Introduction
Gastric cancer (GC) is a highly prevalent malignant
tumor worldwide. According to GLOBOCAN 2022 data released by the
International Agency for Research on Cancer, the incidence and
mortality rates of GC rank fifth among all malignant neoplasms
globally, with an estimated 970,000 new cases and 660,000 deaths
annually (1). Because most patients
are diagnosed at advanced or metastatic stages, thereby forfeiting
the opportunity for curative surgical intervention, there is an
urgent need to develop more effective and less toxic
chemotherapeutic agents for GC management (2,3).
Cisplatin, a first-line chemotherapeutic agent,
plays a crucial role in treating GC by inducing DNA cross-linking
damage and generating reactive oxygen species (ROS) (4,5).
However, its clinical application is often associated with
pronounced nephrotoxicity and neurotoxicity, which substantially
constrain its prolonged use and optimal dosing (6-8).
In recent years, the combination of small-molecule compounds with
cisplatin has attracted considerable attention because of their
synergistic and toxicity-reducing properties. This approach can
lower the required dose of cisplatin while enhancing tumor cell
sensitivity to chemotherapy (9-11).
Therefore, identifying small-molecule compounds capable of
eliciting synergistic effects at low concentrations represents a
promising approach to enhancing cisplatin-based therapeutic
regimens in patients with GC. Hesperadin is a small-molecule kinase
inhibitor identified through high-throughput screening (12-14).
It has been demonstrated that Hesperadin possesses dual functions:
Antitumor and cardioprotective properties. First, by targeting and
inhibiting Aurora B kinase, which regulates mitosis and mediates
antitumor activity, as well as calcium/calmodulin-dependent protein
kinase II-δ (CaMKII-δ), which is involved in the oxidative stress
response, Hesperadin has exhibited significant antiproliferative
and proapoptotic effects in various tumor models. Second, the
inhibition of CaMKII-δ can mitigate the damage caused by
chemotherapeutic drugs to cardiac tissues (15). These properties, along with their
ability to reduce toxicity, offer unique advantages for clinical
application. Hesperadin exerts its antitumor activity through dual
inhibition of Aurora B and CaMKII-δ kinases, and its remarkable
potency is reflected in its exceptionally low
IC50values. Consistent with a previous study in
pancreatic cancer (16), the
present findings demonstrated that Hesperadin exhibits potent
antitumor effects at nanomolar concentrations in GC cells.
The dual functional properties of Hesperadin offer a
novel therapeutic avenue for combination chemotherapy. By targeting
and modulating downstream molecular activities, Hesperadin
increases tumor cell sensitivity to chemotherapeutic agents while
simultaneously mitigating treatment-associated toxicity. However,
its specific role in GC and its potential synergistic interactions
with cisplatin remain incompletely understood. In the present
study, it was demonstrated that hesperadin induces NOX1-dependent
ROS generation, thereby increasing the sensitivity of GC cells to
cisplatin. These findings not only establish a connection between
Hesperadin and the efficacy of cisplatin but also suggest new
possibilities for combination chemotherapy regimens in the
treatment of advanced GC.
Materials and methods
Cell culture
The human GC cell lines AGS and HGC-27 were procured
from the Cell Resource Center of the Chinese Academy of Medical
Sciences in collaboration with Peking Union Medical College. The
cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM;
Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10%
heat-inactivated fetal bovine serum (FBS; Wisent, Inc.). The cells
were maintained at 37˚C in a humidified incubator with 5%
CO2 (Thermo Fisher Scientific, Inc.).
Drugs and reagents
Hesperadin (cat. no. HY-12054; MedChemExpress) and
cisplatin (cat. no. 232120; MilliporeSigma) were dissolved in
dimethyl sulfoxide (DMSO) and normal saline, respectively, and
stored at -20˚C in light-protected conditions. The NOX1 inhibitor
ML171 (2-acetylphenothiazine; cat. no. HY-12805; MedChemExpress)
was also dissolved in DMSO before use. The Cell Counting Kit-8
(CCK-8; cat. no. C0038), Annexin V-FITC apoptosis detection kit
(cat. no. C1062M), ROS detection kit (cat. no. S0033S), JC-1
mitochondrial membrane potential (MMP; cat. no. C2006) assay kit
and DNA damage detection kit (cat. no. C2035S) were obtained from
Beyotime Institute of Biotechnology.
Cell proliferation and cytotoxicity
assays
To evaluate cytotoxicity, AGS and HGC-27 GC cells
were seeded in 96-well plates at a density of 6,000 cells per well
and exposed to Hesperadin (0-90 nM) or cisplatin (0-120 µM) for 24
h. After 48 h of treatment, CCK-8 reagent (10 µl per well) was
added and incubated at 37˚C for 2 h. The absorbance at 450 nm was
then measured using a SpectraMax iD5 microplate reader (Molecular
Devices, LLC) to determine the half-maximal inhibitory
concentration (IC50) values using statistical
software.
For the proliferation inhibition assay, cells were
seeded at the same density and treated with Hesperadin alone or in
combination with cisplatin for 48 h. The procedures for CCK-8
reagent incubation and absorbance measurement at 450 nm were
performed as described for the cytotoxicity assay.
RNA sequencing (RNA-seq) and
bioinformatics
RNA extraction, library preparation, and sequencing
were performed by Shanghai Applied Protein Technology Co., Ltd.
Briefly, total RNA was extracted from AGS cells using
TRIzol® Reagent (cat. no. R0016; Beyotime Institute of
Biotechnology). RNA integrity was verified (RIN >8.0).
Sequencing libraries were constructed using the VAHTS Universal V6
RNA-seq Library Prep Kit for Illumina (Vazyme Biotech Co., Ltd.)
and sequenced on an Illumina NovaSeq 6000 platform to generate 150
bp paired-end reads. Subsequent bioinformatic analysis was
conducted as follows: Raw reads were quality-filtered using Fastp.
De novo assembly was performed with Trinity. Transcripts
were annotated against NR (https://www.ncbi.nlm.nih.gov/refseq/about/nonredundantproteins/),
SwissProt (https://www.uniprot.org/uniprotkb?facets=reviewed%3Atrue),
Pfam (http://pfam.xfam.org/), Gene Ontology
(GO; http://geneontology.org/) and Kyoto
Encyclopedia of Genes and Genomes (KEGG; https://www.genome.jp/kegg/) databases. Coding
sequences were predicted using TransDecoder. Gene expression (FPKM)
was quantified, and differential expression analysis was performed
using DESeq2 (threshold: |log2FC|>1, padj <0.05). GO and KEGG
enrichment analyses were conducted using clusterProfiler (version
4.16.0; Bioconductor).
Identification of differentially
expressed genes (DEGs) and functional enrichment analysis
With |log2-fold change (FC)|>1 and P<0.05 as
screening criteria, DEGs from bulk sequencing were identified via
the ‘Limma’ package (version 3.64.3; Bioconductor) of R. DEGs were
illustrated via volcano maps and heatmaps, which were plotted via
the ‘ggplot2’ and ‘Pheatmap’ packages of R, respectively. In
addition, KEGG and GO analyses were performed via the
‘clusterProfiler’ R package, which aims to assess gene-related
biological processes (BP), molecular functions, cellular
components, and gene-related signaling pathways (KEGG). The
conditions for screening significantly enriched GO terms and KEGG
pathways were Min overlap=three and Min enrichment=1.5. The
enrichment significance threshold was set to an adjusted P-value
below 0.05. In addition, gene set variation analysis (GSVA) is a
non-parametric and unsupervised method for estimating changes in
specific gene sets. The activities of the hallmark pathways were
quantified with the GSVA R package according to the ‘Hallmarks
geneset’ downloaded from the MsigDB database. Finally, gene set
enrichment analysis (GSEA) was employed to identify DEG functions
on the basis of P<0.05 and a false discovery rate (FDR) value of
0.25.
Flow cytometric analysis of apoptosis
and necrosis
Apoptosis was assessed using an Annexin
V-FITC/propidium iodide (PI) apoptosis detection kit (cat. no.
C1062M; Beyotime Institute of Biotechnology). Briefly, after drug
treatment, the cells were collected, washed twice with ice-cold
PBS, and resuspended in 1X binding buffer at a density of
1x106 cells/ml. Subsequently, cells were stained with
Annexin V-FITC and PI for 15 min at room temperature in the dark
according to the manufacturer's instructions. To establish
appropriate gating and compensation, unstained cells and
single-stained controls (cells stained with Annexin V-FITC only or
PI only) were included in each experiment. Flow cytometric analysis
was immediately performed on a Beckman CytoFLEX flow cytometer
(Beckman Coulter, Inc.). The data were analyzed using FlowJo
software (version 10.8.1; FlowJo LLC). The percentages of cells in
early apoptosis (Annexin V+/PI-) and late
apoptosis/necrosis (Annexin V+/PI+) were
quantified.
Detection of intracellular ROS
levels
For ROS detection, following the respective
treatments, cells were collected, washed twice with pre-warmed
phosphate-buffered saline (PBS), and then incubated with 10 µM
CM-H2DCFDA diluted in serum-free medium at 37˚C in the dark for 25
min. After incubation, the cells were washed to remove the excess
probe and analyzed using a fluorescence microscope (MShot MF53-N,
FITC channel), and the captured images were quantified using ImageJ
software (version 1.53k; National Institutes of Health). The
fluorescence intensity was quantified via flow cytometry (FITC
channel).
MMP assay
The MMP was assessed via a JC-1 assay kit (cat. no.
C2006; Beyotime Institute of Biotechnology). The cells were
incubated with JC-1 staining solution for 20 min at 37˚C in the
dark for 20 min. The fluorescence intensity ratios (red/green) were
subsequently analyzed via fluorescence microscopy (MShot MF53-N).
JC-1 aggregates (red fluorescence) in healthy mitochondria with
high MMP and JC-1 monomers (green fluorescence) in depolarized
mitochondria with low MMP were detected using the TRITC and FITC
channels, respectively. For quantitative analysis, the fluorescence
intensity of the red and green channels from the captured images
was measured using ImageJ software (version 1.53k; National
Institutes of Health).
DNA damage assay
The cells were fixed for 15 min and subsequently
incubated overnight at 4˚C with the γ-H2AX primary antibody (cat.
no. C2035S; Beyotime Institute of Biotechnology). After three
gentle rinses with phosphate-buffered saline (PBS), the cells were
incubated for 1 h with an Alexa Fluor 488-conjugated goat
anti-rabbit secondary antibody. Nuclei were counterstained with
DAPI (10 µg/ml) for 5 min. Fluorescence images were captured via a
fluorescence microscope (MShot MF53-N). The number of γ-H2AX foci
per cell was quantified via the ‘Analyze Particles’ function in
ImageJ software (version 1.53k).
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR) analysis
Total RNA was extracted from cultured AGS and HGC-27
cells using an RNA extraction kit (Omega Bio-Tek, Inc.). cDNA was
synthesized from 1 µg RNA using the SweScript RT SuperMix (Wuhan
Servicebio Technology Co., Ltd.). with the following thermal
protocol: 25˚C for 5 min, 42˚C for 15-30 min, and 85˚C for 5 sec.
qPCR was performed on a QuantStudio 5 system (Applied Biosystems;
Thermo Fisher Scientific, Inc.) using Servicebio SYBR Green Master
Mix. The thermocycling protocol was as follows: 95˚C for 10 min; 40
cycles of 95˚C for 15 sec and 60˚C for 1 min. The relative
expression of NOX1 was normalized to GAPDH and calculated using the
2-ΔΔCq method (17). The
sequences of primers used were as follows: NOX1 forward,
5'-TCTGGTTGTTTGGTTAGGGCT-3' and reverse, 5'-CGGCTGCAAAACCCAAGGA-3';
and GAPDH forward, 5'-CGGAGTCAACGGATTTGGTCGTAT-3' and reverse,
5'-AGCCTTCTCCATGGTGGTGAAGAC-3'.
Colony formation assay
The GC cell lines were seeded into culture plates.
After adhering, the cells were treated with the designated
concentrations for 10 days, followed by staining with 0.1% crystal
violet for 30 min. The plates were rinsed with ultrapure water,
air-dried, and scanned. Colonies containing more than 50 cells were
automatically quantified using ImageJ software (version 1.53k;
National Institutes of Health).
Western blot analysis
Cells were lysed in RIPA buffer (cat. no. P0013B;
Beyotime Institute of Biotechnology) supplemented with 1% PMSF. The
protein concentration was determined using a BCA Protein Assay Kit
(cat. no. P0010; Beyotime Institute of Biotechnology). In total,
~20-30 µg of total protein per lane was separated on 12.5% SDS-PAGE
gels (Epizyme, Inc.) and transferred onto polyvinylidene fluoride
(PVDF) membranes (MilliporeSigma). The membranes were blocked with
5% non-fat milk in TBST (0.1% Tween-20) for 2 h at room temperature
and then incubated overnight at 4˚C with the following primary
antibodies: anti-BCL-2 (1:2,000; cat. no. 12789-1-AP; Proteintech
Group, Inc.), anti-BAX (1:20,000; cat. no. 50599-2-Ig; Proteintech
Group, Inc.), anti-caspase-3/cleaved caspase-3 (1:1,000; cat. no.
19677-1-AP; Proteintech Group, Inc.), anti-NOX1 (1:1,000; cat. no.
671300; Chengdu Zhengneng Biotechnology, Co., Ltd.), anti-β-ACTIN
(1:50,000; cat. no. 66009-1-Ig; Proteintech Group, Inc.) and
anti-GAPDH (1:20,000; cat. no. 10494-1-AP; Proteintech Group,
Inc.). After washing, the membranes were incubated with
HRP-conjugated Goat Anti-Rabbit IgG (1:5,000; cat. no. SA00001-2;
Proteintech Group, Inc.) or HRP-conjugated Goat Anti-Mouse IgG
(1:5,000; cat. no. SA00001-1; Proteintech Group, Inc.) secondary
antibodies for 1 h at room temperature. Protein bands were
visualized using an Enhanced Chemiluminescence (ECL) Substrate
(cat. no. WBKLS0500; MilliporeSigma) and captured digitally. The
band intensities were quantified by densitometry using ImageJ
software (version 1.53k; National Institutes of Health).
Small interfering RNA (siRNA)
transfection
For siRNA transfection, cells were cultured in
6-well plates until they reached ~70-90% confluence. Specific
siRNAs were then transfected via Lipofectamine 2000 reagent (cat.
no. 11668-027; Thermo Fisher Scientific, Inc.). A final
concentration of 20 µM of siRNA was used for each transfection. The
transfection was carried out in serum-free Opti-MEM medium and
incubated with the cells at 37˚C for 4 h, after which the medium
was replaced with complete growth medium. The efficiency of
siRNA-mediated interference was verified 48 h post-transfection via
western blotting to ensure effective knockdown of the target genes.
All siRNAs, including the non-targeting negative control (si-NC),
were designed and synthesized by Anhui General Biotech Co., Ltd.
The siRNA sequences utilized in the present study were as follows:
siNOX1#1 (homo-ID27035 siRNA-318) forward,
5'-AUGAGAAGGCCGACAAAUATT-3' and reverse,
5'-AUUUGCGCAGGCUCUUUGCTT-3'; siNOX1#2 (homo-ID27035 siRNA-1315)
forward, 5'-CAUAAGGGCUUUCGAACAATT-3' and reverse,
5'-UUGUUCGAAAGCCCUUAUGTT-3'; and si-NC forward,
5'-UUCUCCGAACGUGUCACGUTT-3' and reverse,
5'-ACGUGACACGUUCGGAGAATT-3'.
Statistical analysis
The data are presented as the mean ± SDs of three
independent biological replicates. Statistical comparisons between
two groups were performed using an unpaired Student's t test. For
comparisons among more than two groups, one-way analysis of
variance (ANOVA) was employed, followed by Tukey's post hoc test.
P<0.05 was considered to indicate a statistically significant
difference. All statistical analyses were performed via GraphPad
Prism software (version 9.5.1; Dotmatics).
Results
Hesperadin induces cell death and
inhibits proliferation in GC cells
To assess the antitumor activity of Hesperadin, its
IC50 in GC cell lines was first determined. The IC50 values of
Hesperadin in AGS and HGC-27 cells were 52.49 nM and 46.37 nM,
respectively (Fig. 1A). Next,
concentrations of 10 and 20 nM were selected for AGS cells and 8
and 16 nM for HGC-27 cells, all of which are below the IC50 values
for subsequent experiments. Compared with the control, Hesperadin
significantly inhibited the proliferation of GC cells in a
concentration-dependent manner (Fig.
1B).
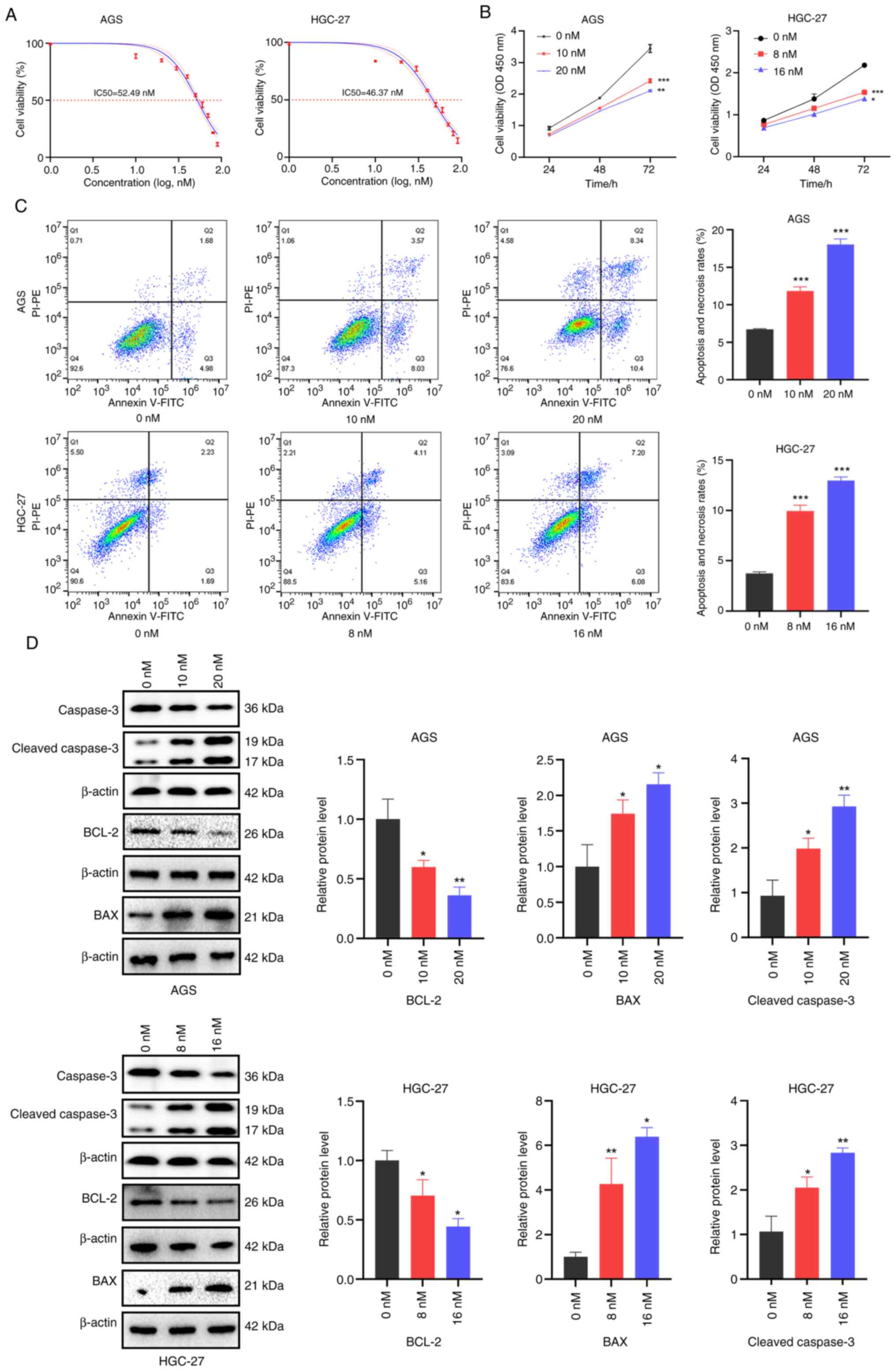 | Figure 1Hesperadin inhibits proliferation and
induces death in gastric cancer cells. (A) After the cells were
treated with graded concentrations of hesperadin (0, 10, 20, 30,
40, 50, 60, 70, 80 and 90 nM) for 48 h, cell viability was assessed
via the CCK-8 assay, and the IC50 was calculated. (B) A
CCK-8 assay was used to evaluate the viability of AGS and HGC-27
cells following 24, 48 and 72 h of Hesperadin treatment. (C) Flow
cytometric analysis was conducted to assess cell mortality after 48
h of Hesperadin treatment. (D) The expression levels of the
apoptosis-related proteins BCL-2, BAX and cleaved caspase-3 were
analyzed via western blotting. β-ACTIN was used as a loading
control. Compared with the control group, the high-concentration
group was compared with the low-concentration group;
*P<0.05, **P<0.01 and
***P<0.001. CCK-8, Cell Counting Kit. |
To further examine the growth-inhibitory effects of
Hesperadin on GC cells, Annexin V-FITC/PI double staining followed
by flow cytometry was performed to evaluate its effect on cell
death. Flow cytometry revealed increased cell death after 48 h of
treatment (Fig. 1C). Western blot
analysis demonstrated that hesperadin significantly downregulated
the expression of the antiapoptotic protein BCL-2 while
upregulating the expression of the proapoptotic proteins BAX and
cleaved caspase-3. These findings indicated that Hesperadin
promotes cell death through activation of the mitochondrial
apoptosis pathway (Fig. 1D). Taken
together, these results demonstrated that Hesperadin suppresses GC
cell proliferation and induces cell death in a dose-dependent
manner via activating the mitochondrial apoptosis pathway.
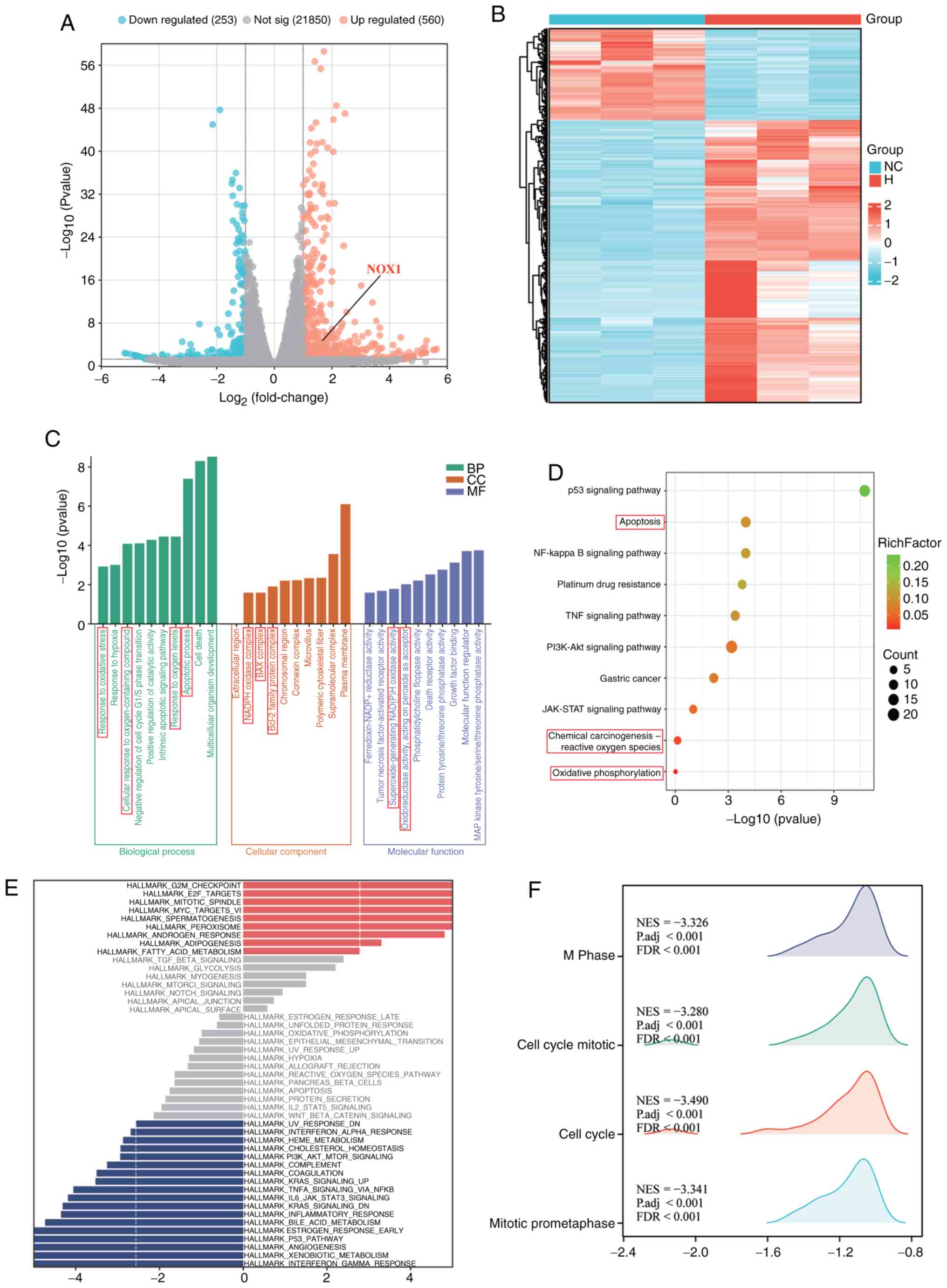
Transcriptomic analysis suggests that
Hesperadin may induce GC cell death through the apoptosis and
oxidative stress pathways
Inducing the apoptosis of tumor cells is one of the
important ways to inhibit cell proliferation (18). To elucidate the molecular mechanisms
driving Hesperadin-induced cell death in GC, RNA-seq was conducted
on AGS cells treated with 10 nM Hesperadin for 48 h. A complete
list of all DEGs is provided in Table
SII. The DEGs identified from the RNA-seq results are
illustrated in Fig. 2A and B. GO, KEGG, GSEA and GSVA enrichment
analyses revealed that the DEGs were significantly enriched in BP
such as apoptosis and the oxidative stress response (Fig. 2C-F). While the top 10 upregulated
DEGs were associated with key cancer pathways such as cell cycle
arrest (for example, CDKN1A), p53 signaling (for example, BTG2) and
cell adhesion (for example, PTPRU) (Table SI), the pronounced enrichment in
oxidative stress pathways particularly captured our attention.
Specifically, GO analysis identified significant enrichment in the
‘cellular response to oxidative stress’ (GO: 0045489) (Fig. 2C) and KEGG analysis confirmed
enrichment in the ‘Oxidative phosphorylation’ pathway (hsa00190)
(Fig. 2D). This transcriptional
profile aligns with the documented mechanism of Hesperadin, which
induces mitochondrial dysfunction and a ROS surge in other cancer
models (16). Consequently, DEGs
within these oxidative stress-related pathways were prioritized for
further investigation. Among them, NADPH Oxidase 1 (NOX1) emerged
as a compelling candidate. Although its fold-change ranked outside
the top 10, NOX1 encodes a critical catalytic subunit of the NADPH
oxidase complex, a dedicated superoxide generator and a master
regulator of oxidative stress (19,20).
Its significant upregulation (log2FC >1.5, P<0.01)
following Hesperadin treatment (Fig.
2A) suggested a functionally central role in mediating the
observed oxidative stress phenotype. NOX1 was therefore selected
for subsequent functional validation.
Hesperadin promotes GC cell death by
activating the oxidative stress pathway through the induction of
ROS generation
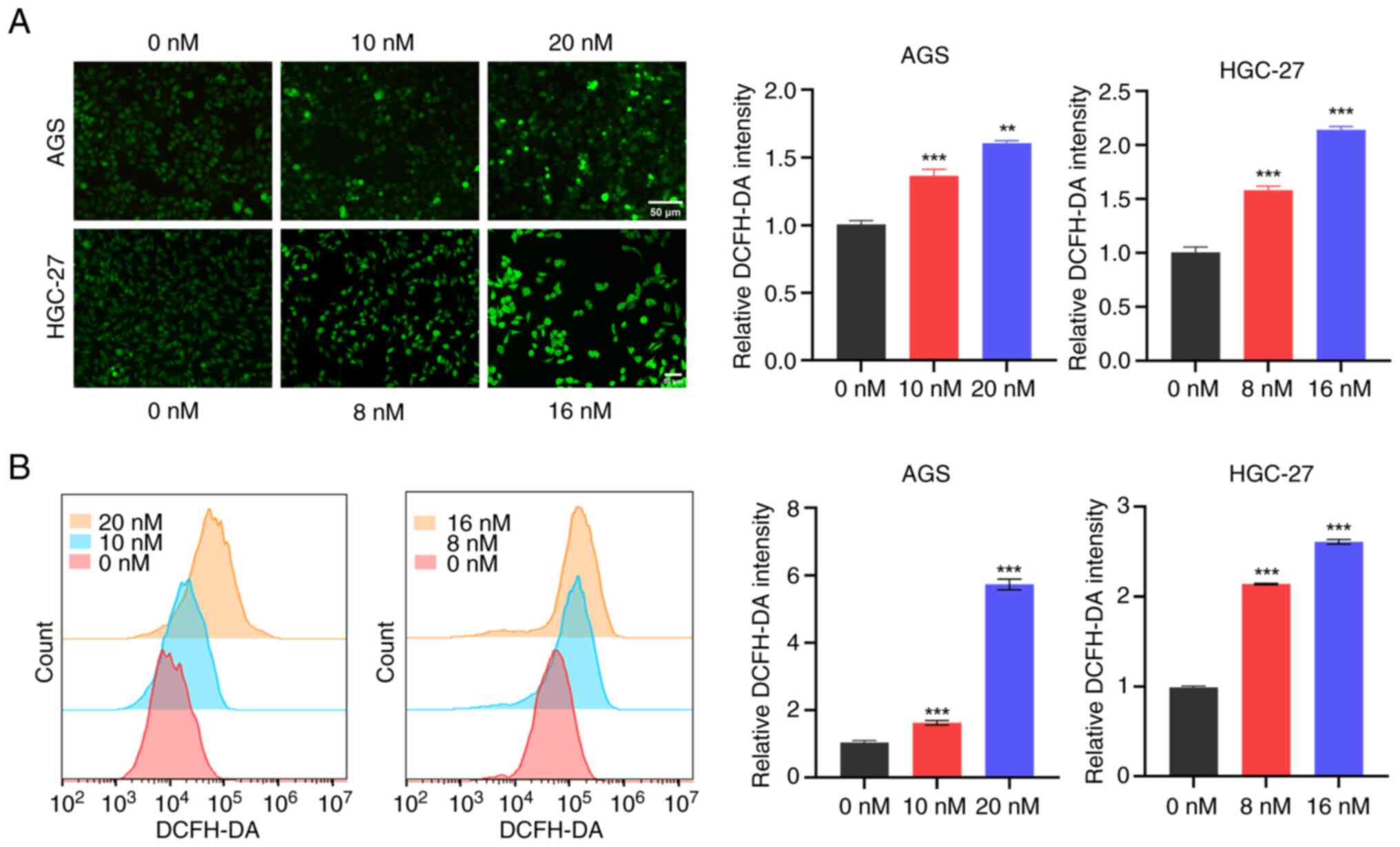
Excessive production of ROS induces oxidative stress
and mitochondrial dysfunction, ultimately leading to cell death
(18,19). To investigate the impact of
Hesperadin on ROS generation and oxidative stress pathways in GC
cells, intracellular ROS levels in AGS and HGC-27 cells were
measured following Hesperadin treatment. The results showed a
significant, concentration-dependent increase in ROS fluorescence
intensity in the Hesperadin-treated cells compared with the control
group (Fig. 3A). Further
quantitative analysis by flow cytometry revealed that Hesperadin
increased ROS levels in GC cells in a dose-dependent manner
(Fig. 3B). In summary, it can be
concluded that Hesperadin induces cell death in GCs by increasing
the generation of ROS, which leads to oxidative stress and
activates the mitochondrial apoptotic pathway.
Hesperadin increases the sensitivity
of GC cells to cisplatin
Cisplatin is a key chemotherapeutic agent widely
used in the treatment of GC (21,22).
Considering the mechanistic similarities between cisplatin and
Hesperadin in modulating tumor-associated pathways, the present
study aimed to evaluate the potential therapeutic benefits of
combining both agents in GC cell models. To investigate the
potential combinatorial effect of Hesperadin and cisplatin, the
IC50 for each agent alone was first determined in AGS
and HGC-27 cells (Figs. 1A and
4A). To distinguish true synergy
from additive toxicity, sub-IC50 concentrations were
selected for all combination treatments. Based on dose-response
profiles and preliminary validation, the chosen concentrations (20
nM Hesperadin and 10 µM cisplatin for AGS cells, and 16 nM
Hesperadin and 8 µM cisplatin for HGC-27 cells) represent ~30-40%
of their respective single-agent IC50 values. These
doses induce partial biological effects (Fig. 1B and C) and moderate proliferation inhibition
without causing overwhelming cell death, thereby providing a clear
window to observe enhanced efficacy upon combination. Under these
conditions, the combinatorial effects were evaluated. The results
revealed that when AGS and HGC-27 cells were treated with a
combination of Hesperadin and cisplatin, the IC50 values
for cisplatin decreased from 33.66 and 26.88 µM to 24.99 and 20.47
µM, respectively (Fig. 4A).
Furthermore, the GC cell survival rate in the Hesperadin-cisplatin
combination treatment group was significantly lower than that in
the control and single-drug groups (Fig. 4B), whereas the clonogenic ability
was significantly reduced (Fig.
4C), and the mortality rate was significantly elevated
(Fig. 4D). Additionally, western
blot analysis revealed that the expression levels of the
proapoptotic proteins BAX and cleaved caspase-3 were increased,
whereas the expression of the antiapoptotic protein BCL-2 was
decreased in the combination treatment group (Fig. 4E). These results indicated that
Hesperadin enhances the antiproliferative effect of cisplatin,
promotes apoptosis, and thereby potentiates the cytotoxic effect of
cisplatin on GC cells.
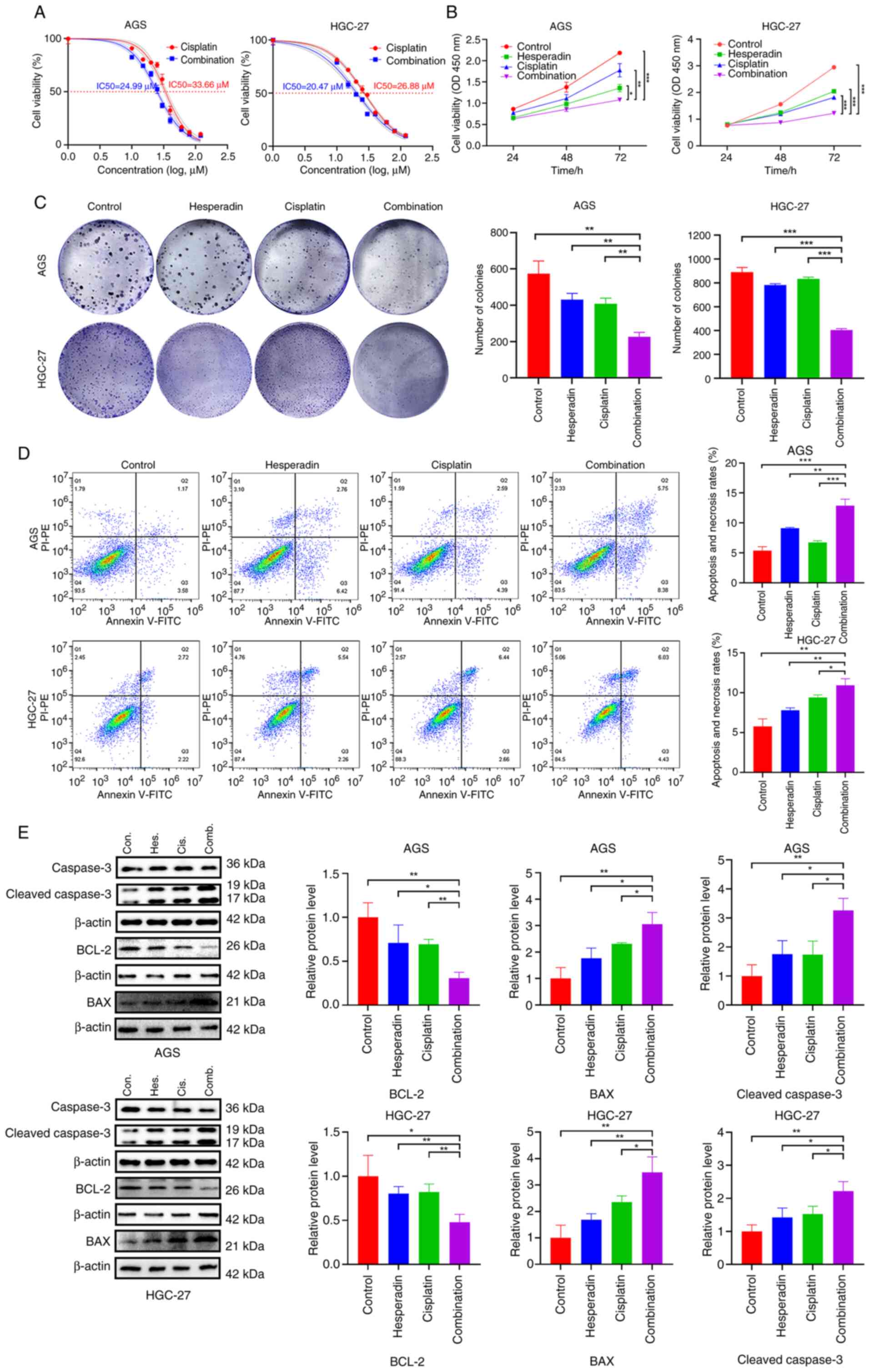 | Figure 4Hesperadin enhances the sensitivity
of gastric cancer cells to cisplatin. (A) Changes in the
IC50 of cisplatin in AGS and HGC-27 cells after 48 h of
cotreatment with Hesperadin (10 nM) and various concentrations of
cisplatin (0, 10, 15, 20, 25, 30, 40, 60, 80, or 120 nM). (B) Cell
Counting Kit-8 assay for cell viability after 24, 48 and 72 h of
cotreatment with Hesperadin and cisplatin. (C) Colony formation
assay to assess the ability of cells to form colonies after 10 days
of cotreatment. (D) Flow cytometric analysis showing cell mortality
after cotreatment with Hesperadin and cisplatin. (E) Western blot
analysis was used to evaluate the expression of apoptosis-related
proteins (BCL-2, BAX and cleaved caspase-3). β-ACTIN was used as a
loading control. Hesperadin concentrations were 20 nM for AGS cells
and 16 nM for HGC-27 cells, whereas cisplatin concentrations were
10 µM for AGS cells and 8 µM for HGC-27 cells. Compared with all
the other groups (Control, Hes. alone, Cis. alone);
*P<0.05, **P<0.01 and
***P<0.001. Con., Control; Hes., Hesperadin; Cis.,
Cisplatin; Comb., Combination. |
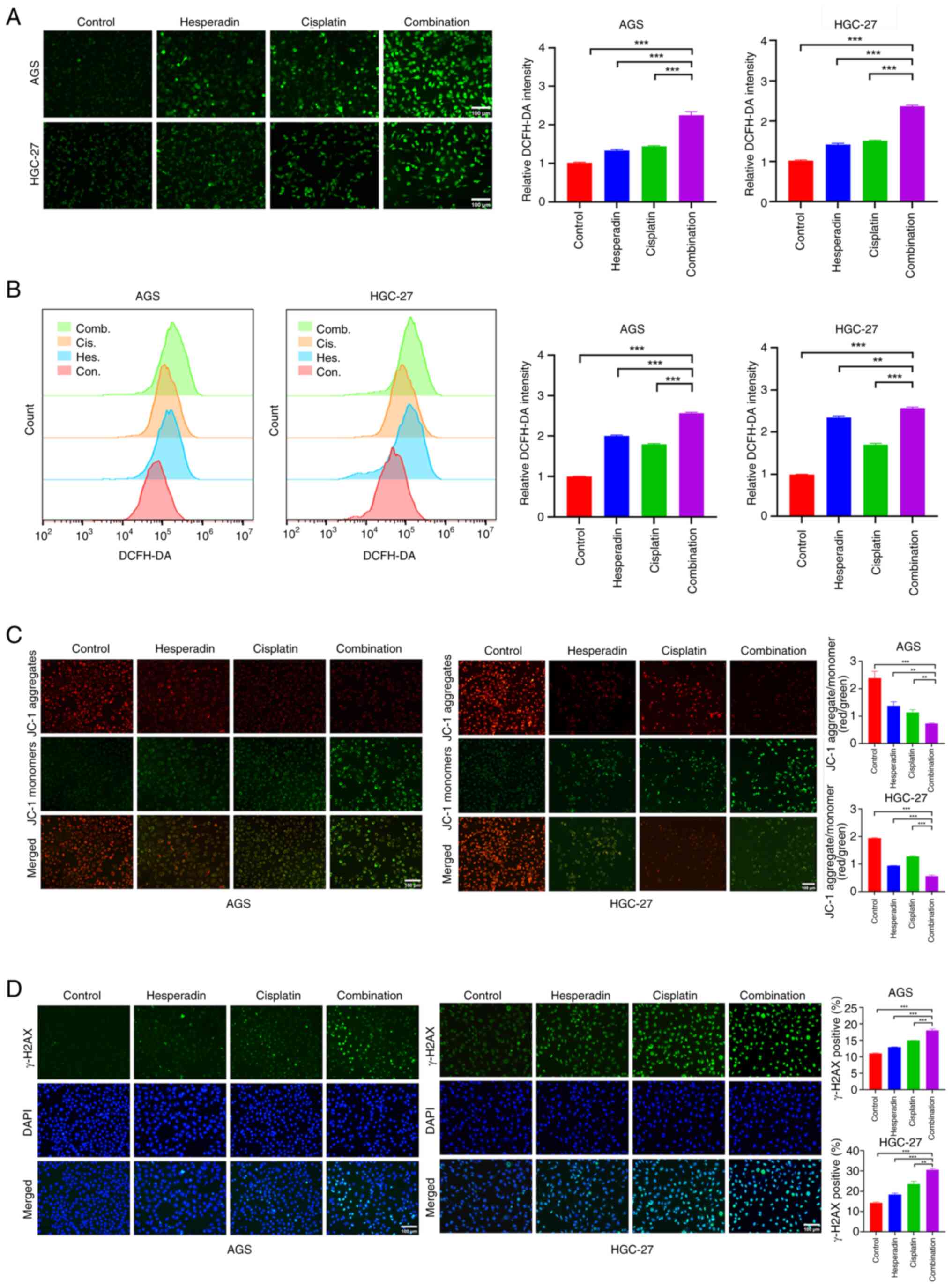
Hesperadin enhances the sensitivity of
GC cells to cisplatin via oxidative stress
Cisplatin exerts its anticancer effect mainly by
binding to DNA to form cross-links and induce the production of
ROS, thereby leading to apoptosis (4,5). The
present study revealed that Hesperadin significantly inhibits GC
cell proliferation and induces cell death by inducing ROS
generation (Figs. 1C and 3). Therefore, it was next explored whether
Hesperadin could increase the sensitivity of GC cells to cisplatin
through oxidative stress. First, the fluorescence intensity in the
Hesperadin-cisplatin combination treatment group was significantly
greater than that in the single-drug treatment groups (Fig. 5A); the same results were obtained
through quantitative flow cytometric analysis (Fig. 5B). Furthermore, it was found that
both Hesperadin alone and cisplatin significantly reduced the MMP,
whereas the combined treatment caused a further significant
reduction in the MMP (Fig. 5C).
Additionally, the fluorescence intensity of the DNA damage marker
γ-H2AX was measured. The results revealed that γ-H2AX fluorescence
intensity in the single-drug treatment groups (Hesperadin or
cisplatin) was significantly higher than in the control group,
whereas the combination treatment group demonstrated a further
significant increase in γ-H2AX fluorescence intensity (Fig. 5D), indicating more extensive DNA
damage following combination treatment. Collectively, these
findings indicated that Hesperadin enhances the sensitivity of GC
cells to cisplatin by promoting ROS generation, disrupting MMP,
exacerbating DNA damage, and thereby facilitating cell death.
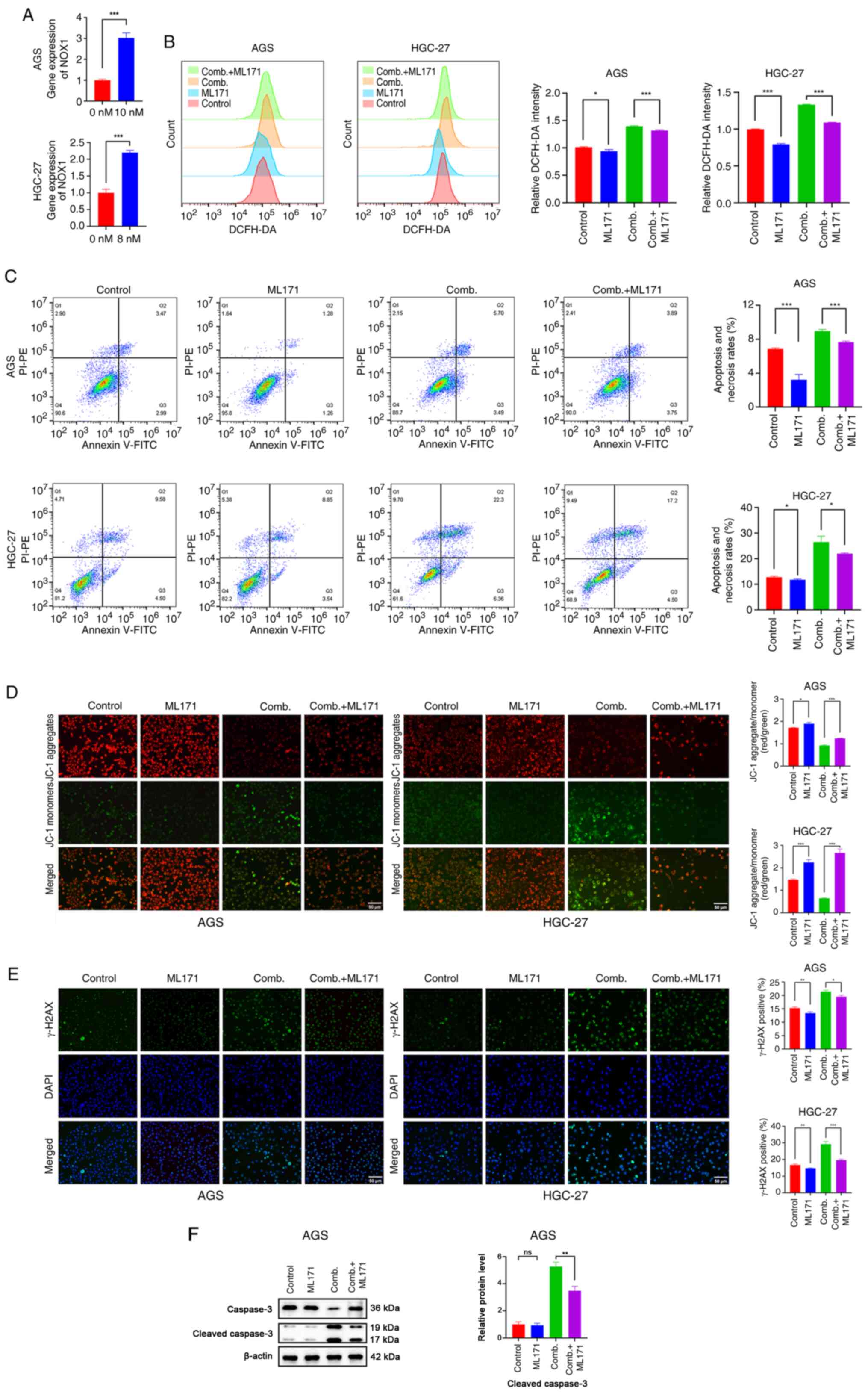
Hesperadin enhances the sensitivity of
GC cells to cisplatin by activating NOX1-induced oxidative
stress
To directly investigate whether the
chemo-sensitizing effect of Hesperadin toward cisplatin was
mediated through the NOX1/ROS axis, as suggested by our
transcriptomic data, the upregulation of NOX1 expression was first
confirmed. Through RT-qPCR analysis, it was found that NOX1
expression was significantly upregulated upon Hesperadin treatment
in GC (Fig. 6A). In addition,
western blot analysis confirmed that the Hesperadin alone,
cisplatin alone, and combination treatments significantly increased
the protein expression level of NOX1 in AGS cells. In HGC-27 cells,
the increase in NOX1 protein following cisplatin treatment did not
reach statistical significance, highlighting a cell-type-dependent
difference in response. However, the combination treatment with
Hesperadin robustly increased NOX1 levels in both cell lines
(Fig. S1C), which was consistent
with the observed transcriptional changes. To validate the
functional role of NOX1, siRNA was subsequently used to knock down
NOX1 expression in AGS and HGC-27 cells. Compared with si-NC
transfection, NOX1-specific siRNA transfection significantly
reduced NOX1 protein levels (Fig.
S1A). ML171, a highly selective NOX1 inhibitor, was
subsequently employed to evaluate the role of NOX1 in
Hesperadin-induced oxidative stress and its impact on cisplatin
sensitivity in GC cells. The results revealed that both ML171
treatment and NOX1 knockdown markedly attenuated the generation of
ROS induced by the combination of cisplatin and Hesperadin
(Figs. 6B and S1B). Furthermore, NOX1 inhibition rescued
the cell death caused by the combined treatment (Fig. 6C). MMP analysis revealed that NOX1
inhibition partially reversed the MMP reduction induced by the
combination treatment (Fig. 6D).
Additionally, γ-H2AX immunofluorescence analysis revealed that NOX1
inhibition significantly attenuated DNA damage in the combination
treatment group (Fig. 6E). To
provide molecular substantiation for the flow cytometry results in
AGS cells, western blot analysis for Cleaved Caspase-3 was
performed using the same treatments. Consistent with the functional
data, the combination treatment significantly increased the level
of Cleaved Caspase-3, which was significantly reversed by
co-treatment with the NOX1 inhibitor ML171 (Fig. 6F). Collectively, these findings
indicated that Hesperadin promotes ROS generation through
NOX1-mediated oxidative stress in GC cells.
Discussion
Hesperadin was initially synthesized as a novel
indolinone compound during anti-proliferative screening and was
subsequently identified as a potent Aurora B kinase inhibitor that
disrupts mitosis by impairing chromosome segregation and spindle
assembly checkpoint function (23).
Its mechanisms of action not only include G2/M phase
cell cycle arrest through Aurora B inhibition but also involve the
induction of DNA damage, oxidative stress (ROS generation),
endoplasmic reticulum stress, and mitochondrial dysfunction,
ultimately promoting cancer apoptosis (16,24).
Additionally, Hesperadin has been identified as a selective
inhibitor of CaMKII-δ, suggesting potential cardioprotective
effects (15). It also acts as a
broad-spectrum anti-influenza agent effective against both
influenza A and B viruses, including oseltamivir-resistant strains
(25). However, its application and
mechanism in the treatment of GC have not yet been extensively
studied.
In the present study, it was confirmed that
Hesperadin significantly inhibits GC cell proliferation and induces
apoptosis while enhancing the chemosensitivity of GC cells to
cisplatin. To elucidate the underlying mechanism, transcriptomic
analysis and identified NOX1 was performed, a key member of the
NADPH oxidase family, as an upstream regulator of Hesperadin's
action. Combined treatment with Hesperadin and cisplatin
significantly upregulated NOX1 expression. Importantly, inhibition
of NOX1 substantially reversed the ROS generation and cytotoxic
effects induced by the combination treatment. These results
demonstrated that Hesperadin enhances cisplatin sensitivity in GC
cells primarily through NOX1-mediated ROS overproduction. Notably,
Hesperadin can modulate NOX1 expression, and cisplatin has also
been shown to do so in other models. Research by Kim et al
(26) in auditory cells showed that
cisplatin upregulates NOX1 expression by inducing the release of
pro-inflammatory cytokines (for example, TNF-α, IL-1β and IL-6) and
activating the MAPK/ERK signaling pathway. However, the specific
mechanism by which cisplatin regulates NOX1 in GC requires further
investigation. On the other hand, as a dual inhibitor of Aurora B
and CAMKII-δ, Hesperadin may influence ROS responses through
multiple pathways. Aurora B, a key mitotic regulator, is involved
in DNA damage response and cell survival under genotoxic stress.
Its inhibition may compromise DNA repair fidelity, thereby
amplifying cisplatin-induced DNA damage and ROS accumulation
(27,28). Meanwhile, CAMKII-δ is known to
participate in redox signaling and mitochondrial regulation in
various cell types (29,30). Although its role in GC remains
unclear, it was hypothesized that CAMKII-δ may similarly regulate
NOX1 activity or mitochondrial ROS generation in GC cells.
Elucidating the specific mechanisms and potential crosstalk between
Hesperadin and cisplatin in regulating NOX1 will be a focus of our
future research. However, the present study has certain
limitations. First, the present findings require further validation
in in vivo animal models to confirm the therapeutic efficacy
and safety profile of the Hesperadin and cisplatin combination.
Second, it is important to note that our mechanistic focus on NOX1
and oxidative stress was influenced by prior literature (31,32)
and the established role of ROS in Hesperadin's action in other
cancers (16). While the present
RNA-seq data supported this focus (Fig.
2C and D), this targeted
approach may have introduced a selection bias, potentially
overlooking other DEGs with higher fold changes or unknown
functions in GC (Table SI) that
could also contribute to the observed phenotype. Moreover, the
precise molecular mechanisms by which Hesperadin regulates NOX1
expression and mediates oxidative stress warrant deeper
investigation. Specifically, the upstream regulatory mechanisms
(for example, how Hesperadin activates NOX1
transcription/translation through specific signaling pathways) and
the downstream effector protein interaction network following
NOX1-mediated ROS generation remain insufficiently elucidated.
Research indicates that the regulation of NOX1 by cisplatin in
auditory cells is associated with inflammatory signaling, whereas
the mechanism by which Hesperadin modulates NOX1 in GC cells
remains unclear. Although the present RNA-seq analysis implicated
potential involvement of pathways such as p53, NF-κB and PI3K-Akt
(Fig. 2D), these preliminary
findings await future experimental validation to define their
precise roles in this regulatory axis.
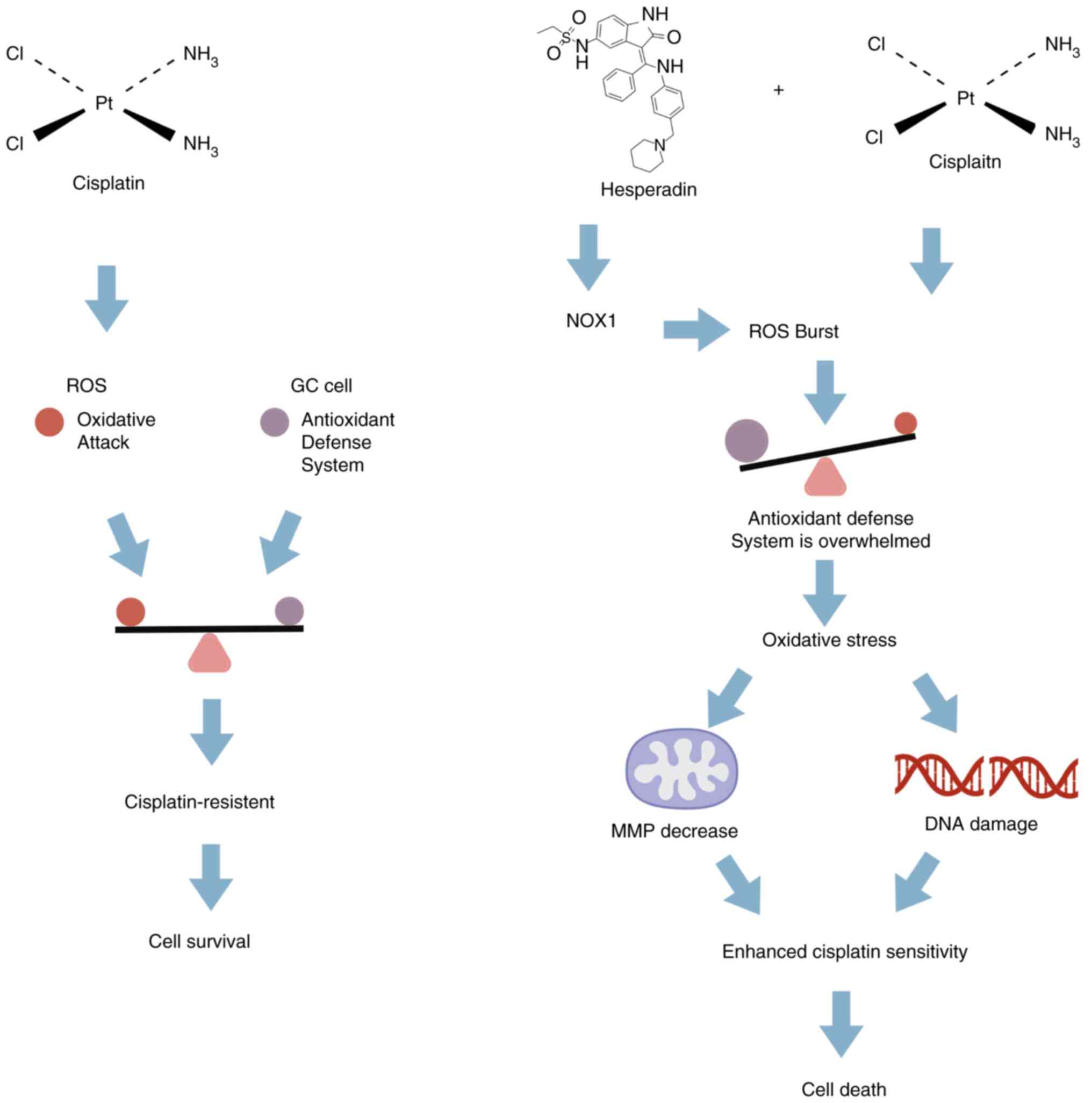
In summary, the present study demonstrated that
Hesperadin sensitizes GC cells to cisplatin by triggering
NOX1-mediated oxidative stress and mitochondrial
dysfunction-induced apoptosis (Fig.
7). Additionally, the current findings indicated that the
combination of Hesperadin and cisplatin represents a promising
therapeutic strategy for the treatment of GC.
Supplementary Material
Transcriptomic profiling and
validation of the NOX1-mediated mechanism. (A) Efficiency of NOX1
knockdown using two independent siRNAs. Western blot analysis
showing NOX1 protein expression in AGS and HGC-27 cells transfected
with si-NC or two different NOX1-specific siRNAs (si-NOX1#1 and
si-NOX1#2). GAPDH served as the loading control.
*P<0.05, **P<0.01 and
***P<0.001 vs. the si-NC group. (B) Genetic
inhibition of NOX1 abrogates Hesperadin-induced ROS generation.
DCFH-DA fluorescence staining (green fluorescence represents ROS;
scale bar, 50 μm) and quantitative analysis showing ROS
levels in AGS and HGC-27 cells transfected with si-NC or si-NOX1
(#1 or #2) and treated with or without Hesperadin.
*P<0.05, **P<0.01 and
***P<0.001 vs. the si-NC + Hesperadin group; ns: not
significant (P≥0.05) vs. the si-NC group. (C) Western blot analysis
of NOX1 protein expression in AGS and HGC-27 cells following
treatment with Hesperadin, cisplatin, or their combination for 48
h. GAPDH was used as a loading control. Data were compared among
all groups. *P<0.05, **P<0.01 and
***P<0.001. siRNA, small interfering RNA; si-NC,
negative control siRNA; ROS, ROS, reactive oxygen species; ns, not
significant (P≥0.05).
Top 10 DEGs identified by
RNA-sequencing analysis.
DEGs in Hesperadin-treated AGS
cells.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Natural Science
Foundation of Bengbu Medical College (grant no. 2022byfy002), the
Graduate Research Innovation Program of Bengbu Medical College
(grant no. Byycx24005), the University Synergy Innovation Program
of Anhui (grant no. GXXT-2022-065) and the Natural Science Program
of Bengbu Medical University (grant no. 2023byzd030).
Availability of data and materials
The data generated in the present study may be found
in the NCBI Sequence Read Archive under accession numbers
SRR35296175, SRR35296174, SRR35296173, SRR35296169, SRR35296168 and
SRR35296167 or at the following URL: https://www.ncbi.nlm.nih.gov/sra/?term=SRR35296175;
https://www.ncbi.nlm.nih.gov/sra/?term=SRR35296174;
https://www.ncbi.nlm.nih.gov/sra/?term=SRR35296173;
https://www.ncbi.nlm.nih.gov/sra/?term=SRR35296169;
https://www.ncbi.nlm.nih.gov/sra/?term=SRR35296168;
https://www.ncbi.nlm.nih.gov/sra/?term=SRR35296167.
The data generated in the present study may be requested from the
corresponding author.
Authors' contributions
WJZ, HZW and YLZ conceptualized the study. ZLJ, QYL,
WJZ and DW analyzed and curated data. ZLJ, QYL, JYC and WJZ
conducted investigation. JYC designed and established the
experimental methodology. WJZ, ZLJ, QYL and DW wrote the original
draft. WJZ and HZW revised the manuscript. All authors wrote,
reviewed and edited the manuscript. All authors read and approved
the final version of the manuscript. WJZ and HZW confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Laversanne M, Sung H, Ferlay J,
Siegel RL, Soerjomataram I and Jemal A: Global cancer statistics
2022: GLOBOCAN estimates of incidence and mortality worldwide for
36 cancers in 185 countries. CA Cancer J Clin. 74:229–263.
2024.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Digklia A and Wagner AD: Advanced gastric
cancer: Current treatment landscape and future perspectives. World
J Gastroenterol. 22:2403–2414. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lei ZN, Teng QX, Tian Q, Chen W, Xie Y, Wu
K, Zeng Q, Zeng L, Pan Y, Chen ZS and He Y: Signaling pathways and
therapeutic interventions in gastric cancer. Signal Transduct
Target Ther. 7(358)2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Huang R and Zhou PK: DNA damage repair:
Historical perspectives, mechanistic pathways and clinical
translation for targeted cancer therapy. Signal Transduct Target
Ther. 6(254)2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Peña Q, Wang A, Zaremba O, Shi Y, Scheeren
HW, Metselaar JM, Kiessling F, Pallares RM, Wuttke S and Lammers T:
Metallodrugs in cancer nanomedicine. Chem Soc Rev. 51:2544–2582.
2022.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Ruhlmann CH, Iversen TZ, Okera M, Muhic A,
Kristensen G, Feyer P, Hansen O and Herrstedt J: Multinational
study exploring patients' perceptions of side-effects induced by
chemo-radiotherapy. Radiother Oncol. 117:333–337. 2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Shahid M, Subhan F, Ahmad N and Sewell
RDE: The flavonoid 6-methoxyflavone allays cisplatin-induced
neuropathic allodynia and hypoalgesia. Biomed Pharmacother.
95:1725–1733. 2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Yu C, Dong H, Wang Q, Bai J, Li YN, Zhao
JJ and Li JZ: Danshensu attenuates cisplatin-induced nephrotoxicity
through activation of Nrf2 pathway and inhibition of NF-κB. Biomed
Pharmacother. 142(111995)2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wang H, Zhang Z, Wei X and Dai R:
Small-molecule inhibitor of Bcl-2 (TW-37) suppresses growth and
enhances cisplatin-induced apoptosis in ovarian cancer cells. J
Ovarian Res. 8(3)2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Shi S, Bai X, Ji Q, Wan H, An H, Kang X
and Guo S: Molecular mechanism of ion channel protein TMEM16A
regulated by natural product of narirutin for lung cancer adjuvant
treatment. Int J Biol Macromol. 223:1145–1157. 2022.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Bai X, Liu Y, Cao Y, Ma Z, Chen Y and Guo
S: Exploring the potential of cryptochlorogenic acid as a dietary
adjuvant for multi-target combined lung cancer treatment.
Phytomedicine. 132(155907)2024.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Morahan BJ, Abrie C, Al-Hasani K, Batty
MB, Corey V, Cowell AN, Niemand J, Winzeler EA, Birkholtz LM,
Doerig C and Garcia-Bustos JF: Human Aurora kinase inhibitor
hesperadin reveals epistatic interaction between Plasmodium
falciparum PfArk1 and PfNek1 kinases. Commun Biol.
3(701)2020.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Wu X, Wu J, Hu W, Wang Q, Liu H, Chu Z, Lv
K and Xu Y: MST4 kinase inhibitor Hesperadin attenuates autophagy
and behavioral disorder via the MST4/AKT pathway in intracerebral
hemorrhage mice. Behav Neurol. 2020(2476861)2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Chhajer R, Bhattacharyya A and Ali N: Cell
death in Leishmania donovani promastigotes in response to Mammalian
Aurora kinase B inhibitor-Hesperadin. Biomed Pharmacother.
177(116960)2024.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zhang J, Liang R, Wang K, Zhang W, Zhang
M, Jin L, Xie P, Zheng W, Shang H, Hu Q, et al: Novel CaMKII-δ
inhibitor Hesperadin exerts dual functions to ameliorate cardiac
ischemia/reperfusion injury and inhibit tumor growth. Circulation.
145:1154–1168. 2022.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zhang Y, Wu J, Fu Y, Yu R, Su H, Zheng Q,
Wu H, Zhou S, Wang K, Zhao J, et al: Hesperadin suppresses
pancreatic cancer through ATF4/GADD45A axis at nanomolar
concentrations. Oncogene. 41:3394–3408. 2022.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Carneiro BA and El-Deiry WS: Targeting
apoptosis in cancer therapy. Nat Rev Clin Oncol. 17:395–417.
2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Li J, Jia YC, Ding YX, Bai J, Cao F and Li
F: The crosstalk between ferroptosis and mitochondrial dynamic
regulatory networks. Int J Biol Sci. 19:2756–2771. 2023.PubMed/NCBI View Article : Google Scholar
|
|
20
|
To TL, Cuadros AM, Shah H, Hung WHW, Li Y,
Kim SH, Rubin DHF, Boe RH, Rath S, Eaton JK, et al: A compendium of
genetic modifiers of mitochondrial dysfunction reveals
intra-organelle buffering. Cell. 179:1222–1238.e17. 2019.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wagner AD, Syn NL, Moehler M, Grothe W,
Yong WP, Tai BC, Ho J and Unverzagt S: Chemotherapy for advanced
gastric cancer. Cochrane Database Syst Rev.
8(CD004064)2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Al-Batran SE, Homann N, Pauligk C, Goetze
TO, Meiler J, Kasper S, Kopp HG, Mayer F, Haag GM, Luley K, et al:
Perioperative chemotherapy with fluorouracil plus leucovorin,
oxaliplatin, and docetaxel versus fluorouracil or capecitabine plus
cisplatin and epirubicin for locally advanced, resectable gastric
or gastro-oesophageal junction adenocarcinoma (FLOT4): A
randomised, phase 2/3 trial. Lancet. 393:1948–1957. 2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Hauf S, Cole RW, LaTerra S, Zimmer C,
Schnapp G, Walter R, Heckel A, van Meel J, Rieder CL and Peters JM:
The small molecule Hesperadin reveals a role for Aurora B in
correcting kinetochore-microtubule attachment and in maintaining
the spindle assembly checkpoint. J Cell Biol. 161:281–294.
2003.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Ladygina NG, Latsis RV and Yen T: Effect
of the pharmacological agent hesperadin on breast and prostate
tumor cultured cells. Biomed Khim. 51:170–176. 2005.PubMed/NCBI(In Russian).
|
|
25
|
Hu Y, Zhang J, Musharrafieh R, Hau R, Ma C
and Wang J: Chemical genomics approach leads to the identification
of Hesperadin, an Aurora B kinase inhibitor, as a broad-spectrum
influenza antiviral. Int J Mol Sci. 18(1929)2017.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Kim HJ, Lee JH, Kim SJ, Oh GS, Moon HD,
Kwon KB, Park C, Park BH, Lee HK, Chung SY, et al: Roles of NADPH
oxidases in cisplatin-induced reactive oxygen species generation
and ototoxicity. J Neurosci. 30:3933–3946. 2010.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Chen TM, Huang CM, Setiawan SA, Hsieh MS,
Sheen CC and Yeh CT: KDM5D histone demethylase identifies
platinum-tolerant head and neck cancer cells vulnerable to mitotic
catastrophe. Int J Mol Sci. 24(5310)2023.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Han X, Zhang JJ, Han ZQ, Zhang HB and Wang
ZA: Let-7b attenuates cisplatin resistance and tumor growth in
gastric cancer by targeting AURKB. Cancer Gene Ther. 25:300–308.
2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Erickson JR, Joiner ML, Guan X, Kutschke
W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O'Donnell SE,
Aykin-Burns N, et al: A dynamic pathway for calcium-independent
activation of CaMKII by methionine oxidation. Cell. 133:462–474.
2008.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Anderson ME, Brown JH and Bers DM: CaMKII
in myocardial hypertrophy and heart failure. J Mol Cell Cardiol.
51:468–473. 2011.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Zhang Z, Zhao X, Gao M, Xu L, Qi Y, Wang J
and Yin L: Dioscin alleviates myocardial infarction injury via
regulating BMP4/NOX1-mediated oxidative stress and inflammation.
Phytomedicine. 103(154222)2022.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Drummond GR, Selemidis S, Griendling KK
and Sobey CG: Combating oxidative stress in vascular disease: NADPH
oxidases as therapeutic targets. Nat Rev Drug Discov. 10:453–471.
2011.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Jiang S, Li H, Zhang L, Mu W, Zhang Y,
Chen T, Wu J, Tang H, Zheng S, Liu Y, et al: Generic diagramming
platform (GDP): A comprehensive database of high-quality biomedical
graphics. Nucleic Acids Res. 53 (D1):D1670–D1676. 2025.PubMed/NCBI View Article : Google Scholar
|