. The TrioMix tool
was employed to visualize relevant single nucleotide polymorphisms
across the genome of the patient and to infer paternal UPiD of the
p arm of chromosome 2 as observed in Fig. 2.
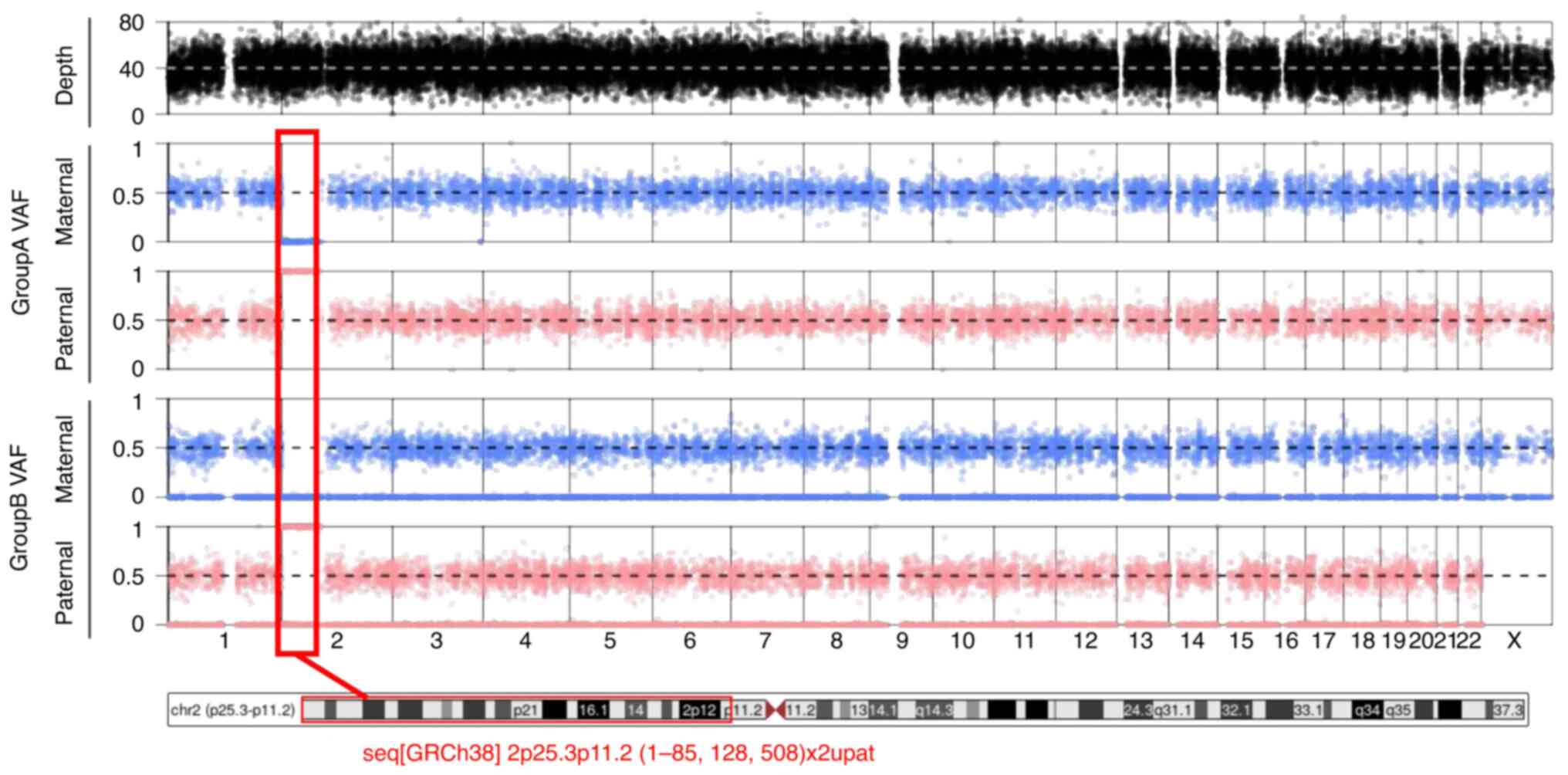 | Figure 2TrioMix tool displays the VAF for SNPs
on each chromosome, separated by maternal and paternal
contributions. In Group A SNPs (where only SNPs that are homozygous
in either parent are considered), the maternal allele frequency
plot for SNPs on the short arm of chromosome 2 revealed a value of
~0, while the paternal plot registered at ~1. This indicates that
the patient inherited parental homozygous SNPs on the p arm of
chromosome 2 solely from the father. In Group B SNPs (where only
SNPs that are heterozygous in either parent are considered), the
VAF of SNPs on the p arm of chromosome 2 exhibits a maternal allele
frequency of 0, coupled with a paternal plot showing allele
frequencies predominantly at 0 and 1, with an absence of
heterozygous SNPs (0.5) along the p arm. This pattern suggests
paternal uniparental isodisomy of most of the p arm of chromosome
2, wherein the patient inherited two identical copies of this
chromosomal segment from the father, with no contribution from the
mother. VAF, variant allele frequencies; SNPs, single nucleotide
polymorphisms. |
Discussion
UPD is rare and occurs in only ~0.2% of the
population (16). There are
numerous forms of UPD, but all result in the inheritance of two
alleles of a gene from one parent. Typically, whole chromosomes
from the same parent, or complete disomy, are observed in these
cases. However, rare occurrences of mitotic recombination can lead
to segmental UPD in which only a part of the chromosome is
identically inherited (17). The
patient in the present case report exhibited uniparental paternal
segmental isodisomy in the p arm of chromosome 2. Therefore, it is
possible that a double-stranded DNA break near the centromere
occurred, leading to mitotic recombination as an attempted repair,
resulting in a daughter cell with segmental UPiD of the p arm of
chromosome 2.
The father of the patient is a carrier of the
ABCG5 c.904+1G>A variant that has previously been
reported to cause sitosterolemia with compound heterozygosity
(14,18). Although the mother did not have
sitosterolemia and the father is an asymptomatic carrier, the
2-year-old female patient inherited two copies of the father's
mutated ABCG5 gene through segmental UPiD, resulting in a
homozygous recessive genotype and manifestation of sitosterolemia.
Although both parents reported at-risk levels of high cholesterol,
they were likely as a result of lifestyle factors as their onset
was late with only moderately high lipid levels. They also did not
present xanthomas and no genetic causes were found, supporting the
sitosterolemia diagnosis resulting from UPiD in the patient. To
date, this is the first reported case of sitosterolemia caused by
UPD of any kind.
With cases of UPD, gene imprinting may also cause
phenotypic abnormalities as some genes are only expressed when
inherited maternally or paternally. However, there are currently no
known paternally imprinted genes on chromosome 2 and no other
phenotypic abnormalities were reported for this patient (19). Furthermore, patients with UPiD can
be prone to homozygosity when the parent could be a carrier for
autosomal recessive disorders. However, in this case, no other
homozygous pathogenic variants in genes on the short arm of
chromosome 2 were present.
This genetic testing was essential to diagnosis. As
is common for patients with sitosterolemia, including this patient,
numerous individuals are initially misdiagnosed with FH due to the
lack of plant sterol-specific testing and early genetic testing.
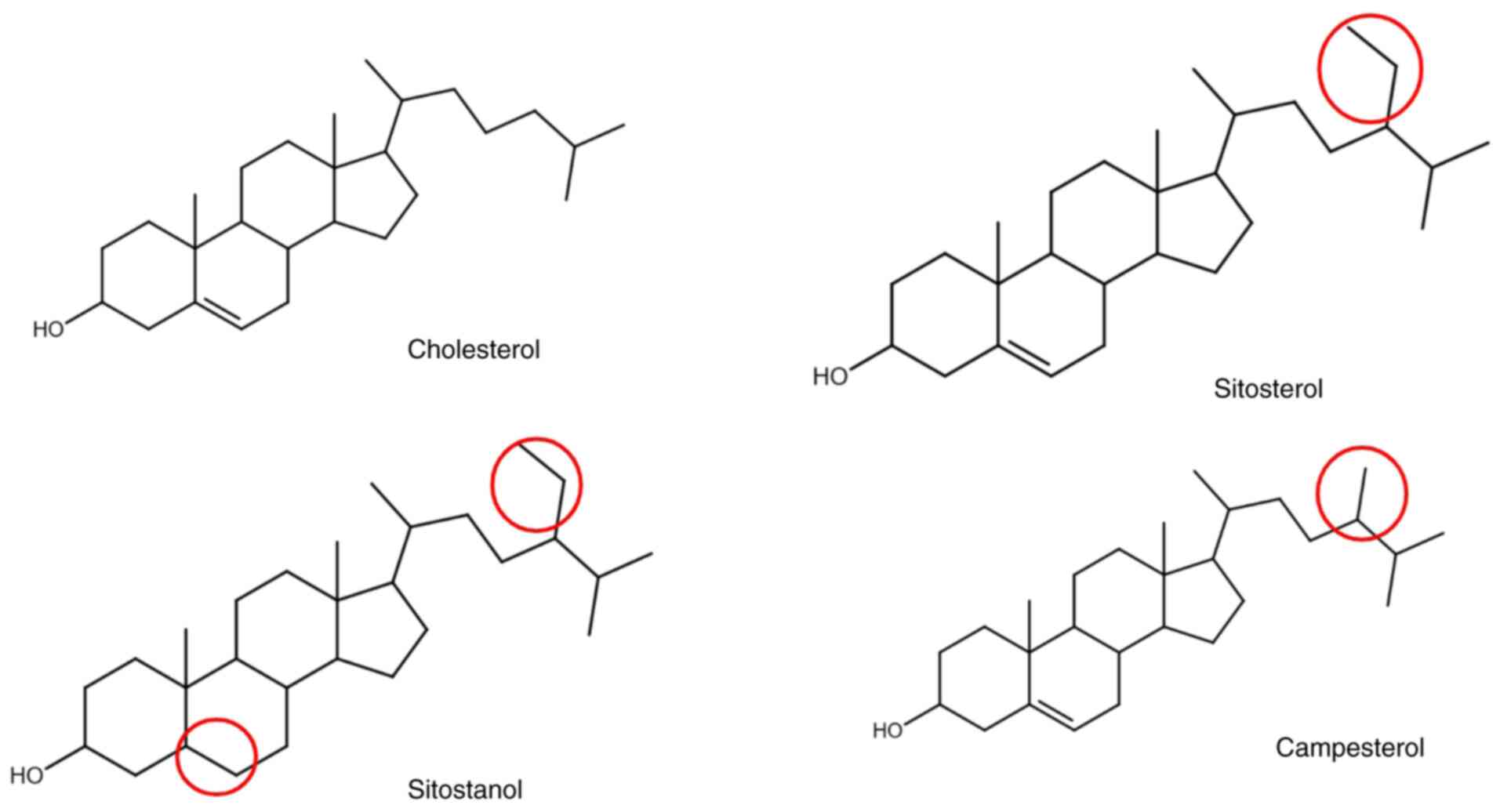
More specifically, total cholesterol tests cannot differentiate
between cholesterol and phytosterols due to their similar
structures, resulting in high total cholesterol levels in patients
with sitosterolemia (Fig. 3). LDL
levels may also be high particularly in children such as this
patient due to immature intestinal cells and breast milk
consumption (1). Suspected patients
with FH are typically advised to shift from saturated animal fat
sources to monounsaturated fats, such as phytosterols, to help
control their cholesterol levels. As a result, the patient
generally avoided animal fat, which is high in saturated fat and
cholesterol and instead consumed plant-based sources of fat such as
canola and olive oil. The patient also reported eating numerous
fruits, vegetables, avocado, nuts, whole grains, and legumes rather
than animal protein-based and processed snacks. However, these
plant-based foods typically contain phytosterols, which patients
with sitosterolemia are unable to excrete properly. As a result,
this consumption likely resulted in the deterioration of the
condition of the patient at the time.
However, after receiving genetic testing and the
sitosterolemia diagnosis, the diet and management plan of the
patient were better curated to the condition. Dietary removal of
phytosterols along with the addition of ezetimibe, which inhibits
the absorption of cholesterol and phytosterols from the small
intestine, significantly reduced the total and LDL cholesterol to
their lowest levels since infancy in only 4 months of this new
treatment plan. Had the patient known of this diagnosis sooner, the
cholesterol levels and symptoms experienced by the patient, likely
could have been improved earlier.
This is not the only case where genetic testing has
proven useful for patients with treatment-resistant hyperlipidemia.
A previous case of a 3-year-old female patient with xanthomas and
high total and LDL cholesterol was initially misdiagnosed with FH
and struggled to show improvement over time (20). After the patient received genetic
testing, a correct diagnosis of sitosterolemia due to ABCG5
variants was made. After starting ezetimibe and removing
phytosterols from the diet, the lipid levels and xanthomas of the
patient significantly improved. Similarly, two 7- and 9-year-old
patients with high cholesterol and xanthomas were also initially
misdiagnosed with FH (21).
However, on an FH low-cholesterol diet, lipid levels did improve
significantly in one patient, and xanthomas were still present in
the other. However, after receiving genetic testing, variants in
ABCG5 were identified, leading to a correct sitosterolemia
diagnosis. Following subsequent phytosterol removal from the diet,
significant xanthoma regression and reduced lipid levels were
observed. In all three patients, genetic diagnosis resulted in a
change of treatment and improvements to health (20,21).
Thus, genetic testing is an important aspect of
diagnosis, especially for those with treatment-resistant
hyperlipidemia and rarer manifestations such as periarthritis, as
it can provide important information on the conditions of patients
that can significantly alter their treatment plan and ultimately
lead to an improved quality of life for a number of these
patients.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Health Systems
Research Institute (grant no. 67-095).
Availability of data and materials
The data generated in the present study may be found
in the NCBI Sequence Read Archive under the BioProject accession
no. PRJNA1281833. The in-house Thai databases, Thai Exome Database
(ThEx) and Thai Long Read Genome Database (Th-LR), both developed
by the Center of Excellence for Medical Genomics, Chulalongkorn
University (Bangkok, Thailand) may be requested from the
corresponding author.
Authors' contributions
HK was involved with the case analysis and was the
primary author of the manuscript. CC edited the manuscript and was
involved with analysis and variant identification. SK analyzed the
bioinformatics data for the creation of Fig. 2. KS analyzed and identified the
variant responsible for the disease. SC was the pediatrician
nutrition specialist involved with the treatment of the patient,
and edited the manuscript. RT was the clinical geneticist involved
with the treatment of the patient. VS designed and supervised the
study, analyzed patient data, and obtained funding. VS and SK
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved (approval no. 264/62)
by the Institutional Review Board of the Faculty of Medicine,
Chulalongkorn University (Bangkok, Thailand). Written informed
consent from the patient and both parents of the patient was
obtained.
Patient consent for publication
Written informed consent was obtained from the
patient and both parents of the patient for the publication of any
patient data and associated images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Yoo EG: Sitosterolemia: A review and
update of pathophysiology, clinical spectrum, diagnosis, and
management. Ann Pediatr Endocrinol Metab. 21:7–14. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Tada H, Nomura A, Ogura M, Ikewaki K,
Ishigaki Y, Inagaki K, Tsukamoto K, Dobashi K, Nakamura K, Hori M,
et al: Diagnosis and management of sitosterolemia 2021. J
Atheroscler Thromb. 28:791–801. 2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Akioyamen LE, Genest J, Shan SD, Reel RL,
Albaum JM, Chu A and Tu JV: Estimating the prevalence of
heterozygous familial hypercholesterolaemia: A systematic review
and meta-analysis. BMJ Open. 7(e016461)2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Kojima N, Tada H, Nomura A, Usui S, Sakata
K, Hayashi K, Nohara A, Inazu A, Kawashiri MA and Takamura M:
Putative pathogenic variants of ABCG5 and ABCG8 of sitosterolemia
in patients with hyper-low-density lipoprotein cholesterolemia. J
Lipid Atheroscler. 13:53–60. 2024.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Expert Panel on Integrated Guidelines for
Cardiovascular Health and Risk Reduction in Children and
Adolescents; National Heart, Lung and Blood Institute. Expert panel
on integrated guidelines for cardiovascular health and risk
reduction in children and adolescents: Summary report. Pediatrics.
128 (Suppl 5):S213–S256. 2011.PubMed/NCBI View Article : Google Scholar
|
|
6
|
McKenna A, Hanna M, Banks E, Sivachenko A,
Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly
M and DePristo MA: The genome analysis toolkit: A MapReduce
framework for analyzing next-generation DNA sequencing data. Genome
Res. 20:1297–1303. 2010.PubMed/NCBI View Article : Google Scholar
|
|
7
|
McLaren W, Gil L, Hunt SE, Riat HS,
Ritchie GR, Thormann A, Flicek P and Cunningham F: The ensembl
variant effect predictor. Genome Biol. 17(122)2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Liu X, Jian X and Boerwinkle E: dbNSFP: A
lightweight database of human nonsynonymous SNPs and their
functional predictions. Hum Mutat. 32:894–899. 2011.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Liu X, Li C, Mou C, Dong Y and Tu Y:
dbNSFP v4: A comprehensive database of transcript-specific
functional predictions and annotations for human nonsynonymous and
splice-site SNVs. Genome Med. 12(103)2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Karczewski KJ, Francioli LC, Tiao G,
Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A,
Birnbaum DP, et al: The mutational constraint spectrum quantified
from variation in 141,456 humans. Nature. 581:434–443.
2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Ioannidis NM, Rothstein JH, Pejaver V,
Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E,
Karyadi D, et al: REVEL: An ensemble method for predicting the
pathogenicity of rare missense variants. Am J Hum Genet.
99:877–885. 2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Jaganathan K, Kyriazopoulou
Panagiotopoulou S, McRae JF, Darbandi SF, Knowles D, Li YI,
Kosmicki JA, Arbelaez J, Cui W, Schwartz GB, et al: Predicting
splicing from primary sequence with deep learning. Cell.
176:535–548.e24. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Yoon CJ, Kim SY, Nam CH, Lee J, Park JW,
Mun J, Park S, Lee S, Yi B, Min KI, et al: Estimation of
intrafamilial DNA contamination in family trio genome sequencing
using deviation from Mendelian inheritance. Genome Res.
32:2134–2144. 2022.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Ba H, Peng H, He X, Cheng L, Lin Y, Li X,
Wang H and Qin Y: Sitosterolemia with atherosclerosis in a child: A
case report. Front Pediatr. 9(668316)2021.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Scuffins J, Keller-Ramey J, Dyer L,
Douglas G, Torene R, Gainullin V, Juusola J, Meck J and Retterer K:
Uniparental disomy in a population of 32,067 clinical exome trios.
Genet Med. 23:1101–1107. 2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Papenhausen PR, Kelly CA, Harris S,
Caldwell S, Schwartz S and Penton A: Clinical significance and
mechanisms associated with segmental UPD. Mol Cytogenet.
14(38)2021.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Su SQ, Xiong DS, Ding XM, Kuang JA and Lin
YC: Pediatric patients with familially inherited sitosterolemia:
Two case reports. Front Cardiovasc Med. 9(927267)2022.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Tan X, Liu B, Yan T, Wei X, Qin Y, Zeng D
and Yuan D: Prenatal diagnosis of paternal uniparental disomy for
chromosome 2 in two fetuses with intrauterine growth restriction.
Mol Cytogenet. 16(20)2023.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Alquraishi AS and Rayees S: Sitosterolemia
with two heterozygous variants including a novel mutation
c.1800T>A in the ABCG5 Gene: A case report of a rare condition
in a young Saudi girl. Cureus. 16(e63088)2024.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Huang D, Zhou Q, Chao YQ and Zou CC:
Clinical features and genetic analysis of childhood sitosterolemia:
Two case reports and literature review. Medicine (Baltimore).
98(e15013)2019.PubMed/NCBI View Article : Google Scholar
|