1. Introduction
Degenerative joint diseases include osteoarthritis
(OA), intervertebral disc (IVD) degeneration (IVDD) and rheumatoid
arthritis (RA). The gradual structural and functional deterioration
of articular cartilage, subchondral bone and surrounding joint
tissues are common features, which lead to joint pain, stiffness
and functional impairment (1-3).
It is reported that degenerative joint diseases affect >250
million individuals worldwide (4).
In OA, progressive degradation of the cartilage matrix occurs, with
notable reductions in the main components of the extracellular
matrix (ECM), such as type II collagen and proteoglycans; in later
stages, osteophytes (which are outgrowths of bone at joint margins
that develop in response to cartilage loss) often form, and joint
deformities may develop (2,5). In IVDD, the nucleus pulposus (NP)
cells and annulus fibrosus cells progressively lose their viability
and function; over time, the intervertebral discs become
dehydrated, collapse and deform, causing various clinical issues
such as lower back pain (6).
Furthermore, in RA, an autoimmune disease, chronic inflammation and
synovial hyperplasia erode the articular cartilage and subchondral
bone, typically manifesting as marked joint swelling, morning
stiffness and cartilage destruction (7,8).
Regardless of the specific form, inflammation and oxidative stress
both serve roles in the pathogenesis of these diseases.
Oxidative stress is the excessive accumulation of
reactive oxygen species (ROS) and reactive nitrogen species that
surpass the scavenging capacity of endogenous antioxidant systems
(such as superoxide dismutase, catalase, glutathione peroxidase and
peroxiredoxins). Oxidative stress causes cellular damage and can
lead to apoptosis or necrosis (9,10). In
tissues such as articular cartilage and intervertebral discs,
oxidative stress induces the expression of matrix-degrading enzymes
[such as matrix metalloproteinases (MMPs; MMP-1, -3 and -13) and
aggrecanases (such as a disintegrin and metalloproteinase with
thrombospondin motifs-4 and -5)], which accelerates the degradation
of the ECM (2,11,12).
Moreover, oxidative stress can activate associated signaling
pathways (such as the NLRP3 inflammasome), exacerbating local
inflammation (13,14). With aging or disease progression,
the capacity of joint cells to defend against free radical damage
declines. This results in an increased likelihood that oxidative
damage will accumulate, which further exacerbates joint or disc
degeneration (2,15).
Mitochondria, the organelles responsible for
oxidative phosphorylation and energy supply in cells, are the
primary sites of endogenous ROS production (1,16).
During oxidative phosphorylation in the mitochondrial electron
transport chain, superoxide anions (O2-) are
generated and then converted to H2O2, OH·,
singlet oxygen and peroxynitrite by antioxidant systems such as
superoxide dismutases (SODs; such as SOD2) (17). If the levels of the
O2- exceeds this regulatory capacity, damage
on the mitochondrial proteins, membrane lipids and DNA may occur
(16). Previous studies reveal that
mitochondrial dysfunction and excess mitochondrial ROS production
are involved in numerous degenerative diseases, including OA, RA
and IVDD (5,8,18).
Therefore, protecting mitochondria and improving redox homeostasis
is a notable research focus and may be a strategy in the prevention
and treatment of degenerative joint diseases.
Mitochondrial oxidative stress results in tissue
damage in degenerative joint diseases; however, therapeutic
strategies aimed at restoring the mitochondrial redox balance are
scarce. Although conventional antioxidants (such as vitamin C,
vitamin E and N-acetylcysteine) and anti-inflammatory drugs (for
example, non-steroidal anti-inflammatory drugs such as ibuprofen
and indomethacin, or corticosteroids such as prednisone) can
transiently alleviate symptoms, they cannot mitigate the
mitochondria-derived ROS at the source (2). Despite growing evidence that aberrant
mitochondrial ROS contribute to cartilage degradation, NP cell
senescence and synovial inflammation, only a small number of
therapeutic strategies target mitochondrial oxidative stress. At
present, the existing antioxidant approaches rely on
non-mitochondria-specific compounds, such as vitamin C, vitamin E,
N-acetylcysteine, glutathione and polyphenols (for example,
resveratrol), and clinical translation is limited (2). Therefore, an evaluation of the
mitochondria-targeted antioxidants and quality-control mechanisms
was needed in order to assess this translational gap and highlight
the novel disease-modifying treatments for OA, IVDD and RA.
2. Methods
Literature search strategy
A systematic search of PubMed (https://pubmed.ncbi.nlm.nih.gov/), Web of Science
(https://www.webofscience.com/) and the
Cochrane Library (https://www.cochranelibrary.com/) was carried out for
articles published between 2000 and June 2025. Search terms
included combinations of ‘osteoarthritis’ OR ‘intervertebral disc
degeneration’ OR ‘rheumatoid arthritis’ with ‘mitochondria’,
‘oxidative stress’, ‘mitophagy’, ‘MitoQ’, ‘SS-31’ and
‘mitochondrial antioxidant’. Boolean operators and Medical Subject
Headings were used as appropriate. Reference lists of relevant
reviews and clinical guidelines were also reviewed to identify
additional studies.
The inclusion criteria used were as follows: i)
Primary research or review articles written in English; ii) in
vitro, in vivo or ex vivo models of OA, IVDD or
RA; iii) interventions that directly targeted mitochondrial
function or oxidative stress; and iv) quantifiable outcomes
regarding the levels of ROS, cell survival, ECM integrity or
functional scores.
The exclusion criteria used were as follows: i)
Publications not written in English; ii) conference abstracts or
editorials; iii) duplicate reports; or iv) studies with
insufficient methodological detail.
3. Biological basis of mitochondria-targeted
antioxidant strategies in degenerative joint diseases
Production and scavenging of
mitochondrial ROS
Within mitochondria, 1-5% of the oxygen taken in by
the cell may ‘leak’ in the form of ROS during the activity of the
electron transport chain (19,20).
Under normal conditions, SODs located in the mitochondria (such as
SOD2, which is primarily located in the mitochondrial matrix),
peroxiredoxins and glutathione peroxidases convert or remove these
ROS, which keeps the levels of the ROS within a reasonable range
(typically at nM to low µM concentrations, such as superoxide in
the 10-100 nM range and hydrogen peroxide in the 1-10 µM range,
which act as signaling molecules) and allows them to function in
cell signal transduction and normal metabolic processes, such as
MAPK (ERK, JNK and p38), NF-κB, PI3K-Akt, hypoxia-inducible
factor-1α and Wnt/β-catenin signaling pathways (2,21)
(Fig. 1). However, when
mitochondrial function is impaired (due to exogenous harmful
stimuli, aging or gene mutations that inactivate complexes involved
in the electron transport chain) excessive levels of ROS can
accumulate, resulting in lipid peroxidation, oxidative protein
modifications and breaks in the DNA (16,22).
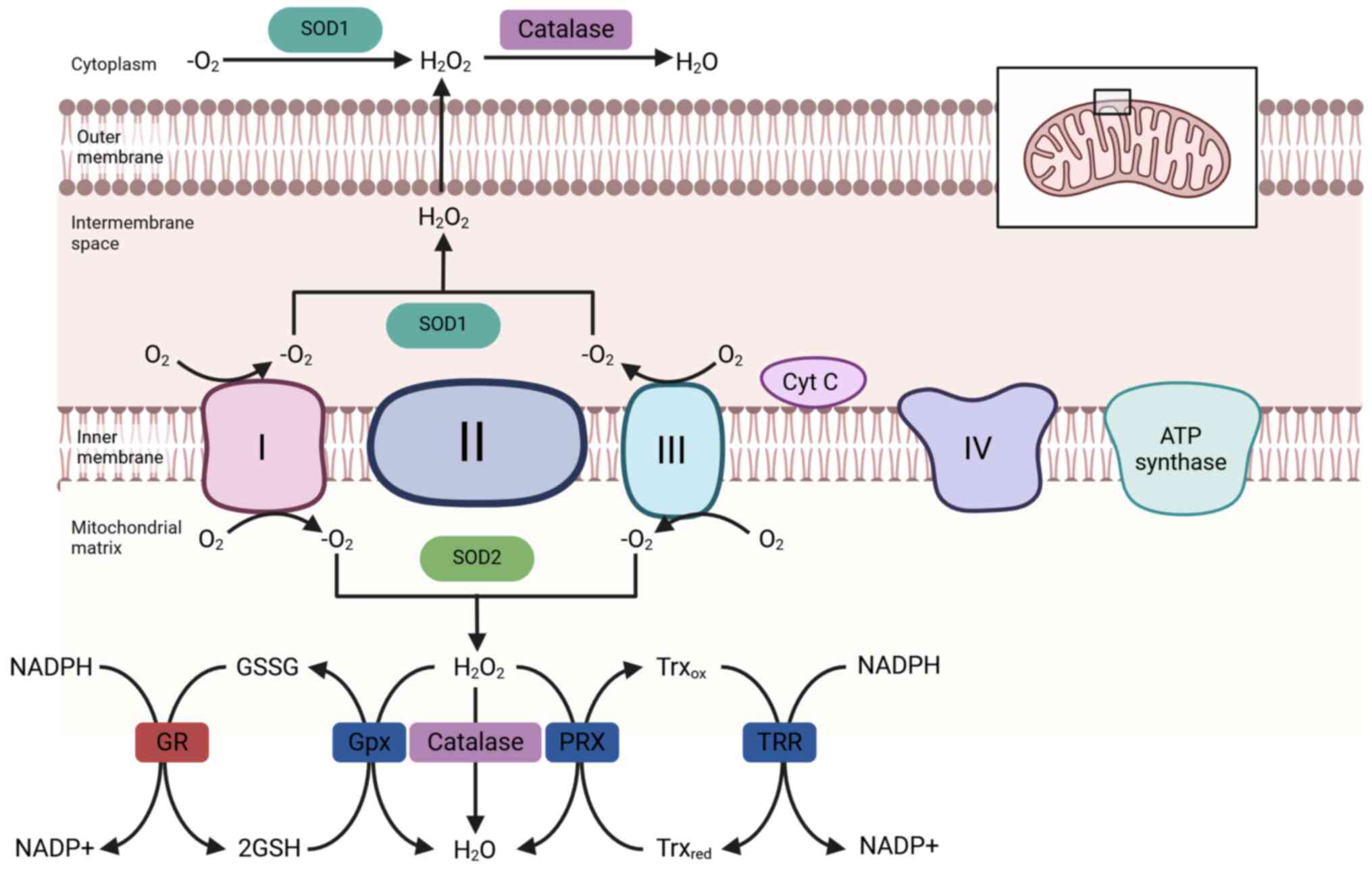 | Figure 1Schematic illustration of
mitochondrial reactive oxygen species generation and the
antioxidant defense systems. During mitochondrial respiration,
electron leakage from the electron transport chain leads to the
formation of superoxide anions, which are subsequently converted to
hydrogen peroxide and hydroxyl radicals through enzymatic and
non-enzymatic reactions. To counteract oxidative stress,
mitochondria utilize several antioxidant mechanisms, such as SOD
that catalyzes the dismutation of superoxide into
H2O2, which is further detoxified by Gpx and
PRX. The glutathione system, composed of reduced GSH, oxidized GSSG
and GR, maintains redox homeostasis. In parallel, the Trx/TRR
system reduces oxidized proteins and supports peroxiredoxin
activity. Cyt C is also a key electron carrier and its release into
the cytosol can trigger apoptotic signaling. Gpx, glutathione
peroxidase; PRX, peroxiredoxin; SOD, superoxide dismutase; Cyt C,
cytochrome c; GR, glutathione reductase; GSSG, glutathione
disulfide; GSH, glutathione; TRR, thioredoxin reductase; Trx,
thioredoxin; ox, oxidized; red, reduced. |
Mechanisms linking mitochondrial
dysfunction with degenerative changes in joints
Mitochondrial dysfunction is associated with
degenerative changes in joint tissues by promoting apoptosis,
pyroptosis and inflammatory responses, as well as undermining
cartilage matrix integrity and normal cellular functions (23,24).
The excessive generation of mitochondrial ROS and the loss of the
mitochondrial membrane potential can cause a release of cytochrome
c. This can lead to the subsequent activation of the caspase
cascade, which induces apoptosis (13,25).
Furthermore, elevated ROS levels can promote the assembly of the
NLRP3 inflammasome, triggering pyroptosis and increasing the
inflammatory damage within joint tissues (13,26).
In addition to the aforementioned direct mechanisms
of cell death, oxidative stress activates inflammatory pathways
such as NF-κB and MAPK. These inflammatory pathways increase the
production of proinflammatory factors (such as IL-1β and TNF-α) and
MMPs, which together accelerate cartilage matrix degradation
(7,27).
As mitochondrial damage worsens, cells face an
energy deficit. An overabundance of ROS further impairs
proteoglycans and collagen, increasing the breakdown of the ECM in
the cartilage (15,28). For example, in RA, mitochondrial
dysfunction in fibroblast-like synoviocytes increases both local
inflammation and synovial hyperplasia (7). Moreover, the accumulation of ROS may
lead to cell cycle arrest, reduced chondrocyte proliferation and
the emergence of cellular senescence phenotypes (such as increased
p16 and p21 expression levels), which are observed in conditions
such as osteoporosis, OA and IVDD (29-31).
4. Research progress on
mitochondria-targeted antioxidants in degenerative joint
diseases
Mitoquinone (MitoQ)
MitoQ is a mitochondria-targeted antioxidant formed
by conjugating a ubiquinone analog with triphenylphosphonium (TPP)
through an alkyl chain. It demonstrates certain protective effects
in models of both OA and IVDD (2,32)
(Table I).
 | Table IComparison of mitochondria-targeted
antioxidants in degenerative joint diseases. |
Table I
Comparison of mitochondria-targeted
antioxidants in degenerative joint diseases.
| Antioxidant | Structure and
targeting | Mechanism of
action | Evidence in OA | Evidence in
IVDD | Evidence in RA |
|---|
| MitoQ | Ubiquinone and
TPP+ | Scavenges mtROS,
stabilizes the mitochondrial membrane and downregulates
NF-κB/NLRP3 | Reduces cartilage
damage and inflammation (demonstrated in in vitro studies
and models of mice with OA) (25,32) | Reduces NP cell
apoptosis and increases ECM stability (demonstrated in rat models)
(8,40) | Limited |
| MitoTEMPO | TEMPO and
TPP+ | Removes
mitochondrial superoxide, inhibits the expression of JNK/AP-1 and
NF-κB, and promotes mitophagy | Reduces MMPs
expression levels and cartilage degeneration (demonstrated in mice
with OA) | Reduces
caspase-induced pyroptosis and increases PINK1/Parkin-induced
mitophagy (demonstrated in NP cell models) | Reduces synovial
inflammation and ROS (demonstrated in vitro) |
| SkQ1 | Plastoquinone and
TPP+ | Induces apoptosis
in neutrophils, and reduces synovial oxidative stress | Not reported | Not reported | Reduces joint
damage and the levels of cytokines (demonstrated in a rat model of
RA) |
| SS-31
(Elamipretide) | Peptide binding to
cardiolipin | Stabilizes the ETC,
reduces lipid peroxidation and preserves mitochondrial
dynamics | Increases the
synthesis of the ECM by chondrocytes and reduces senescence
(demonstrated in in vitro cellular models) | Reduces NP
apoptosis and increases mitochondrial stability (demonstrated in
in vitro LPS- and oxidative stress-induced cellular
models) | Limited |
Research in OA
In vitro studies, using chondrocyte models
induced with oxidative stress, demonstrate that MitoQ mitigates
mitochondrial damage and ECM degradation, while enhancing the
expression of anti-inflammatory genes, such as IL-10 and
TGF-β, as well as antioxidant genes including SOD2,
catalase, glutathione peroxidase 1 and
NAD(P)H:quinone oxidoreductase 1 (2,31).
In vivo, MitoQ alleviates cartilage degeneration and reduces
the levels of inflammatory cytokines, which slows the progression
of OA (32,33).
Research in IVDD. Studies indicate that under
mechanical overload or inflammatory stimulation, intervertebral
disc tissue exhibits notable accumulation of ROS, elevated
apoptosis and impaired ECM synthesis (34,35).
MitoQ inhibits the activation of inflammatory signaling pathways
such as the NLRP3 inflammasome and NF-κB, and prevents the
excessive secretion of key cytokines, such as IL-1β, TNF-α and IL-6
(30,32,36).
In a rat IVDD model, MitoQ administration maintains disc height and
hydration; therefore, delaying degenerative changes (6,30,36).
These findings suggest that MitoQ confers protective
effects on both articular cartilage and intervertebral discs,
potentially due to its ability to reduce mitochondrial oxidative
damage and stabilize energy metabolism.
MitoTEMPO
MitoTEMPO is a mitochondria-targeted antioxidant
formed by coupling the superoxide scavenger TEMPO with the
TPP+ cationic group, which removes mitochondrial
superoxide radicals (37,38). Multiple in vivo and in
vitro studies highlight its beneficial role in counteracting
oxidative stress and apoptosis in joint cells (27,37).
Research in OA
Under high-glucose conditions or inflammatory
stimulation, ROS levels in chondrocytes increase. Previous studies
report that MitoTEMPO blocks the JNK/activator protein-1 and NF-κB
pathways that are activated by ROS, leading to a reduction in the
expression of MMPs compared with the expression of MMPs without
MitoTEMPO treatment (27,37). Furthermore, intra-articular
injection of MitoTEMPO in mice with cholesterol-induced OA reduces
MMP-13 gene expression levels compared with vehicle-treated OA
controls. Additionally, this intervention alleviates cartilage
lesions and leads to a notable reduction in cartilage degeneration
(2).
Research in IVDD. MitoTEMPO also demonstrates
protective effects in IVDD. In NP cells with IL-1β exposure or
endplate chondrocytes with H2O2 exposure,
MitoTEMPO notably reduces the pathological increase in
mitochondrial ROS compared with the stressor-only groups without
MitoTEMPO treatment. This suppresses the activation of the caspase
cascade and pyroptosis (25,39).
When mitochondrial ROS are suppressed, PTEN-induced kinase 1
(PINK1)/Parkin-mediated mitophagy is maintained, reducing the
accumulation of damaged mitochondria in cells (25,39).
This process improves the function of NP cells and the homeostasis
of the ECM, exerting an interventional effect on IVDD.
Research in RA. Although the research on
MitoTEMPO in RA is limited, studies in synovial cell models suggest
that exogenous mitochondrial ROS scavenging can attenuate the
inflammatory cascade and ROS bursts (7,40).
Therefore, MitoTEMPO exhibits broad-spectrum antioxidant protection
by targeting mitochondria in various degenerative joint
diseases.
Plastoquinonyl-decyl-triphenylphosphonium (SkQ1)
SkQ1 is a mitochondria-targeted antioxidant derived
from plastoquinone linked to TPP (8). In a RA model, low doses of SkQ1 (on
the nM scale) suppresses the progression of arthritis and reduces
the pathological damage to cartilage and the synovium (8). Its mechanism is associated with
inducing neutrophil apoptosis, alleviating oxidative stress and
improving the joint microenvironment. Although direct evidence in
OA and IVDD is scarce, the robust mitochondrial antioxidant
properties of SkQ1 and its efficacy in autoimmune arthritis models
suggest it may hold potential value for degenerative joint diseases
as well.
Szeto-Schiller-31 (SS-31)
SS-31 (also known as Elamipretide) is a
mitochondria-targeting short peptide that binds to cardiolipin on
the inner mitochondrial membrane. This helps to stabilize the
electron transport chain and reduce the generation of ROS (41). Studies demonstrate that SS-31
improves the mitochondrial membrane potential, inhibits
inflammation and promotes the synthesis of the ECM in both
intervertebral disc cells and chondrocytes (6,42).
Additionally, SS-31 reduces mitochondrial lipid
peroxidation, a key factor in oxidative damage, and partially
reverses stress-induced cellular senescence phenotypes (43). Its ability to ameliorate
mitochondrial function also contributes to mitigating inflammatory
signaling and promoting the stability of the ECM, which is critical
for preventing tissue degeneration (44).
5. Other strategies targeting mitochondrial
regulatory molecules in degenerative joint diseases
Apart from the direct use of mitochondria-targeted
antioxidants, recent studies increasingly focus on mitochondrial
dynamics, mitophagy and mitochondrial biogenesis as indirect
methods for reducing mitochondrial ROS accumulation, which may
offer a therapeutic potential in degenerative joint diseases
(Fig. 2) (45,46).
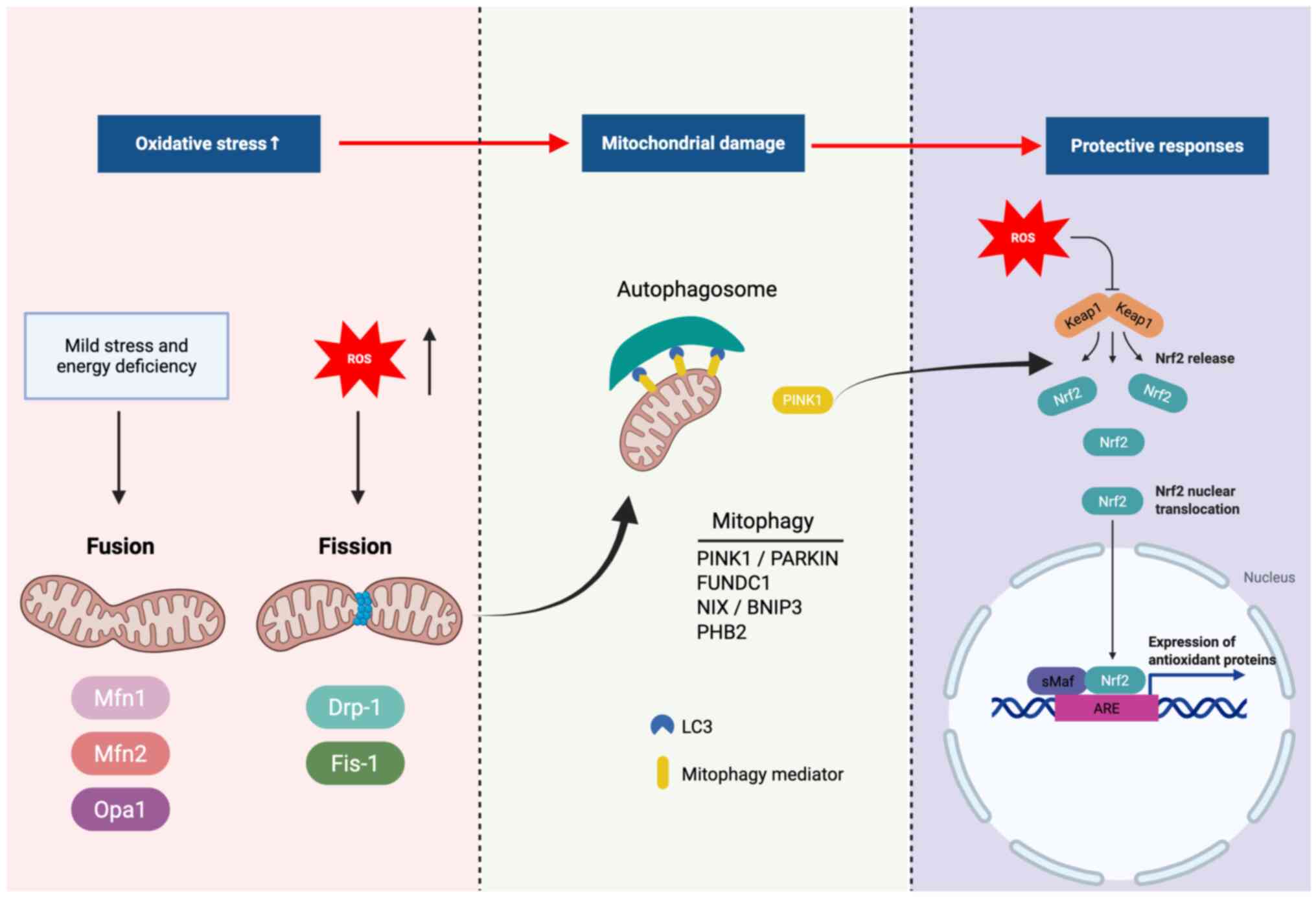 | Figure 2Mitochondrial stress responses
following ROS-induced damage and the protective signaling pathways
that maintain mitochondrial quality. ROS overproduction disrupts
mitochondrial dynamics, leading to altered fusion (mediated by Mfn1
and 2, and Opa1) and fission (promoted by Drp1 and Fis1). Damaged
mitochondria activate mitophagy, in which PINK1 accumulates on the
outer mitochondrial membrane to recruit PARKIN, an E3 ubiquitin
ligase, facilitating ubiquitination of outer membrane proteins.
Receptors such as FUNDC1, NIX and BNIP3 interact with LC3 to
promote autophagosome formation, while PHB2 functions as an inner
membrane mitophagy receptor. Additionally, the Keap1-Nrf2-ARE
signaling axis provides antioxidant defense, with sMaf proteins
serving as transcriptional partners of Nrf2 in activating
antioxidant response elements. These pathways together coordinate
mitochondrial turnover and cytoprotective responses. ROS, reactive
oxygen species; Mfn1, mitofusin 1; Mfn2, mitofusin 2; Opa1, optic
atrophy 1; Drp1, dynamin-related protein 1; Fis1, fission protein
1; PINK1, PTEN-induced kinase 1; PARKIN, E3 ubiquitin ligase
involved in mitophagy; FUNDC1, FUN14 domain-containing protein 1;
NIX, BCL2/adenovirus E1B 19 kDa protein-interacting protein 3-like;
BNIP3, BCL-2/adenovirus E1B 19 kDa protein-interacting protein 3;
LC3, microtubule-associated protein 1 light chain 3; Nrf2, nuclear
factor erythroid 2-related factor 2; sMaf, small musculoaponeurotic
fibrosarcoma oncogene homologs that dimerize with Nrf2; Keap1,
Kelch-like ECH-associated protein 1; ARE, antioxidant response
element; PHB2, prohibitin 2. |
Regulation of PINK1/Parkin-mediated
mitophagy
The PINK1/Parkin pathway is an essential cellular
mechanism for clearing damaged mitochondria. When the mitochondrial
membrane potential decreases, PINK1 accumulates on the outer
mitochondrial membrane, recruiting Parkin to the mitochondrial
surface. Parkin then ubiquitinates several outer mitochondrial
membrane substrates, including mitofusins 1/2, voltage-dependent
anion channel 1 and translocase of outer mitochondrial membrane 20,
which facilitates the recognition and clearance of damaged
mitochondria through autophagy. Enhancing PINK1/Parkin-mediated
mitophagy can reduce the release of mitochondrial ROS, which
protects cells from damage (47).
Previous studies demonstrate that inhibition of leucine-rich repeat
kinase 2 can restore Parkin-mediated mitophagy and alleviate IVDD
(48,49). Taurine, a naturally occurring amino
acid derivative, promotes mitophagy by activating the PINK1/Parkin
pathway, which ameliorates IVDD (50). Additionally, knocking down the
expression of early growth response protein 1 activates
PINK1-Parkin-dependent mitophagy, which suppresses NP cell
senescence and mitochondrial damage, and slows disc degeneration
(51). Furthermore, a recent study
reveals that cellular repressor of E1A-stimulated genes 1 can
alleviate NP cell pyroptosis through the PINK1/Parkin-associated
mitophagy pathway, which improves IVDD (52).
Maintenance of mitochondrial dynamics
balance (fission and fusion)
An imbalance in mitochondrial fission and fusion
also leads to the excessive generation of ROS (36). Under pathological stimuli such as
mechanical compression, excessive mitochondrial fission (mediated
by dynamin-related protein 1) separates damaged mitochondrial
segments; however, impaired fusion processes (mediated by mitofusin
1/2 and optic atrophy 1) result in the accumulation of fragmented
and dysfunctional mitochondria, causing a persistent elevation of
ROS (36). Antioxidants such as
MitoQ not only reduce ROS levels but also attenuate excessive
fission and promote fusion of functionally intact mitochondria
(36,53). Therefore, the modulation of proteins
involved in mitochondrial dynamics or the combined use with
mitochondrial antioxidants may be a future therapeutic direction
for joint degeneration.
Nuclear factor erythroid 2-related
factor 2 (Nrf2)-Kelch-like ECH-associated protein 1 (Keap1)
antioxidant signaling pathway and mitochondria
Nrf2, a central transcription factor for cellular
antioxidant responses, induces the expression of antioxidant genes
including heme oxygenase-1 (HO-1), NAD(P)H quinone
dehydrogenase 1 and SOD2(32).
When mitochondrial ROS levels increase, oxidative modification of
Keap1 leads to the stabilization and nuclear translocation of Nrf2,
which then activates antioxidant gene expression to alleviate
oxidative stress and maintain cellular homeostasis (54). A recent study demonstrates that both
pharmacological and genetic activation of the Nrf2 pathway serves a
crucial role in mitigating inflammatory responses and apoptosis in
joint cartilage and IVD tissues (49). For example, Kartogenin-loaded
hydrogel promotes IVD repair by protecting mesenchymal stem cells
from oxidative stress via the Nrf2/Thioredoxin-interacting
protein/NLRP3 axis, which reduces inflammation and cellular
apoptosis (55). Additionally,
astaxanthin activates the Nrf2/HO-1 pathway, which suppresses
oxidative stress and prevents cartilage endplate degeneration by
inhibiting apoptosis and the degradation of the ECM (56).
Other regulatory molecules or
bioactive substances. Deacetylases [such as sirtuin (SIRT)3 and
1]
SIRT family proteins serve essential roles in
mitochondrial homeostasis, redox balance and the regulation of
energy metabolism (25,28). For example, SIRT3, localized in
mitochondria, deacetylates and activates key antioxidant enzymes
such as manganese superoxide dismutase (SOD2), isocitrate
dehydrogenase 2 and catalase, which enhances mitochondrial ROS
detoxification (28). Upregulation
of SIRT3 suppresses cellular senescence and inflammation in joint
cells.
A study by Guo et al (57) reveals that dysregulation of SIRT3
and its metabolic network, including downstream effectors such as
isocitrate dehydrogenase 2 and manganese SOD, serves a role in
mitochondrial oxidative stress and cartilage degradation in OA.
This suggests that restoring the activity of SIRT3 may serve as a
therapeutic approach to delay joint degeneration (57).
AMP-activated protein kinase (AMPK)-peroxisome
proliferator-activated receptor γ coactivator 1-α (PGC-1α)
pathway. The AMPK-PGC-1α signaling axis promotes mitochondrial
biogenesis and the expression of antioxidant enzymes. A study by
Yang et al (58)
demonstrates that exposure to advanced glycation end-products
represses the AMPKα-SIRT1-PGC-1α pathway in osteoarthritic
chondrocytes, with reductions in PGC-1α expression levels compared
with untreated control cells, resulting in impaired mitochondrial
biogenesis and antioxidant defense. However, the study by Li et
al (59) demonstrates that
adipokine omentin-1 can activate the AMPK-PGC-1α pathway, promoting
mitochondrial biogenesis in chondrocytes and slowing OA
progression. The study by Yang et al (58) demonstrates that advanced glycation
end products inhibit the AMPK-SIRT1-PGC-1α pathway, which induces
mitochondrial dysfunction and inflammation in chondrocytes. This
suggests the antioxidant and anti-aging properties of the
AMPK-SIRT1-PGC-1α pathway (58).
Additionally, the study by Guo et al
(57) reveals that activation of
the AMPK-PGC-1α pathway not only restores energy metabolism but
also reduces the accumulation of ROS and the expression of
proinflammatory cytokines, including IL-1β, IL-6 and TNF-α, in
degenerative joint diseases. This demonstrates its potential role
as a therapeutic axis (57).
Itaconate derivatives (such as 4-octyl
itaconate). A previous study indicates that exogenous itaconate
reduces inflammatory responses and inhibits the generation of
mitochondrial ROS (54). Zinc-based
metal-organic super containers, such as 4-octyl itaconate
(4-OI)@Zn-NH-pyr, demonstrates enhanced mitochondrial targeting and
ROS-scavenging activity compared with free 4-OI alone, which
alleviates proinflammatory states in arthritis models (54).
Additionally, novel mitochondria-targeted
nanomedicines such as Mn3O4/UIO-TPP nanozymes
scavenge mitochondrial ROS and restore mitochondrial function in OA
cartilage, demonstrating potential therapeutic effects in rat
models with anterior cruciate ligament transection induced OA
(60,61). These nanozymes represent a promising
material-based extension of mitochondrial antioxidant therapy and
support the feasibility of precision mitochondria-targeted
intervention (62).
Crosstalk between mitochondria and the
endoplasmic reticulum (ER) in joint degeneration
Mitochondria and the ER are functionally connected
via mitochondria-associated membranes, which coordinate calcium
signaling, lipid exchange and cellular stress responses. In
degenerative joint diseases, this interplay becomes dysregulated,
which contributes to oxidative injury and inflammation (63).
ER stress promotes the release of Ca2+
through inositol 1,4,5-trisphosphate receptors, while excessive
uptake of Ca2+ by the mitochondria via the mitochondrial
calcium uniporter leads to the overproduction of ROS, mitochondrial
dysfunction and apoptosis (14).
Furthermore, ER stress activates proapoptotic pathways (such as
protein kinase R-like ER kinase-activating transcription factor
4-CHOP) and promotes the activation of the NLRP3 inflammasome
(64).
In addition, ion channels (such as transient
receptor potential cation channel, subfamily V, member 4)
participate in mechano-oxidative signaling in joint cells, and
their dysregulation may further exacerbate mitochondrial stress
(65). Targeting ER-mitochondria
interactions or associated Ca2+ channels may offer new
therapeutic avenues in OA, IVDD and RA.
6. Crosstalk in immunometabolism:
Mitochondrial oxidative stress and the immune system in
degenerative joint diseases
Mitochondrial oxidative stress not only contributes
to direct tissue damage but also serves a role in modulating immune
responses, which are involved in the pathogenesis of OA, IVDD and
RA. Both innate immune cells, such as macrophages, dendritic cells,
neutrophils and natural killer cells, and adaptive immune cells,
including T cells and B cells, respond to and are regulated by
mitochondrial metabolism and ROS signaling, forming an
interconnected immunometabolic axis (66,67).
Innate immunity and mitochondrial
ROS
Innate immune cells, such as macrophages and
neutrophils, are sensitive to mitochondrial ROS. In RA and IVDD,
macrophages polarize toward a proinflammatory (M1-like) phenotype
under oxidative stress conditions, leading to an enhanced
production of TNF-α, IL-1β and IL-6, which further exacerbates
tissue inflammation and matrix degradation (68). Mitochondrial ROS act as secondary
messengers to activate the NLRP3 inflammasome, a key inflammatory
complex that promotes caspase-1 activation and IL-1β/IL-18
maturation, which accelerates pyroptosis in disc and synovial
tissues (69).
Neutrophils also exhibit enhanced respiratory bursts
and NETosis in the presence of mitochondrial ROS, which contributes
to synovial inflammation and cartilage degradation in RA (70). Toll-like receptors (TLRs),
especially TLR2 and 4, recognize damage-associated molecular
patterns (such as mitochondrial DNA and cardiolipin) that are
released from stressed mitochondria. This activates the myeloid
differentiation primary response protein 88-dependent signaling
pathway and NF-κB-induced transcription of proinflammatory
cytokines, including TNF-α, IL-1β and IL-6 (71,72).
Adaptive immunity and oxidative
microenvironment
T cell differentiation and function are dependent on
metabolism. Under elevated ROS conditions, mitochondrial
dysfunction promotes a T helper 17 phenotype in CD4+ T
cells, promoting proinflammatory responses in RA and OA synovium
(73,74). Regulatory T cells, which rely on
oxidative phosphorylation for their function, are suppressed in
oxidative microenvironments, which further amplifies inflammation
(73).
B cells are also influenced by mitochondrial
metabolism, with mitochondrial ROS promoting autoantibody
production and immune complex deposition in RA (75). Mitochondria-targeted antioxidants
such as MitoQ and SkQ1 dampen the activation of T cells and the
secretion of cytokines, suggesting that the modulation of
mitochondrial ROS may re-establish immune homeostasis in
degenerative joint diseases (76).
Cytokines, chemokines and the
mitochondrial stress response
Cytokines, such as IL-1β, TNF-α and IL-6, are not
only induced by mitochondrial ROS but also feedback to further
disrupt mitochondrial integrity and function. These cytokines
impair mitochondrial membrane potential, increase the generation of
ROS and inhibit mitophagy, forming a positive feedback loop of
oxidative inflammation (77).
However, anti-inflammatory cytokines (such as IL-10) may promote
mitophagy and mitochondrial quality control by activating pathways
such as SIRT3 or AMPK-PGC-1α (78).
Chemokines, such as chemokine (C-C motif) ligand 2
and chemokine (C-X-C motif) ligand 8, recruit immune cells into
inflamed joints and are also modulated by mitochondrial oxidative
stress (70). Therefore,
therapeutic strategies targeting mitochondrial pathways may reduce
tissue damage and immune cell infiltration.
7. Clinical prospects and challenges
Limitations of animal models and
preclinical studies
Although mitochondria-targeted antioxidants such as
MitoQ, MitoTEMPO and SkQ1 reveal notable protective effects on
joints in animal models (including rodents such as mice and rats,
mid-sized models such as rabbits, and large animals such as dogs
and sheep), the pathological processes in animal models are more
acute or controlled compared with clinical populations. Therefore,
these animal models do not fully replicate the chronic, long-term
degenerative processes observed in humans (79). Additionally, there are notable
differences in pharmacokinetics and drug sensitivity in different
species (80). Therefore,
translating results from animal studies into clinical applications
necessitates further validation through extensive clinical trials
(8,18,32).
Administration methods and safety
considerations
The majority of studies on mitochondria-targeted
antioxidants rely on intra-articular injections or local
administration (79,81). Although several
mitochondria-targeted antioxidants, such as MitoQ, SkQ1 (Visomitin)
and to an extent MitoTEMPO, can be administered orally, systemic
delivery raises concerns regarding bioavailability, mitochondrial
targeting efficiency, and systemic safety profiles (2,32).
Local injections also have risks of tissue trauma and infection.
Advancements in drug delivery systems, such as hydrogels and
nanocarriers combined with mitochondria-targeting ligands, may
overcome these limitations. However, achieving efficient, sustained
and localized drug release at the joint is still challenging at
present (3,6,82).
Potential side effects of long-term
medication
While drugs such as MitoQ and SkQ1 demonstrate good
tolerability at low doses [for example, SkQ1 at 0.25-1.25
nmol/kg/day (0.13-0.70 µg/kg/day) in rat models and MitoQ at ~20
mg/day in human trials] (8),
long-term administration (defined as continuous dosing for 3-6
months or longer in preclinical and clinical studies) requires
vigilant monitoring to avoid causing redox imbalances in normal
tissues. Additionally, moderate levels of ROS serve critical
physiological roles in signaling and tissue repair in degenerative
joint diseases. Excessive clearance of ROS may disrupt essential
cellular signaling pathways (1,15).
Therefore, calibrating the dose and duration of
mitochondria-targeted antioxidants is crucial for clinical
application.
Combination therapies
Numerous studies increasingly advocate for combining
mitochondria-targeted antioxidants (such as MitoQ, SkQ1 and
MitoTEMPO) with autophagy regulators (such as apigenin and
rapamycin), inflammasome inhibitors (such as MCC950 and CTSB
inhibitors), growth factors (such as TGF-β and IGF-1) or
conventional anti-inflammatory medications (such as non-steroidal
anti-inflammatory drugs and disease-modifying anti-rheumatic drugs)
in order to achieve synergistic therapeutic effects on degenerative
joint diseases (7,62). For example, combining MitoTEMPO with
autophagy activators, such as salidroside, honokiol or urolithin A,
simultaneously reduces excessive ROS and increases the clearance
rate of damaged mitochondria, which potentially inhibits apoptosis
in cartilage or intervertebral discs (83).
In addition, cellular and organelle-level strategies
enhance mitochondrial restoration. One such approach is
mitochondrial transplantation, in which healthy exogenous
mitochondria are delivered into dysfunctional cells to rescue
energy production and reduce oxidative stress (84). Recent studies demonstrate that
mitochondrial transplantation improves joint tissue homeostasis and
attenuates degeneration in in vitro chondrocyte and NP cell
models, as well as in vivo mice with destabilization of the
medial meniscus-induced OA and rat with needle puncture-induced
IVDD models (85,86). This strategy may be biologically
complementary to chemical antioxidant therapies.
Furthermore, future therapeutic paradigms may
involve mitochondrial genome editing to correct mitochondrial DNA
mutations that are associated with joint degeneration. Emerging
tools such as mitochondria-targeted zinc finger nucleases and
mitochondria-targeted transcription activator-like effector
nucleases demonstrate potential in restoring mitochondrial
integrity and function in tissues with OA (87,88).
Although these technologies are still mostly experimental, they
could potentially be used in precision mitochondrial medicine for
degenerative joint diseases in the future (87).
8. Conclusions
Mitochondrial dysfunction and an excessive
generation of ROS are central pathological mechanisms underlying
degenerative joint diseases, including OA, IVDD and RA. The present
study demonstrated the role of mitochondrial oxidative damage in
promoting disease progression, from the degradation of the
cartilage ECM in OA and NP cell death in IVDD, to chronic synovitis
and immune activation in RA. Mitochondria-targeted antioxidants,
such as MitoQ, MitoTEMPO, SkQ1 and SS-31, demonstrate potential in
mitigating oxidative stress and slowing disease deterioration.
These findings provided theoretical support for a paradigm shift
from symptom-based management toward mitochondria-centered
interventions.
At present, despite promising preclinical data, the
evidence is mostly limited to in vitro and small-animal
studies, such as mice and rats, with scarce validation in human
tissues or large-scale clinical settings. Additionally, the
heterogeneity of degenerative joint diseases further complicates
the interpretation of therapeutic efficacy. In particular,
differences in mitochondrial activity, ROS levels and drug
responsiveness among OA, IVDD and RA are yet to be fully
characterized. Furthermore, the lack of multicenter randomized
clinical trials evaluating dosing, safety profiles and delivery
methods for mitochondria-targeted antioxidants is a notable
limitation. Additionally, the majority of studies overlook
long-term outcomes and potential off-target effects, especially in
elderly or comorbid populations.
Future studies should investigate the temporal and
spatial regulation of mitochondrial ROS in specific joint tissues
and immune cell subpopulations. In particular, the association
between mitochondrial dysfunction and epigenetic regulation,
cytokine signaling or metabolic reprogramming warrants further
investigation. Elucidating how mitochondrial stress interacts with
cartilage calcification, subchondral bone remodeling and synovial
hyperplasia may reveal additional therapeutic checkpoints. The use
of advanced models such as patient-derived organoids, single-cell
mitochondrial profiling and spatial transcriptomics may aid
mechanistic investigations. Additionally, mitochondrial-nuclear
crosstalk or sex-specific mitochondrial responses should also be
investigated in the future.
Clinical translation should prioritize the
development of delivery systems, including hydrogels, nanoparticles
and scaffold-based platforms that enable a localized, sustained
release of mitochondria-targeted antioxidants (such as MitoQ,
MitoTEMPO or SkQ1). Additionally, their combination with autophagy
enhancers (such as rapamycin, spermidine or resveratrol) or
regenerative factors [such as bone morphogenetic proteins (BMP)-2
and -7] or TGF-β) at degenerative sites should be investigated.
Furthermore, integrating mitochondria-targeted antioxidants with
mitophagy enhancers (such as rapamycin, urolithin A and
salidroside), immune modulators (such as IL-17A antibodies, Treg
enhancers and fumaric acid derivatives) or regenerative therapies
(such as stem cells or exosome-based systems) may also offer a
therapeutic effect. Such combination regimens may synergistically
halt or reverse disease progression. Furthermore, precision
medicine frameworks leveraging genetic, metabolic or imaging
biomarkers may allow for personalized dosing and treatment
selection, which may also enhance therapeutic outcomes.
Mitochondria-centered therapies involve
multi-disciplinary teams from orthopedics, immunology, geriatrics
and bioengineering. However, a number of controversies remain
unresolved. For example, contradictory findings regarding the role
of ROS as only deleterious vs. the role of ROS as signaling
mediators in joint homeostasis. Moreover, the possibility that
prolonged ROS suppression might impair physiological defense
mechanisms or tissue remodeling should be investigated.
Interdisciplinary dialogue and standardized methodological
approaches will likely be required to address these issues and
guide the rational design of future therapies.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
YH designed the study and drafted the manuscript. XY
summarized the comments from the reviewers, drafted the responses
and contributed to manuscript revision and language editing. TZ
contributed to the literature review and assisted in data
interpretation. YG revised the manuscript. JS designed the present
study, revised the manuscript and supervised the project. All
authors read and approved the final version of the manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Martin JA, Martini A, Molinari A, Morgan
W, Ramalingam W, Buckwalter JA and McKinley TO: Mitochondrial
electron transport and glycolysis are coupled in articular
cartilage. Osteoarthritis Cartilage. 20:323–329. 2012.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Bolduc JA, Collins JA and Loeser RF:
Reactive oxygen species, aging and articular cartilage homeostasis.
Free Radic Biol Med. 132:73–82. 2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Chen Q, Qian Q, Xu H, Zhou H, Chen L, Shao
N, Zhang K, Chen T, Tian H, Zhang Z, et al: Mitochondrial-targeted
metal-phenolic nanoparticles to attenuate intervertebral disc
degeneration: alleviating oxidative stress and mitochondrial
dysfunction. ACS Nano. 18:8885–8905. 2024.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Hunter DJ and Bierma-Zeinstra S:
Osteoarthritis. Lancet. 393:1745–1759. 2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Nasto LA, Robinson AR, Ngo K, Clauson CL,
Dong Q, St Croix C, Sowa G, Pola E, Robbins PD, Kang J, et al:
Mitochondrial-derived reactive oxygen species (ROS) play a causal
role in aging-related intervertebral disc degeneration. J Orthop
Res. 31:1150–1157. 2013.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Wang Y, Deng M, Wu Y, Zheng C, Zhang F,
Guo C, Zhang B, Hu C, Kong Q and Wang Y: A multifunctional
mitochondria-protective gene delivery platform promote
intervertebral disc regeneration. Biomaterials.
317(123067)2025.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Al-Azab M, Qaed E, Ouyang X, Elkhider A,
Walana W, Li H, Li W, Tang Y, Adlat S, Wei J, et al:
TL1A/TNFR2-mediated mitochondrial dysfunction of fibroblast-like
synoviocytes increases inflammatory response in patients with
rheumatoid arthritis via reactive oxygen species generation. FEBS
J. 287:3088–3104. 2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Andreev-Andrievskiy AA, Kolosova NG,
Stefanova NA, Lovat MV, Egorov MV, Manskikh VN, Zinovkin RA, Galkin
II, Prikhodko AS, Skulachev MV and Lukashev AN: Efficacy of
mitochondrial antioxidant plastoquinonyl-decyl-triphenylphosphonium
bromide (SkQ1) in the rat model of autoimmune arthritis. Oxid Med
Cell Longev. 2016(8703645)2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Saeidnia S and Abdollahi M: Toxicological
and pharmacological concerns on oxidative stress and related
diseases. Toxicol Appl Pharmacol. 273:442–455. 2013.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Burton GJ and Jauniaux E: Oxidative
stress. Best Pract Res Clin Obstet Gynaecol. 25:287–299.
2011.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Cao G, Yang S, Cao J, Tan Z, Wu L, Dong F,
Ding W and Zhang F: the role of oxidative stress in intervertebral
disc degeneration. Oxid Med Cell Longev.
2022(2166817)2022.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wen P, Zheng B, Zhang B, Ma T, Hao L and
Zhang Y: The role of ageing and oxidative stress in intervertebral
disc degeneration. Front Mol Biosci. 9(1052878)2022.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Peng X, Zhang C, Zhou ZM, Wang K, Gao JW,
Qian ZY, Bao JP, Ji HY, Cabral VLF and Wu XT: A20 attenuates
pyroptosis and apoptosis in nucleus pulposus cells via promoting
mitophagy and stabilizing mitochondrial dynamics. Inflamm Res.
71:695–710. 2022.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Li W, Cao T, Luo C, Cai J, Zhou X, Xiao X
and Liu S: Crosstalk between ER stress, NLRP3 inflammasome, and
inflammation. Appl Microbiol Biotechnol. 104:6129–6140.
2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Bartell SM, Kim HN, Ambrogini E, Han L,
Iyer S, Serra Ucer S, Rabinovitch P, Jilka RL, Weinstein RS, Zhao
H, et al: FoxO proteins restrain osteoclastogenesis and bone
resorption by attenuating H2O2 accumulation. Nat Commun.
5(3773)2014.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Kim J, Xu M, Xo R, Mates A, Wilson GL,
Pearsall AW IV and Grishko V: Mitochondrial DNA damage is involved
in apoptosis caused by pro-inflammatory cytokines in human OA
chondrocytes. Osteoarthritis Cartilage. 18:424–432. 2010.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Palma FR, He C, Danes JM, Paviani V,
Coelho DR, Gantner BN and Bonini MG: Mitochondrial superoxide
dismutase: What the established, the intriguing, and the novel
reveal about a key cellular redox switch. Antioxid Redox Signal.
32:701–714. 2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Farnaghi S, Prasadam I, Cai G, Friis T, Du
Z, Crawford R, Mao X and Xiao Y: Protective effects of
mitochondria-targeted antioxidants and statins on
cholesterol-induced osteoarthritis. FASEB J. 31:356–367.
2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Liu Y, Fiskum G and Schubert D: Generation
of reactive oxygen species by the mitochondrial electron transport
chain. J Neurochem. 80:780–787. 2002.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Murphy MP: How mitochondria produce
reactive oxygen species. Biochem J. 417:1–13. 2008.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Valcárcel-Ares MN, Riveiro-Naveira RR,
Vaamonde-García C, Loureiro J, Hermida-Carballo L, Blanco FJ and
López-Armada MJ: Mitochondrial dysfunction promotes and aggravates
the inflammatory response in normal human synoviocytes.
Rheumatology (Oxford). 53:1332–1343. 2014.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Cui H, Kong Y and Zhang H: Oxidative
stress, mitochondrial dysfunction, and aging. J Signal Transduct.
2012(646354)2012.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Blanco FJ, López-Armada MJ and Maneiro E:
Mitochondrial dysfunction in osteoarthritis. Mitochondrion.
4:715–728. 2004.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Early JO, Fagan LE, Curtis AM and Kennedy
OD: Mitochondria in injury, inflammation and disease of articular
skeletal joints. Front Immunol. 12(695257)2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Ma Z, Tang P, Dong W, Lu Y, Tan B, Zhou N,
Hao J, Shen J and Hu Z: SIRT1 alleviates IL-1β induced nucleus
pulposus cells pyroptosis via mitophagy in intervertebral disc
degeneration. Int Immunopharmacol. 107(108671)2022.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Hu Z, Wang Y, Gao X, Zhang Y, Liu C, Zhai
Y, Chang X, Li H, Li Y, Lou J and Li C: Optineurin-mediated
mitophagy as a potential therapeutic target for intervertebral disc
degeneration. Front Pharmacol. 13(893307)2022.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Ansari MY, Ahmad N, Voleti S, Wase SJ,
Novak K and Haqqi TM: Mitochondrial dysfunction triggers a
catabolic response in chondrocytes via ROS-mediated activation of
the JNK/AP1 pathway. J Cell Sci. 133(jcs247353)2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Song Y, Li S, Geng W, Luo R, Liu W, Tu J,
Wang K, Kang L, Yin H, Wu X, et al: Sirtuin 3-dependent
mitochondrial redox homeostasis protects against AGEs-induced
intervertebral disc degeneration. Redox Biol. 19:339–353.
2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Wu C, Luo J, Liu Y, Fan J, Shang X, Liu R,
Ye C, Yang J and Cao H: Doxorubicin suppresses chondrocyte
differentiation by stimulating ROS production. Eur J Pharm Sci.
167(106013)2021.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Li BL, Liu X, Gao M, Zhang F, Chen X, He
Z, Wang J, Tian W, Chen D, Zhou Z and Liu S: Programmed NP cell
death induced by mitochondrial ROS in a one-strike loading disc
degeneration organ culture model. Oxid Med Cell Longev.
2021(5608133)2021.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Shao Y, Zhang H, Guan H, Wu C, Qi W, Yang
L, Yin J, Zhang H, Liu L, Lu Y, et al: PDZK1 protects against
mechanical overload-induced chondrocyte senescence and
osteoarthritis by targeting mitochondrial function. Bone Res.
12(41)2024.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Hou L, Wang G, Zhang X, Lu F, Xu J, Guo Z,
Lin J, Zheng Z, Liu H, Hou Y, et al: Mitoquinone alleviates
osteoarthritis progress by activating the NRF2-Parkin axis.
iScience. 26(107647)2023.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Poudel SB, Ruff RR, Yildirim G, Miller RA,
Harrison DE, Strong R, Kirsch T and Yakar S: Development of primary
osteoarthritis during aging in genetically diverse UM-HET3 mice.
Arthritis Res Ther. 26(118)2024.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Tisherman R, Coelho P, Phillibert D, Wang
D, Dong Q, Vo N, Kang J and Sowa G: NF-κB signaling pathway in
controlling intervertebral disk cell response to inflammatory and
mechanical stressors. Phys Ther. 96:704–711. 2016.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Wang F, Cai F, Shi R, Wang XH and Wu XT:
Aging and age related stresses: A senescence mechanism of
intervertebral disc degeneration. Osteoarthritis Cartilage.
24:398–408. 2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Kang L, Liu S, Li J, Tian Y, Xue Y and Liu
X: The mitochondria-targeted anti-oxidant MitoQ protects against
intervertebral disc degeneration by ameliorating mitochondrial
dysfunction and redox imbalance. Cell Prolif.
53(e12779)2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Laiguillon MC, Courties A, Houard X,
Auclair M, Sautet A, Capeau J, Fève B, Berenbaum F and Sellam J:
Characterization of diabetic osteoarthritic cartilage and role of
high glucose environment on chondrocyte activation: Toward
pathophysiological delineation of diabetes mellitus-related
osteoarthritis. Osteoarthritis Cartilage. 23:1513–1522.
2025.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Ansari MY, Ball HC, Wase SJ, Novak K and
Haqqi TM: Lysosomal dysfunction in osteoarthritis and aged
cartilage triggers apoptosis in chondrocytes through BAX mediated
release of Cytochrome c. Osteoarthritis Cartilage. 29:100–112.
2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Kang L, Liu S, Li J, Tian Y, Xue Y and Liu
X: Parkin and Nrf2 prevent oxidative stress-induced apoptosis in
intervertebral endplate chondrocytes via inducing mitophagy and
anti-oxidant defenses. Life Sci. 243(117244)2020.PubMed/NCBI View Article : Google Scholar
|
|
40
|
McGarry T, Biniecka M, Gao W, Cluxton D,
Canavan M, Wade S, Wade S, Gallagher L, Orr C, Veale DJ and Fearon
U: Resolution of TLR2-induced inflammation through manipulation of
metabolic pathways in Rheumatoid arthritis. Sci Rep.
7(43165)2017.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Szeto HH, Liu S, Soong Y, Alam N, Prusky
GT and Seshan SV: Protection of mitochondria prevents high-fat
diet-induced glomerulopathy and proximal tubular injury. Kidney
Int. 90:997–1011. 2016.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Peng X, Wang K, Zhang C, Bao JP, Vlf C,
Gao JW, Zhou ZM and Wu XT: The mitochondrial antioxidant SS-31
attenuated lipopolysaccharide-induced apoptosis and pyroptosis of
nucleus pulposus cells via scavenging mitochondrial ROS and
maintaining the stability of mitochondrial dynamics. Free Radic
Res. 55:1080–1093. 2021.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Zhang X, Eliasberg CD and Rodeo SA:
Mitochondrial dysfunction and potential mitochondrial protectant
treatments in tendinopathy. Ann N Y Acad Sci. 1490:29–41.
2021.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Siekacz K, Piotrowski WJ, Iwański MA,
Górski P and Białas AJ: The role of interaction between
mitochondria and the extracellular matrix in the development of
idiopathic pulmonary fibrosis. Oxid Med Cell Longev.
2021(9932442)2021.PubMed/NCBI View Article : Google Scholar
|
|
45
|
An F, Zhang J, Gao P, Xiao Z, Chang W,
Song J, Wang Y, Ma H, Zhang R, Chen Z and Yan C: New insight of the
pathogenesis in osteoarthritis: the intricate interplay of
ferroptosis and autophagy mediated by mitophagy/chaperone-mediated
autophagy. Front Cell Dev Biol. 11(1297024)2023.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Wu J, Zhou X, Xu X and Xie J: A molecular
chemical perspective: mitochondrial dynamics is not a bystander of
cartilage diseases. ACS Pharmacol Transl Sci. 8:1473–1497.
2025.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Xiao B, Goh JY, Xiao L, Xian H, Lim KL and
Liou YC: Reactive oxygen species trigger Parkin/PINK1
pathway-dependent mitophagy by inducing mitochondrial recruitment
of Parkin. J Biol Chem. 292:16697–16708. 2017.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Lin J, Zheng X, Zhang Z, Zhuge J, Shao Z,
Huang C, Jin J, Chen X, Chen Y, Wu Y, et al: Inhibition of LRRK2
restores parkin-mediated mitophagy and attenuates intervertebral
disc degeneration. Osteoarthritis Cartilage. 29:579–591.
2021.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Zeng Z, Zhou X and Wang Y, Cao H, Guo J,
Wang P, Yang Y and Wang Y: Mitophagy-A new target of bone disease.
Biomolecules. 12(1420)2022.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Lin S, Li T, Zhang B and Wang P: Taurine
rescues intervertebral disc degeneration by activating mitophagy
through the PINK1/Parkin pathway. Biochem Biophys Res Commun.
739(150587)2024.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Wu ZL, Wang KP, Chen YJ, Song W, Liu Y,
Zhou KS, Mao P, Ma Z and Zhang HH: Knocking down EGR1 inhibits
nucleus pulposus cell senescence and mitochondrial damage through
activation of PINK1-Parkin dependent mitophagy, thereby delaying
intervertebral disc degeneration. Free Radic Biol Med. 224:9–22.
2024.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Zhang Y, Xing D, Liu Y, Sha S, Xiao Y, Liu
Z, Yin Q, Gao Z and Liu W: CREG1 attenuates intervertebral disc
degeneration by alleviating nucleus pulposus cell pyroptosis via
the PINK1/Parkin-related mitophagy pathway. Int Immunopharmacol.
147(113974)2025.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Cheung C, Tu S, Feng Y, Wan C, Ai H and
Chen Z: Mitochondrial quality control dysfunction in
osteoarthritis: Mechanisms, therapeutic strategies & future
prospects. Arch Gerontol Geriatr. 125(105522)2024.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Luchkova A, Mata A and Cadenas S: Nrf2 as
a regulator of energy metabolism and mitochondrial function. FEBS
Lett. 598:2092–2105. 2024.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Wang F, Guo K, Nan L, Wang S, Lu J, Wang
Q, Ba Z, Huang Y and Wu D: Kartogenin-loaded hydrogel promotes
intervertebral disc repair via protecting MSCs against reactive
oxygen species microenvironment by Nrf2/TXNIP/NLRP3 axis. Free
Radic Biol Med. 204:128–150. 2023.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Yang G, Liu X, Jing X, Wang J, Wang H,
Chen F, Wang W, Shao Y and Cui X: Astaxanthin suppresses oxidative
stress and calcification in vertebral cartilage endplate via
activating Nrf-2/HO-1 signaling pathway. Int Immunopharmacol.
119(110159)2023.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Guo P, Alhaskawi A, Adel Abdo Moqbel S and
Pan Z: Recent development of mitochondrial metabolism and
dysfunction in osteoarthritis. Front Pharmacol.
16(1538662)2025.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Yang Q, Shi Y, Jin T, Duan B and Wu S:
Advanced glycation end products induced mitochondrial dysfunction
of chondrocytes through repression of AMPKα-SIRT1-PGC-1α pathway.
Pharmacology. 107:298–307. 2022.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Li Z, Zhang Y, Tian F, Wang Z, Song H,
Chen H and Wu B: Omentin-1 promotes mitochondrial biogenesis via
PGC1α-AMPK pathway in chondrocytes. Arch Physiol Biochem.
129:291–297. 2023.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Zhang S, Cai J, Yao Y, Huang L, Zheng L
and Zhao J: Mitochondrial-targeting Mn3O4/UIO-TPP nanozyme scavenge
ROS to restore mitochondrial function for osteoarthritis therapy.
Regen Biomater. 10(rbad078)2023.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Wang SH, Xu XL and Chen W: How do
organelle-targeting nanotherapeutics treat inflammatory diseases? A
comprehensive review of the literature. Int J Nanomedicine.
20:7133–7152. 2025.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Chen X, Li C, Cao X, Jia X, Chen X, Wang
Z, Xu W, Dai F and Zhang S: Mitochondria-targeted supramolecular
coordination container encapsulated with exogenous itaconate for
synergistic therapy of joint inflammation. Theranostics.
12:3251–3272. 2022.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Kan S, Duan M, Liu Y, Wang C and Xie J:
Role of mitochondria in physiology of chondrocytes and diseases of
osteoarthritis and rheumatoid arthritis. Cartilage. 13
(2_suppl):1102S–1121S. 2021.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Liu X, Chen Y, Wang H, Wei Y, Yuan Y, Zhou
Q, Fang F, Shi S, Jiang X, Dong Y and Li X: Microglia-derived IL-1β
promoted neuronal apoptosis through ER stress-mediated signaling
pathway PERK/eIF2α/ATF4/CHOP upon arsenic exposure. J Hazard Mater.
417(125997)2021.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Yan Z, He Z, Jiang H and Zhang Y, Xu Y and
Zhang Y: TRPV4-mediated mitochondrial dysfunction induces
pyroptosis and cartilage degradation in osteoarthritis via the
Drp1-HK2 axis. Int Immunopharmacol. 123(110651)2023.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Li P, Zhou M, Wang J, Tian J, Zhang L, Wei
Y, Yang F, Xu Y and Wang G: Important role of mitochondrial
dysfunction in immune triggering and inflammatory response in
rheumatoid arthritis. J Inflamm Res. 17:11631–11657.
2024.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Hu K, Song M, Song T, Jia X and Song Y:
Osteoimmunology in osteoarthritis: Unraveling the interplay of
immunity, inflammation, and joint degeneration. J Inflamm Res.
18:4121–4142. 2025.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Dou Y, Zhang Y, Liu Y, Sun X, Liu X, Li B
and Yang Q: Role of macrophage in intervertebral disc degeneration.
Bone Res. 13(15)2025.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Wang S, Wang H, Feng C, Li C, Li Z, He J
and Tu C: The regulatory role and therapeutic application of
pyroptosis in musculoskeletal diseases. Cell Death Discov.
8(492)2022.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Wright HL, Lyon M, Chapman EA, Moots RJ
and Edwards SW: Rheumatoid arthritis synovial fluid neutrophils
drive inflammation through production of chemokines, reactive
oxygen species, and neutrophil extracellular traps. Front Immunol.
11(584116)2021.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Wu B, Ni H, Li J, Zhuang X, Zhang J, Qi Z,
Chen Q, Wen Z, Shi H, Luo X and Jin B: The impact of circulating
mitochondrial DNA on cardiomyocyte apoptosis and myocardial injury
after TLR4 activation in experimental autoimmune myocarditis. Cell
Physiol Biochem. 42:713–728. 2017.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Mukherjee S, Patra R, Behzadi P, Masotti
A, Paolini A and Sarshar M: Toll-like receptor-guided therapeutic
intervention of human cancers: Molecular and immunological
perspectives. Front Immunol. 14(1244345)2023.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Chávez MD and Tse HM: Targeting
mitochondrial-derived reactive oxygen species in T cell-mediated
autoimmune diseases. Front Immunol. 12(703972)2021.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Masoumi M, Alesaeidi S, Khorramdelazad H,
Behzadi M, Baharlou R, Alizadeh-Fanalou S and Karami J: Role of T
cells in the pathogenesis of rheumatoid arthritis: Focus on
immunometabolism dysfunctions. Inflammation. 46:88–102.
2023.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Weyand CM, Wu B, Huang T, Hu Z and Goronzy
JJ: Mitochondria as disease-relevant organelles in rheumatoid
arthritis. Clin Exp Immunol. 211:208–223. 2022.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Jing W, Liu C, Su C, Liu L, Chen P, Li X,
Zhang X, Yuan B, Wang H and Du X: Role of reactive oxygen species
and mitochondrial damage in rheumatoid arthritis and targeted
drugs. Front Immunol. 14(1107670)2023.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Michalak KP and Michalak AZ: Understanding
chronic inflammation: couplings between cytokines, ROS, NO, Cai2+,
HIF-1α, Nrf2 and autophagy. Front Immunol.
16(1558263)2025.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Sung JY, Kim SG, Park SY, Kim JR and Choi
HC: Telomere stabilization by metformin mitigates the progression
of atherosclerosis via the AMPK-dependent p-PGC-1α pathway. Exp Mol
Med. 56:1967–1979. 2024.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Zhi ZY and Wang PC: The mitochondrial
targeting drug SkQ1 attenuates the progression of post-traumatic
osteoarthritis through suppression of mitochondrial oxidative
stress. Curr Mol Pharmacol. 17(e18761429383749)2024.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Xiong Z, Liao Y, Zhang Z, Wan Z, Liang S
and Guo J: Molecular insights into oxidative-stress-mediated
cardiomyopathy and potential therapeutic strategies. Biomolecules.
15(670)2025.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Wu J, Xu J, Zhang M, Zhong J, Gao W and Wu
M: Chondrocyte mitochondrial quality control: A novel insight into
osteoarthritis and cartilage regeneration. Adv Wound Care (New
Rochelle): Apr 18, 2025 (Epub ahead of print).
|
|
82
|
Wang Z, Yin F, Xu J, Zhang T, Wang G, Mao
M, Wang Z, Sun W, Han J, Yang M, et al: CYT997(Lexibulin) induces
apoptosis and autophagy through the activation of mutually
reinforced ER stress and ROS in osteosarcoma. J Exp Clin Cancer
Res. 38(44)2019.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Saberi M, Zhang X and Mobasheri A:
Targeting mitochondrial dysfunction with small molecules in
intervertebral disc aging and degeneration. Geroscience.
43:517–537. 2021.PubMed/NCBI View Article : Google Scholar
|
|
84
|
McCully JD, Levitsky S, del Nido PJ and
Cowan DB: Mitochondrial transplantation for therapeutic use. Clin
Trans Med. 5(e16)2016.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Luo H, Lai Y, Tang W, Wang G, Shen J and
Liu H: Mitochondrial transplantation: A promising strategy for
treating degenerative joint diseases. J Transl Med.
22(941)2024.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Lee AR, Woo JS, Lee SY, Na HS, Cho KH, Lee
YS, Lee JS, Kim SA, Park SH, Kim SJ and Cho ML: Mitochondrial
transplantation ameliorates the development and progression of
osteoarthritis. Immune Netw. 22(e14)2022.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Zhong G, Madry H and Cucchiarini M:
Mitochondrial genome editing to treat human osteoarthritis-A
narrative review. Int J Mol Sci. 23(1467)2022.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Yang X, Jiang J, Li Z, Liang J and Xiang
Y: Strategies for mitochondrial gene editing. Comput Struct
Biotechnol J. 19:3319–3329. 2021.PubMed/NCBI View Article : Google Scholar
|














