Introduction
Thalassemia is one of the most common hematological
disorders globally, characterized by impaired synthesis of
hemoglobin (Hb) chains. The primary form of adult hemoglobin is
hemoglobin A (HbA), which accounts for >95% of total Hb and
exists as a tetramer composed of two α-globin chains and two β-like
globin chains (α2β2). Disruption of the
balanced synthesis of these α- and β-globin chains, triggered by
mutations or deletions in the α-globin-encoding genes HBA1
(OMIM *141800) and HBA2 (OMIM *141850), or the
β-globin-encoding gene HBB (OMIM * 141900), leads to red
blood cell damage and hemolysis, ultimately resulting in
α-thalassemia or β-thalassemia, respectively (1,2).
Genetically, the human genome normally contains four functional
copies of HBA gene (genotype: αα/αα) and two functional
copies of the HBB gene (genotype:
βN/βN). The clinical severity of thalassemia
is directly tied to the number of functionally defective HBA
or HBB alleles. Based on this allele dosage effect,
α-thalassemia is classified into four subtypes: α-thalassemia
minima (α+ trait, mildest, often asymptomatic),
α-thalassemia trait (αTT, mild anemia), α-thalassemia intermedia
(αTI, moderate anemia requiring intermittent transfusion), and
α-thalassemia major (αTM, severe, life-threatening anemia);
β-thalassemia is classified into three subtypes: β-thalassemia
trait (βTT, mild asymptomatic anemia), β-thalassemia intermedia
(βTI, moderate anemia with infrequent transfusion needs), and
β-thalassemia major (βTM, severe anemia requiring lifelong regular
transfusions) (3-4).
Among the regions heavily affected by thalassemia
worldwide, China, especially the southern provinces, stands out due
to high disease prevalence and significant associated health
burdens, making it a key focus for thalassemia epidemiological and
clinical research (5). In recent
years, due to population mobility and the growing number of
inter-regional marriages, the thalassemia carrier rate has been
exhibiting a trend towards northward expansion (6).
In accordance with national health policies, Hubei
Province incorporated thalassemia into the newborn screening
program in 2016, and subsequently the screening was extended to the
prenatal stage in 2022. However, the epidemiological data on
thalassemia in Hubei exhibit regional variability. In light of
these disparities, the present study focused on individuals of
reproductive age and the pediatric population in Hubei in an aim to
analyze thalassemia-associated genetic mutations and hematological
changes. It is hoped that the results of the present study can
provide comprehensive and solid data for genetic counseling,
disease prevention and early intervention strategies.
Materials and methods
Study subjects
Between January, 2019 and September, 2024, a total
of 2,604 individuals of reproductive age and 407 pediatric
subjects, who underwent thalassemia gene testing at Renmin Hospital
of Wuhan University (Wuhan, China), were enrolled in the present
study. The individuals of reproductive age, which were
non-selectively screened, had a mean age of 31.39±5.72 years (age
range, 17-60 years) and included 526 males (20.20%; mean age,
33.25±6.36 years) and 2,078 females (79.80%; mean age, 30.93±5.45
years). The pediatric subjects had a mean age of 3.19±4.31 years
(age range, 1 day to 16 years), consisting of 230 males (mean age,
2.72±3.88 years) and 177 females (mean age, 3.78±4.76 years). These
pediatric subjects were enrolled based on clinical indications,
including jaundice, anemia, hemolytic anemia, or a family history
of thalassemia. Hematological and anemia-related biochemical data
collected during their hospital visits were analyzed. Ethical
approval for the present study was obtained from the Medical Ethics
Committee of Renmin Hospital of Wuhan University (Wuhan, China),
with the approval no. WDRY2023-K175. Individual informed consent
was waived by the Ethics Committee of Renmin Hospital of Wuhan
University, as the study involved no additional interventions, and
all data were analyzed in a de-identified manner.
Instruments and reagents
Hematological analysis was performed using the
Sysmex XN-9000 automated hematology analyzer (Sysmex Corporation).
The following reagents were used: the CELLPACK DCL diluent (Sysmex
Corporation) for cell counting and hemoglobin measurement, the
SULFOLYSER lysing reagent (Sysmex Corporation) for hemoglobin
release, and the STROMATOLYSER-FB reagent (Sysmex Corporation) for
fluorescent staining of nucleic acids to perform leukocyte
differential counting. Biochemical assays for serum iron (SI),
total iron binding capacity (TIBC), serum ferritin (SF), folate
(FOL), and vitamin B12 (VITB12) were performed on an ADVIA 1800
analyzer (Siemens Healthineers) using the manufacturer's
corresponding reagent kits, namely the ADVIA Chemistry Iron Assay
(cat. no. 10377510), ADVIA Chemistry TIBC Assay (cat. no.
03940010), ADVIA Centaur Ferritin Assay (cat. no. 10309969), ADVIA
Centaur Folate Assay (cat. no. 124838), and ADVIA Centaur Vitamin
B12 Assay (cat. no. 10330218), respectively. Transferrin saturation
(TS) was calculated from the SI and TIBC results. The GeneRotex 96
nucleic acid extraction system (Xi'an Tianlong Science and
Technology Co., Ltd.) and whole blood genomic DNA extraction kits
(cat. no. T146H; Xi'an Tianlong Science and Technology Co., Ltd.)
were utilized for genomic DNA extraction. A NanoDrop
spectrophotometer (Thermo Fisher Scientific) was used to assess DNA
quality. Thalassemia mutations were analyzed using commercial
detection kits from Shenzhen Yaneng Bioscience, including the
deletion α-thalassemia gene assay kit (cat. no. 20243401205), the
non-deletion α-thalassemia gene assay kit (cat. no. 20193401915),
and the β-thalassemia gene assay kit (cat. no. 20163400463). An
Automatic Hybridization System (Shenzhen Yaneng Bioscience) was
used to identify thalassemia gene mutations. A 3500 Genetic
Analyzer (Thermo Fisher Scientific) and a Veriti 96-well Fast
Thermal Cycler (Thermo Fisher Scientific) were used for further
genetic analysis.
Hematology and biochemical
analyses
Peripheral venous blood (2 ml) was collected in EDTA
anticoagulant tubes for the analysis of red blood cell count (RBC),
Hb, mean corpuscular volume (MCV) and mean corpuscular hemoglobin
(MCH), which were measured using a Sysmex XN-9000 automated
hematology analyzer (Sysmex Corporation). Serum samples (5 ml) were
collected into a serum separator tube with inert gel for the
measurement of SI, TIBC, SF, TS, FOL and VITB12 levels, which were
determined using an ADVIA 1800 clinical chemistry analyzer (Siemens
Healthineers).
Genetic testing
Genomic DNA was first extracted and then quantified
to ensure its concentration ranged from 50-150 ng/µl, with an
OD260/280 ratio between 1.7 and 1.9. Gap-polymerase chain reaction
(gap-PCR) was applied to detect three common α-thalassemia
deletions, namely--SEA, -α3.7 and -α4.2. Reverse dot blot (RDB) was
used to identify non-deletion α-thalassemia mutations (αWS, αQS and
αCS) and 17 β-thalassemia mutations (CD41-2, βE, CD43, CD71-72,
IVS-II-654, -28, -29, -30, -32, CD14-15, CD27-28, CD31, IVS-I-1,
IVS-I-5, CAP, CD17 and the initiation codon). The complete
specifications of all tested mutations, including their Human
Genome Variation Society (HGVS) nomenclature and functional
impacts, are presented in Table I.
Cases were considered discordant if the hematological indices (such
as MCV <80 fl, MCH <27 pg) suggested a thalassemia trait, but
the initial gap-PCR/RDB results were negative, or if
genotype-phenotype inconsistencies were observed (for example
severe anemia with mild genotype). Such cases were prioritized for
Sanger sequencing and research-grade gap-PCR (Shenzhen Yaneng
Bioscience) to confirm rare genotypes.
 | Table ICommon thalassemia mutations assessed
and their molecular features. |
Table I
Common thalassemia mutations assessed
and their molecular features.
| Traditional name | Gene | Transcript | HGVS
nomenclature | Common name | Type | Functional
impact |
|---|
| 31M | HBB | NM_000518.5 | c.94delC
(p.Arg32fs) | CD31(-C) | β0 | Translation
affected |
| CapM | HBB | NM_000518.5 | c.-11_-8delAAAC | CAP+43/+40
(-AAAC) | β+ | Transcription
affected |
| 41-42M | HBB | NM_000518.5 | c.126_129delCTTT
(p.Phe42Leufs*19) | CD 41/42 (-TTCT) | β0 | Translation
affected |
| 43M | HBB | NM_000518.5 | c.130G>T
(p.Glu44*) | CD 43 (G>T) | β0 | Translation
affected |
| 71-72M | HBB | NM_000518.5 | c.216_217insA
(p.Ala73Serfs*13) | CD 71/72 (+A) | β0 | Translation
affected |
| IntM | HBB | NM_000518.5 | c.2T>G
(p.Met1Arg) | Init CD
ATG>AGG | β0 | Translation
affected |
| 654M | HBB | NM_000518.5 | c.316-197C>T | IVS2-654C>T | β+ | RNA processing
affected |
| 14-15M | HBB | NM_000518.5 | c.45_46insG
(p.Trp15Glyfs*2) | CD 14/15 (+G) | β0 | Translation
affected |
| 17M | HBB | NM_000518.5 | c.52A>T
(p.Lys18*) | CD 17
(AAG>TAG) | β0 | Translation
affected |
| -28M | HBB | NM_000518.5 | c.-78A>G | -28A>G | β+ | Transcription
affected |
| -29M | HBB | NM_000518.5 | c.-79A>G | -29A>G | β+ | Transcription
affected |
| 26M (βEM) | HBB | NM_000518.5 | c.79G>A
(p.Glu27Lys) | CD 26 GAG>AAG
[Glu>Lys] | β0 | RNA processing
affected |
| -30M | HBB | NM_000518.5 | c.-80T>C | -30T>C |
β0/β+ | Transcription
affected |
| -32M | HBB | NM_000518.5 | c.-82C>A | -32 (C>A) | β+ | Transcription
affected |
| 27/28M | HBB | NM_000518.5 | c.84_85insC
(p.Leu28Profs*22) | CD 27/28 (+C) | β0 | Translation
affected |
| IVS-I-1M | HBB | NM_000518.5 | c.92+1G>T | IVS 1-1 (G>T) | β0 | RNA processing
affected |
| IVS-1-5M | HBB | NM_000518.5 | c.92+5G>C | IVS1-5G>C | β+ | RNA processing
affected |
| --SEA | HBA1+ HBA2 | NC_000016.9 | g.215400_
234700del19300 | --SEA | α0 | α0
deletion type |
| -α4.2 | HBA2 | NC_000016.9 | g.219817_
224074del4258 | -α4.2 | α+ | α+
deletion type |
| -α3.7 | HBA1 HBA2 | NC_000016.9 | g.223300_
227103del3804 | -α3.7 | α+ | α+
deletion type |
| αWS | HBA2 | NM_000517.6 | c.369C>G
(p.His123Gln) | αWS | α+ | Translation
affected |
| αQS | HBA2 | NM_000517.6 | c.377T>C
(p.Leu126Pro) | αQS | α0 | Translation
affected |
| αCS | HBA2 | NM_000517.6 | c.427T>
(p.Trp143Gln) | αCS | α0 | Translation
affected |
Clinical phenotype classification
Based on consensus guidelines (3,4),
β-thalassemia phenotypes were classified into βTT, βTI, and βTM.
α-thalassemia was categorized as α+ trait, αTT, αTI, αTM and rare
genotypes.
Statistical analysis
Microsoft Excel was utilized to input and organize
the data. Carrier rates and proportions are presented as
percentages (%). For the carrier population data, the direct
counting method was employed, with the number of detected cases
denoted as ‘n’. Hematological and biochemical indicators were
characterized using the mean ± standard deviation (SD). Statistical
analyses were performed using Prism 9.0 (Dotmatics) and SPSS 13.0
(SPSS, Inc.) software. To compare continuous variables, t-tests
were applied. For categorical variables, the chi-squared test was
utilized for assessment. P<0.05 was considered to indicate a
statistically significant difference.
Results
Prevalence and genotype
distribution
The thalassemia carrier rate was 14.94% (389/2604)
in the individuals of reproductive age and 37.35% (152/407) in the
pediatric population. The carrier rates for α-thalassemia,
β-thalassemia and αβ-thalassemia were 7.60% (198/2604), 7.68%
(200/2604) and 0.35% (9/2604) in the individuals of reproductive
age, respectively and 16.46% (67/407), 21.38% (87/407) and 0.49%
(2/407) in the pediatric group, respectively (Table II, Table III and Table IV).
| Table IIPrevalence and genotype distribution
of thalassemia. |
Table II
Prevalence and genotype distribution
of thalassemia.
| | Reproductive age
group | Pediatric
group | Statistics |
|---|
| Clinical
Phenotype | Genotype | n | Proportions
(%) | Carriage rate
(%) | n | Proportions
(%) | Carriage rate
(%) | χ² and P-value |
|---|
| α+
trait | | 86 | 43.43 | 3.30 | 18 | 26.87 | 4.42 | 1.32, 0.25 |
| |
-α3.7/αα | 73 | 36.87 | 2.80 | 16 | 23.88 | 3.93 | |
| | -α4.2
/αα | 9 | 4.55 | 0.35 | 2 | 2.99 | 0.49 | |
| |
αWSα/αα | 4 | 2.02 | 0.15 | 0 | 0.00 | 0.00 | |
| αTT | | 99 | 50.00 | 3.80 | 48 | 71.64 | 11.79 | 48.41,
<0.01a |
| |
--SEA/αα | 93 | 46.97 | 3.57 | 43 | 64.18 | 10.57 | |
| |
αCSα/αα | 3 | 1.52 | 0.12 | 2 | 2.99 | 0.49 | |
| |
αQSα/αα | 1 | 0.51 | 0.04 | 2 | 2.99 | 0.49 | |
| |
-α3.7/-α4.2 | 1 | 0.51 | 0.04 | 1 | 1.49 | 0.25 | |
| |
αQSα/-α3.7 | 1 | 0.51 | 0.04 | 0 | 0.00 | 0.00 | |
| αTI | | 7 | 3.54 | 0.27 | 1 | 1.49 | 0.25 | 0.01, 0.93 |
| |
--SEA/-α3.7 | 4 | 2.02 | 0.15 | 1 | 1.49 | 0.25 | |
| |
αCSα/--SEA | 2 | 1.01 | 0.08 | 0 | 0.00 | 0.00 | |
| |
--SEA/-α4.2 | 1 | 0.51 | 0.04 | 0 | 0.00 | 0.00 | |
| Rare Genotype |
HKαα/αα | 6 | 3.03 | 0.23 | 0 | 0.00 | 0.00 | 0.94, 0.33 |
| |
HKαα/-α3.7 | 5 | 2.53 | 0.19 | 0 | 0.00 | 0.00 | |
| |
HKαα/--SEA | 1 | 0.51 | 0.04 | 0 | 0.00 | 0.00 | |
| Total | | 198 | 100 | 7.60 | 67 | 100 | 16.46 | 34.41,
4.46x10-9a |
| Table IIIDistribution and frequency of
β-thalassemia genotype. |
Table III
Distribution and frequency of
β-thalassemia genotype.
| | Reproductive-age
group (n=2604) | Pediatric group
(n=407) | Statistics |
|---|
| Clinical
phenotype | Genotype | n | Proportions
(%) | Carriage rate
(%) | n | Proportions
(%) | Carriage rate
(%) | χ2 and
P-value |
|---|
| βTT | | 200 | 98.00 | 7.53 | 86 | 96.55 | 20.64 | 74.07,
<0.01a |
| |
βN/β654 | 97 | 48.50 | 3.73 | 41 | 47.13 | 10.07 | |
| |
βN/β41-42 | 55 | 27.50 | 2.11 | 20 | 22.99 | 4.91 | |
| |
βN/β17 | 24 | 12.00 | 0.92 | 10 | 11.49 | 2.46 | |
| |
βN/β-28 | 9 | 4.50 | 0.35 | 3 | 3.45 | 0.74 | |
| |
βN/βE | 6 | 3.00 | 0.23 | 3 | 3.45 | 0.74 | |
| |
βN/β71-72 | 2 | 1.00 | 0.08 | 3 | 3.45 | 0.74 | |
| |
βN/β-29 | 1 | 0.50 | 0.04 | 1 | 1.15 | 0.25 | |
| |
βN/β43 | 1 | 0.50 | 0.04 | 2 | 2.30 | 0.49 | |
| |
βN/βCAP | 1 | 0.50 | 0.04 | 0 | 0.00 | 0.00 | |
| |
βN/βIVS-I-1 | 0 | 0.00 | 0.00 | 1 | 1.15 | 0.25 | |
| |
βN/β27-28 | 3 | 1.50 | 0.12 | 2 | 2.30 | 0.49 | |
| |
βN/β14-15 | 1 | 0.50 | 0.04 | 0 | 0.00 | 0.00 | |
| βTI | | 0 | 0.00 | 0.00 | 1 | 1.15 | 0.25 | 6.40,
0.011a |
| |
β654/β654 | 0 | 0.00 | 0.00 | 1 | 1.15 | 0.25 | |
| Total | | 200 | 100 | 7.68 | 87 | 100 | 21.38 | 76.56,
<0.01a |
| Table IVDistribution and frequency of
αβ-thalassemia genotypes. |
Table IV
Distribution and frequency of
αβ-thalassemia genotypes.
| Genotype | Reproductive Age
Group | Pediatric
Group | Statistics |
|---|
| α-Thalassemia | β-Thalassemia | n | Proportions
(%) | Carriage rate
(%) | n | Proportions
(%) | Carriage rate
(%) | χ2 and
P-value |
|---|
|
--SEA/αα |
βN/β654 | 2 | 22.22 | 0.08 | 0 | 0.00 | 0.00 | |
|
--SEA/αα |
βN/βE | 1 | 11.11 | 0.04 | 1 | 50.00 | 0.25 | |
|
--SEA/αα |
βN/β17 | 1 | 11.11 | 0.04 | 1 | 50.00 | 0.25 | |
|
--SEA/αα |
βN/β-28 | 1 | 11.11 | 0.04 | 0 | 0.00 | 0.00 | |
|
-α3.7/αα |
βN/β654 | 1 | 11.11 | 0.04 | 0 | 0.00 | 0.00 | |
|
-α3.7/αα |
βN/βCAP | 1 | 11.11 | 0.04 | 0 | 0.00 | 0.00 | |
|
-α3.7/αα |
βN/β654 | 1 | 11.11 | 0.04 | 0 | 0.00 | 0.00 | |
|
-α3.7/αα |
βN/β654 | 1 | 11.11 | 0.04 | 0 | 0.00 | 0.00 | |
| Total | | 9 | 100 | 0.35 | 2 | 100 | 0.49 | 0.21, 0.65 |
The overall population carrier rate was 17.97%
(541/3011), with the carrier rates for α-thalassemia and
β-thalassemia being 8.8% (265/3011) and 9.53% (287/3011),
respectively.
Predominant genotypes for
thalassemia
The predominant genotypes observed in both groups
were--SEA/αα and -α3.7/αα. The αTT phenotype was most common,
accounting for 71.64% (48/67) in the pediatric group, which was
significantly higher than that in the individuals of reproductive
age [50.00% (99/198)] (P<0.01) (Table II).
The dominant genotypes in both groups were β654,
β41-42 and β17. The βTT phenotype accounted for 98.00 and 96.55% of
cases in the individuals of reproductive age and pediatric
populations, respectively (Table
III). A total of 11 cases of αβ-thalassemia were identified,
presenting eight distinct genotypes (Table IV).
Regional genotype comparisons
The α-thalassemia genotype distribution in Hubei
(--SEA/αα, -α3.7/αα-α4.2/αα) was consistent with that of
neighboring regions (Hunan, Jiangxi, Chongqing and Guizhou);
however, it differed from high-prevalence regions (Yunnan, Guangxi
and Hainan) (7-14)
(Fig. 1A). Similarly, the primary
β-thalassemia genotypes (βN/β654 and βN/β41-42) in Hubei were
similar to those in Hunan and Jiangxi, although they diverged from
other regions (Fig. 1B).
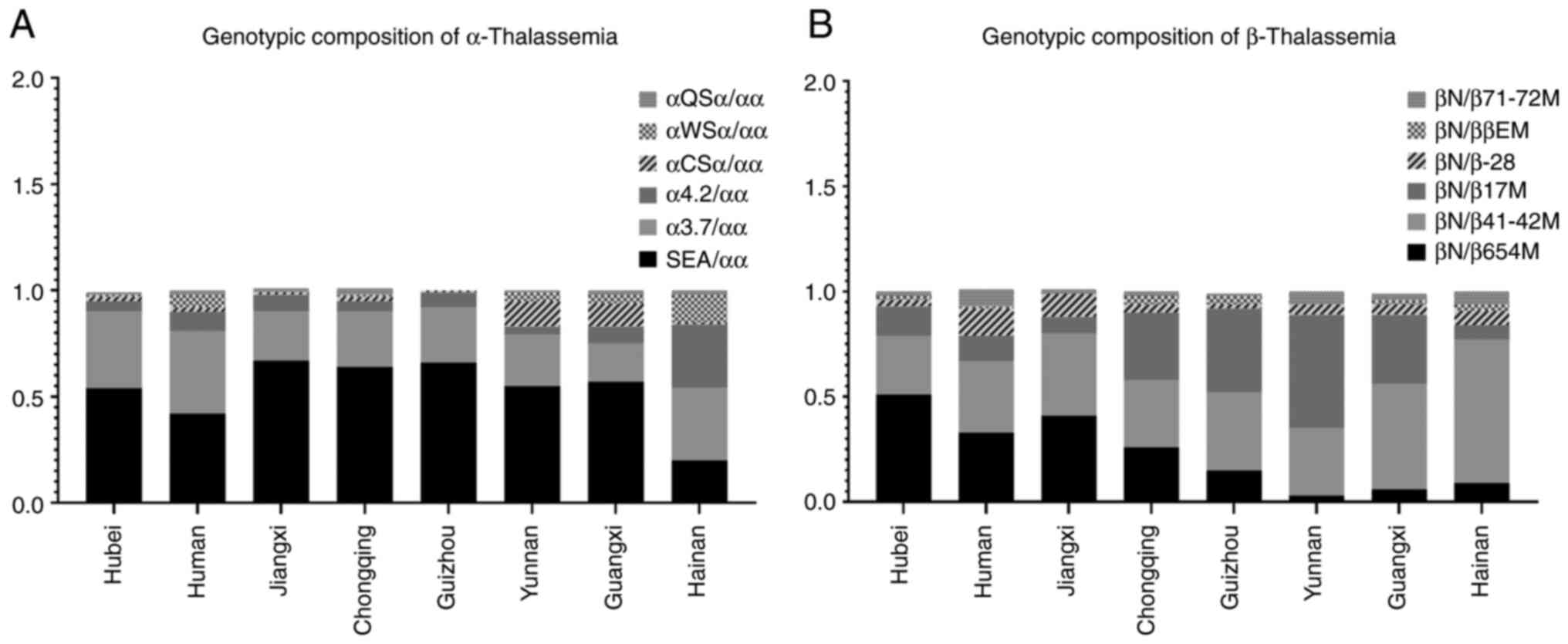 | Figure 1Genotypic composition of
α-/β-thalassemia across geographic regions. (A) Depiction of the
most prevalent genotypic distribution of α-thalassemia, including
deletion variants (--SEA/αα, -α3.7/αα, -α4.2/αα) and non-deletion
variants (αWSα/αα, αQSα/αα, αCSα/αα) in Hubei and other
high-prevalence regions (Hunan, Jiangxi, Chongqing, Guizhou,
Yunnan, Guangxi, Hainan). (B) Illustratrion of the dominant
genotypic composition of β-thalassemia, featuring mutations β654,
β41-42, β17, β-28, βE and β71-72M in the same regions, with βN
denoting wild-type alleles. Bar heights reflect the relative
frequency of each genotype, demonstrating that the genotypes in
Hubei align with neighboring provinces (Hunan, Jiangxi, Chongqing
and Guizhou), but differ from southern high-prevalence areas
(Yunnan, Guangxi and Hainan). |
Characteristics of hematological and
anemia-related biochemical data among different clinical phenotype
groups
Significantly reduced levels of RBC, Hb, MCV and MCH
(P<0.05) were observed across all clinical phenotypes, apart
from rare subtypes. Notably, the reductions in Hb and MCH
parameters were more marked compared to those in RBC and MCV
measurements (Fig. 2).
Concurrently, thalassemia carriers exhibited significantly elevated
SF and vitamin B12 levels (P<0.05 for both biomarkers), as
detailed in Table V.
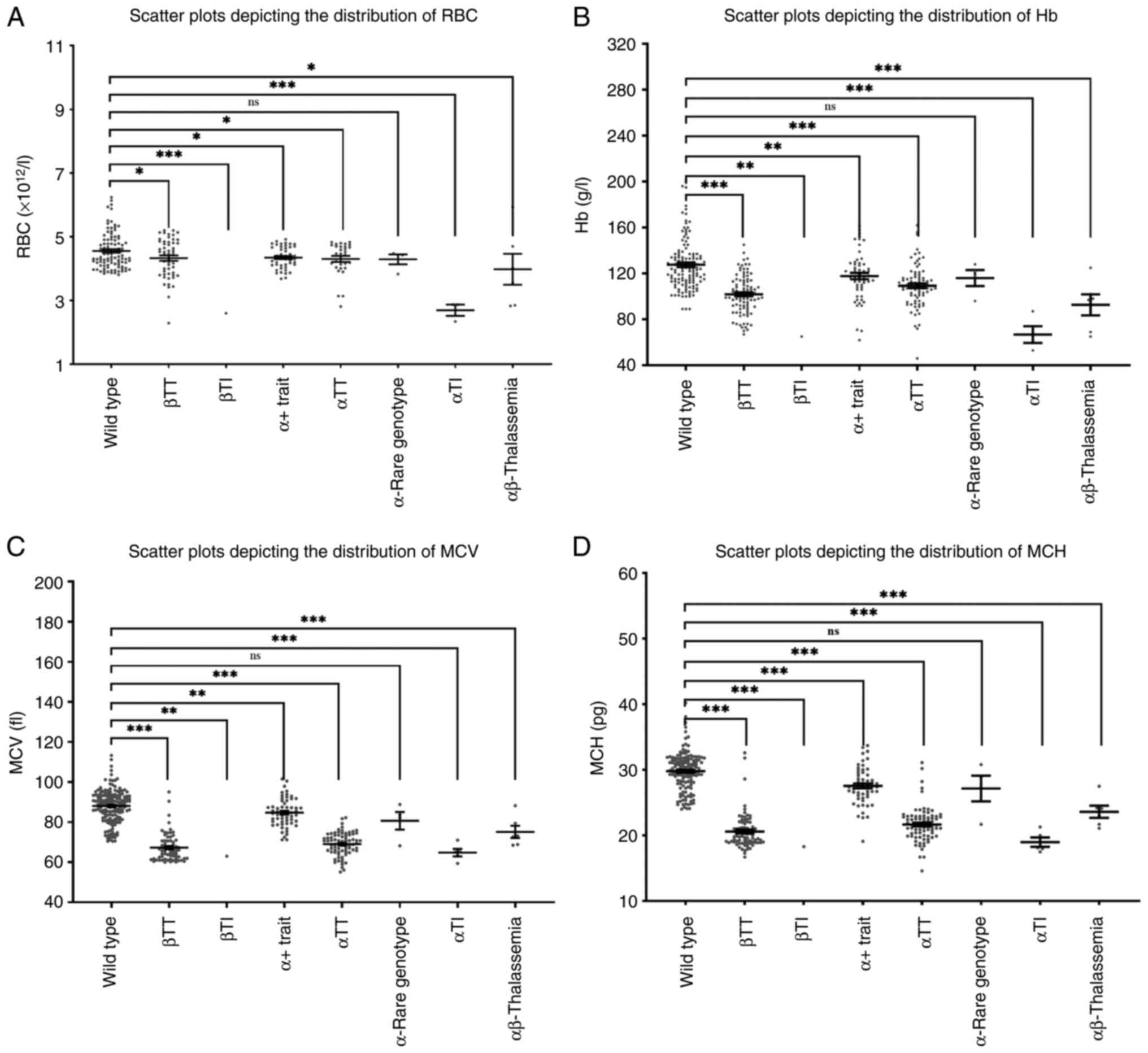 | Figure 2Distribution of RBC, Hb, MCV and MCH
values across thalassemia clinical phenotypes. (A-D) Scatter plots
illustrating individual data points with group medians for (A) RBC
(x1012/l), (B) Hb (g/l), (C) MCV (fl) and (D) MCH (pg)
across thalassemia phenotypes, including βTT, βTI, αTT, αTI and
αβ-thalassemia compared to wild-type controls, with symbols
indicating statistical significance vs. wild type (ns, not
significant; *P≤0.05, **P≤0.01 and
***P≤0.001) and hematological parameters differentiating
phenotypes from controls and correlating with clinical severity.
RBC, red blood cell count; Hb, hemoglobin; MCV, mean corpuscular
volume; MCH, mean corpuscular hemoglobin; βTT, β-thalassemia trait;
βTI, β-thalassemia intermediate; αTT, α-thalassemia trait; αTI,
α-thalassemia intermedia. |
| Table VVariations in iron and folate
metabolism in patients with thalassemia (mean ± SD, N). |
Table V
Variations in iron and folate
metabolism in patients with thalassemia (mean ± SD, N).
| Phenotype
classification biochemical indicators | SF (ng/ml) | FOL (ng/ml) | VITB12 (pg/ml) | SI (µmol/l) | TIBC (µmol/l) | TS (%) |
|---|
| Wild type | 68.94±20.35,
112 | 21.80±3.70, 78 | 287.90±27.06,
89 | 12.85±1.73, 64 | 49.63±3.01, 96 | 25.06±3.97, 82 |
| α+
trait | 74.13±32.21,
18 | 12.28±2.49, 12 | 398.60±44.47,
11 | 13.98±3.10, 10 | 69.40±6.08, 12 | 23.47±4.55, 12 |
| αTT | 90.13±22.20,
40 | 14.00±1.10, 31 | 468.30±44.73,
31 | 22.38±4.40, 26 | 59.95±2.89, 33 | 26.25±3.13, 32 |
| αTI | 313.5±96.34, 6 | 13.41±6.05, 3 | 458.00±50.06,
3 | 24.02±7.33, 5 | 39.20±8.78, 4 | 64.08±11.20, 4 |
| Rare genotypes | 73.13±23.11, 3 | 10.67±4.04, 3 | 471.70±106.50,
3 | 18.33±3.41, 3 | 64.60±9.62, 3 | 30.99±9.14, 3 |
| βTT | 176.00±30.82,
58 | 23.76±9.12, 54 | 521.20±44.33,
54 | 18.97±1.28, 41 | 65.26±5.48, 59 | 29.74±2.00, 54 |
| βTI | 465.40±0.00, 1 | 19.24±0.00, 1 | 527.00±0.00, 1 | 16.40±0.00, 1 | 32.80±0.00, 1 | 50.00±0.00, 1 |
| αβ-Thalassemia | 69.25±5.65, 2 | 14.94±1.22, 3 | 432.30±40.99,
3 | 10.77±2.87, 3 | 51.87±3.24, 3 | 20.34±4.63, 3 |
| χ2 and
P-value | 2.95,
0.01a | 0.26, 0.97 | 4.14,
0.00b | 1.73, 0.11 | 1.95, 0.06 | 1.41, 0.21 |
Discussion
Thalassemia, an inherited hematologic disorder, is
characterized by a broad spectrum of genotypic variations and
diverse clinical manifestations. Its high morbidity and substantial
disease burden pose significant public health challenges. In China,
the disease exhibits marked regional heterogeneity, with
particularly high prevalence rates observed in southern provinces,
including Hainan and Guangdong. Notably, the implementation of
comprehensive prenatal screening programs in these high-endemic
regions has effectively reduced the incidence of severe thalassemia
cases among newborns, demonstrating the importance of preventive
strategies.
The present large-scale genetic screening study
provides comprehensive epidemiological data on thalassemia in Hubei
Province, revealing several critical findings. The observed carrier
rate of 14.94% among individuals of reproductive age aligns with
regional reports from Tongji Hospital (19.08%) and Changsha DIAN
Medical Laboratory (26.18%) (15),
while demonstrating some variation from other published data
(16). This discrepancy may reflect
differences in study populations or methodological approaches, such
as variations in ethnic composition, geographical origins and
sampling methods, as well as methodological differences in genetic
testing and data analysis.
The present study positions Hubei as an emerging
thalassemia-endemic region, with an overall carrier rate of 17.97%,
particularly among pediatric populations (37.35%). The high
pediatric carrier rate approaches that of traditional high-burden
regions, indicating potential inadequacies in current screening
programs (15,17-20).
Given that the pediatric subjects in the present study had specific
clinical indications related to thalassemia, it is likely that the
actual carrier rate in the general pediatric population is lower,
although this may still be concerning. This highlights the urgent
need to strengthen prenatal prevention strategies, including
expanding screening coverage, improving screening methods and
enhancing genetic counseling services.
The genotypic distribution patterns observed in the
present study are consistent with both intraprovincial reports
within Hubei and inter-provincial data from neighboring regions
(21-23).
This suggests relative genetic homogeneity across the province and
shared genetic-environmental influences with adjacent regions. The
predominance of --SEA/αα and CD41-42 mutations aligns with
established genetic drift patterns in central China, likely
reflecting historical population migrations from southern endemic
provinces where these mutations are highly prevalent (6). Understanding these genetic patterns is
crucial for predicting disease prevalence, developing targeted
prevention strategies and providing accurate genetic
counseling.
The findings of the present study strongly support
the primary thalassemia screening strategy in China, which combines
phenotypic screening techniques by using RBC indices (RBC, Hb, MCV
and MCH) with hemoglobin analysis. These parameters demonstrate a
strong association with genetic diagnoses across all clinical
phenotypes, rendering them effective initial screening tools.
However, due to the potential for false negatives in routine blood
tests, especially in high-prevalence regions, it is advisable to
directly conduct universal genetic testing in prenatal care
(24). This approach can improve
the accuracy of diagnosis and enable early intervention, reducing
the risk of severe thalassemia in offspring.
Notably, the present study reveals a previously
unreported positive association between the thalassemia carrier
status and elevated serum vitamin B12 levels (χ2=4.14,
P<0.01). This unexpected finding may be related to compensatory
erythropoiesis, where the body increases the utilization of vitamin
B12 as a substrate for hemoglobin synthesis in response to impaired
hemoglobin production. However, considering the cross-sectional
design of the present study and the limited sample size for this
specific analysis, future research with larger cohorts and
longitudinal measurements of vitamin B12 metabolism is required to
confirm this association and clarify the underlying mechanisms.
In conclusion, the present study establishes Hubei
as an emerging thalassemia-endemic region in central China, with
pediatric carrier rates approaching those of traditionally
high-burden areas. These findings emphasize the urgent need for
targeted public health interventions, including enhanced screening
programs, optimized genetic counseling services and region-specific
prevention strategies (14,25). Although Hubei has implemented
prenatal and newborn screening initiatives, the observed regional
genotypic heterogeneity, such as distinct profiles compared to
southern provinces, highlights the necessity for tailored screening
algorithms. The identification of a substantial carrier rate among
individuals of reproductive age (14.94%) and the utility of key
hematological markers (Hb, MCH) provide actionable insight to
refine local screening protocols, thereby reducing diagnostic gaps
and improving the cost-effectiveness of existing programs.
Integrating hematological and genetic screening approaches remains
critical for effective thalassemia control in Hubei, with the
potential to mitigate disease burden and enhance population health
outcomes. Notably, the higher carrier rate in pediatric subjects
(37.35%) may be attributed to selection bias inherent in
hospital-based sampling, as these individuals were recruited based
on clinical indicators, such as anemia and jaundice, or a family
history of thalassemia, factors strongly associated with increased
thalassemia risk. This recruitment strategy likely overestimates
the true population-level prevalence compared to community-based
screening. Future studies utilizing representative community
samples are therefore warranted to validate these estimates and
provide a more accurate assessment of the regional thalassemia
burden.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Natural Science
Foundation of Hubei Province (grant no. 2024AFC016).
Availability of data and materials
The data generated in the present study are not
publicly available due restrictions that apply to the availability
of these data, which were used under the license from the Ethics
Committee of Renmin Hospital of Wuhan University, but may be
requested from the corresponding author.
Authors' contributions
WY made a substantial contribution to the
acquisition of data by performing all laboratory experiments for
thalassemia diagnosis. YL substantially contributed to the data
acquisition by performing clinical examinations and compiling the
hematological and biochemical profiles. JW was responsible for
reviewing and validating the data analysis results, as well as
drafting the introduction and discussion sections of the
manuscript. TY and YZ confirm the authenticity of all the raw data
and conducted data analysis. Additionally, TY and YZ took primary
responsibility for drafting the initial versions of the
methodology, the results sections and all tables of the manuscript.
JW designed the study and edited the final manuscript. All authors
read and approved the final manuscript.
Ethics approval and consent to
participate
Ethics approval for the present study was obtained
from the Medical Ethics Committee of Renmin Hospital of Wuhan
University (approval no. WDRY2023-K175; Wuhan, China). This
retrospective study utilized anonymized clinical laboratory data
from routine patient care at Renmin Hospital of Wuhan University.
Individual informed consent was waived by the Ethics Committee of
Renmin Hospital of Wuhan University, as the study involved no
additional interventions, and all data were analyzed in a
de-identified manner.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Baird DC, Batten SH and Sparks SK:
Alpha-and beta-thalassemia: rapid evidence review. Am Fam
Physician. 105:272–280. 2022.PubMed/NCBI
|
|
2
|
Lee JS, Im Cho S, Park SS and Seong MW:
Molecular basis and diagnosis of thalassemia. Blood Res. 56
(Suppl):S39–S43. 2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Writing group for practice guidelines for
diagnosis and treatment of genetic diseases. Clinical practice
guidelines for alpha thalassemia. Chin J Med Gene. 235–242.
2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Writing group for practice guidelines for
diagnosis and treatment of genetic diseases. Clinical practice
guidelines for β-thalassemia. Chin J Med Genet. 243–251.
2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Tingting Z and Xiangmin X: Advances in the
Prevention, Diagnosis, and Treatment of Thalassemia Guangdong Med
J: 1189-1193, 2023.
|
|
6
|
Beijing angel mother Charity Foundation
and China Institute of Public Welfare of Beijing normal university
Foundation: BLUE BOOK OF THALASSEMIA IN CHINA: Investigation report
on the prevention and treatment of thalassemia in China (2020),
China Social Publishing House, Beijing, 2021.
|
|
7
|
Zhizhuo H, Yun G, Wei C and Guanda H:
Analysis of screening and genotypes of thalassemia in Liuzhou Area
of Guangxi Province. J Clin Hematol (China). 414–418. 2022.
|
|
8
|
Tao J, Bodan W, Dan H, Desheng Z, Aiqi C,
Ruihong W, Yiming Y and Jie Z (eds.): Thalassemia Screening and
Genetic Diagnosis Results Analysis in Wenshan Zhuang-Miao
Autonomous Prefecture, Yunnan Province. J Kunming Univ Sci Technol
(Natural Sci). 162–167. 2023.
|
|
9
|
Qiuling J, Qi L, Wenye S, Yao Z, Hongjian
C, Huimin H, Weiying L and Yanlin M: The Prevalence and genetic
analysis of Thalassemia in 20450 cases in Hainan. J Practical Med.
1092–1095. 2020.
|
|
10
|
Jincai W, Kaichun W, Kunlan Y and Shuxian
D: Analysis of screening results of 92550 cases of thalassemia in
Puning area. Hainan Med J. 105–108. 2024.
|
|
11
|
Min Y, Caiyun L, Dongzhu L and Haoqing Z:
Application of Next-generation sequencing in screening of
thalassemia gene in 11212 pregnant women in suxian and beihu
districts of Chenzhou city, Hunan province. J Exp Hematol. 188–192.
2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Yuan Y, Lidan Z, Ying Z, Shengyan L, Jin
CYW, Yong Z, Xi XCZ and Yunlong L: Screening results and analysis
on genotype of thalassemia in 108 140 anemia patients in Chongqing.
J Army Med Univ. 1750–1756. 2020.
|
|
13
|
Xiaoxi Z, Xue Y, Guangping L, Daili T, Mei
L, Yaqing J, Bi W and Wenfeng Y: Analysis of gene detection results
of thalassemia in reproductive age population of different ethnic
groups in some areas of Guizhou province. Chin J Birth Health
Heredity. 1235–1241. 2021.
|
|
14
|
Yuanyuan H, Lihua Y, Jun H, Aiqiong J,
Qiaohui L, Xuelian S and Youqiong L: Analysis of gene testing
results for thalassemia in Childbearing-Age population of Laibin
City, Guangxi. J Mod Lab Med. 96–102. 2024.
|
|
15
|
Ying W, Yan X, Si L, Li Z and Rong P:
Analysis of the detection of thalassemia gene in Hunan Province
Practical Preventive Med: 1363-1365, 2021.
|
|
16
|
Runhong X, Hui L, Yayun Q, Yufei J, Meiqi
Y, Guoqiang S, Miaomiao C and Jieping S: Genetic variation and
distribution characteristics of thalassemia in people of
childbearing age in Hubei Province. Chin J Endemiol. 280–285.
2023.
|
|
17
|
Yunjuan W, Yanliang Z, Qiuyue X, Ying Z,
Zhaowu H and Yang S: Gene Screening and Result Analysis of 2376
Cases of Thalassemia. J Kunming Med Univ. 68–71. 2021.
|
|
18
|
Bin L, Min X, Yan K and Meng T: Analysis
of Gene Test Results of Thalassemia in Reproductive Age Population
in Kunming. Bioprocess, 2023.
|
|
19
|
Yan C, Jiejing C, Wen X, Junfang Z and
Canchang L: Analysis of gene detection result of 1165 cases of
thalassemia. Med Diagnosis: 21-26, 2022.
|
|
20
|
Qiyin L, Shu K, Ding W, Qing L, Xiaofang S
and Zhen C: A Study on Genotyping and Screening Misses of
Thalassemia in the Population of Childbearing Age in Guangzhou.
Prog Mod Biomed. 3247–3252. 2023.
|
|
21
|
Yajun D, Yan L, Yi Y, Qi G and Zhijun Z:
Analysis of prenatal screening results of 4 629 pregnant women with
thalassemia in Shiyan area of Hubei Province. J Dalian Med Univ.
509–512. 2023.
|
|
22
|
Miao Y, Yongjie D, Xiaoju P and Jinhua Y:
Analysis of prenatal screening results of thalassemia in 1 872
pregnant women in Yiling area of Hubei province. Appl Preventive
Med. 352–355. 2019.
|
|
23
|
Haiyan K, Sheng L, Yanru Z, Lin L, Hong J
and Anxing F: Analysis of gene types and clinical phenotype in
neonatal disease screening of thalassemia of 30 554 neonates in
Huangshi city. Chin J Child Health Care. 328–331. 2020.
|
|
24
|
Jing Y, Lian Z, Xiaoyi L and Yinhan Z:
Analysis of the Hematological Parameters of Pregnant Women with
Thalassemia. J Mod Labo Med, 2013.
|
|
25
|
Cai A, Liu X, Ma Q, He G, Jing C, He J,
Zeng F and Zhu B: Prevalence, mutation distribution, and economic
burden of thalassemia in China: A systematic review and regional
analysis. Arch Public Health. 83(92)2025.PubMed/NCBI View Article : Google Scholar
|














