Introduction
Tongue squamous cell carcinoma (TSCC), the most
common oral cancer, demonstrates a strong tendency for cervical
lymph node metastasis, even in the early stages (1). According to current National
Comprehensive Cancer Network guidelines (Version 1.2026), standard
treatment for head and neck squamous cell carcinoma, including
TSCC, primarily involves surgical resection, with adjuvant
radiotherapy or chemoradiotherapy for high-risk pathological
features (2). In recent years,
immune checkpoint inhibitors have been incorporated into the
management of very advanced disease (unresectable, recurrent, or
metastatic). Although therapeutic advances have been made, outcomes
remain poor, particularly in patients with advanced or recurrent
disease.
Suramin, a polysulfonated naphthylurea first
developed in 1916 for African trypanosomiasis (3,4), has
been repurposed for oncology due to its ability to block diverse
signaling pathways (TGF-β, bFGF, PDGF, EGF, and VEGF) and cell
cycle regulators (cyclins A2, B1, D1, and E). In addition, it
interferes with the RNA-binding protein HuR, which stabilizes
oncogenic transcripts (5-7).
These pleiotropic actions have renewed the interest
in suramin and its use in hormone-refractory prostate cancer (HRPC)
(8). At low doses, suramin can
function as a chemosensitizer (4),
and nanoparticle formulations combining suramin with doxorubicin
have demonstrated strong efficacy without systemic toxicity in
breast cancer models (9). In TSCC,
suramin suppresses tumor proliferation, migration, and invasion
(7). However, clinical evidence
remains limited, and the mechanisms underlying TSCC remain unclear.
We investigated the effects of suramin on global gene expression in
HSC-3 cells using next-generation sequencing (NGS) and evaluated
its in vivo activity in a luciferase-labeled orthotopic
mouse model to elucidate its therapeutic potential.
Materials and methods
Cell lines, cell culture, suramin
treatment
The human tongue squamous cell carcinoma cell line
HSC-3 (RIKEN, Tsukuba, Japan) and luciferase-expressing HSC-3 cells
(HSC-3-Luc; JCRB Cell Bank, Osaka, Japan) were used. The cells were
maintained in Dulbecco's Modified Eagle's medium (DMEM)
supplemented with fetal bovine serum (FBS). Cells were incubated
for 24 h in serum-free DMEM containing 100 µM suramin (FUJIFILM
Wako Pure Chemical Corporation, Osaka, Japan). Control cells
received an equivalent volume of UltraPure™ distilled water
(Invitrogen; NY, USA), the solvent for suramin. The use of 100 µM
suramin for 24 h was based on our previous report (7), in which this concentration effectively
suppressed HuR-regulated genes, including CCNB1 and
CCNA2, at both the mRNA and protein levels while remaining
non-cytotoxic in HSC-3 cells (IC50=732 µM).
RNA extraction and sequencing
Total RNA was extracted using the RNeasy Mini Kit
(Qiagen; Hilden, Germany), and 1 µg of total RNA was used for
downstream processing. The mRNA was enriched with oligo (dT) beads,
fragmented, and reverse-transcribed to generate cDNA. Library
construction and sequencing were performed by Novogene (Beijing,
China) using an Illumina platform.
Bioinformatics analysis
Raw reads were qualitatively checked and aligned to
the human reference genome. Gene expression was quantified using
HTSeq (v0.6.1, union mode) and normalized to fragments per kilobase
of transcript per million mapped reads to enable comparisons
between samples.
Differentially expressed genes (DEGs) were
identified using DEGseq (10) with
TMM normalization under a Poisson distribution model, and defined
as those with |log2 (fold change)| >1 and q<0.005
(Benjamini-Hochberg false discovery rate). Gene Ontology (GO) and
Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses
were performed using g:Profiler (version e113_eg59_p19_f6a03c19;
database updated May 23, 2025). In GO analysis, terms with a size
>1,500 were excluded, and categories with an intersection size
≥3 were retained; for KEGG, terms with a size <300 and an
intersection size ≥3 were applied. Protein-protein interaction
(PPI) networks were generated using STRING (version 12.0) with a
high-confidence interaction threshold (combined score >0.700),
and hub genes were identified by degree centrality. The DEGs were
annotated using AnimalTFDB v4.0, and only sequence-specific
transcription factors (TFs) were retained, excluding chromatin
regulators and cofactors. Candidate transcription factors were
further validated by confirming the binding motifs in JASPAR 2024
and by cross-referencing the binding activity with the ENCODE
ChIP-seq datasets.
Biological replicates were not included, and this
single-sample design limited statistical robustness; therefore,
conservative DEG thresholds were applied to reduce false-positive
findings.
Orthotopic tongue cancer mouse model and
treatments. HSC-3-Luc cells (1x105) suspended in 30
µl of sterile HBSS (without calcium, magnesium, and phenol red;
Gibco, Thermo Fisher Scientific, Waltham, MA, USA) were
orthotopically injected into the left lateral tongue of 8-week-old
male BALB/cAJcl-nu/nu mice using a 27-gauge needle. Day 0 was
defined as the day of tumor implantation. Mice were anesthetized by
intraperitoneal injection with a combination of medetomidine (Orion
Pharma, Espoo, Finland), midazolam (Astellas Pharma, Tokyo, Japan),
and butorphanol (Meiji Seika Pharma, Tokyo, Japan) (MMB) at doses
of 0.6, 3.2, and 4.0 mg/kg, respectively. Because a previously
reported intraperitoneal regimen of 0.75/4/5 mg/kg (11) resulted in deep anesthesia in
preliminary experiments, an 80% reduced dose was employed in the
present study. Because the procedure involved only intralingual
injection, surgical-level deep anesthesia was not required, and
adequate anesthesia was confirmed by the absence of body movement
or facial response to gentle pinching with forceps or needle
puncture of the tongue. The mice were randomized into three groups:
vehicle (n=4), suramin 20 mg/kg (n=5), and suramin 60 mg/kg (n=4),
which were administered intraperitoneally twice weekly.
Tumor
growth was monitored by bioluminescence imaging (BLI) using an IVIS
Spectrum (PerkinElmer) on days 1, 8, 15, 22, 25, and 29 after
intraperitoneal injection of VivoGlo™ Luciferin (150 mg/kg, 200 µl;
Promega, Madison, WI, USA). Tumor growth was assessed on predefined
imaging days (days 1, 8, 15, 22, and 29). An additional imaging
session on day 25 was performed to assess tumor status because
euthanasia was required at that time point. In this orthotopic
tongue cancer model, anesthesia is associated with a risk of upper
airway compromise due to posterior tongue displacement.
Accordingly, in vivo bioluminescence imaging was performed
at the minimum necessary frequency to reduce anesthesia-related
risk. For IVIS imaging, anesthesia was induced and maintained with
2.0% isoflurane. Photon flux was quantified using fixed ROIs under
standardized settings. Longitudinal analysis was restricted to days
1-15, when all mice were alive, and the signals remained within the
linear range, to minimize signal saturation and attrition bias.
Tumor presence was not assessed by any method other than IVIS
during the experimental period because reliable palpation of mouse
tongue tumors was not feasible. Tumor presence was confirmed only
at the final necropsy, as confirmation of tumor mass formation
required dissection. Mice were monitored daily for general health
status and euthanized if body weight loss exceeded 20% or if severe
debilitation occurred. Euthanasia was performed by cervical
dislocation under isoflurane anesthesia on day 29 or earlier, if
the humane endpoints were met, immediately after the final IVIS
imaging while the mice remained anesthetized, and death was
confirmed by the sustained absence of respiration and heartbeat.
The cervical lymph nodes and lungs were harvested at necropsy,
immersed in luciferin, and subjected to ex vivo BLI to
detect metastases.
Tissues showing detectable ex vivo BLI
signals were fixed in 10% neutral-buffered formalin, embedded in
paraffin, and histologically examined to confirm the presence of
metastatic SCC. Lymph nodes and lungs without detectable ex
vivo BLI signals were not systematically examined. Because
necropsies were performed at different time points owing to
attrition, ex vivo metastasis measurements could not be
temporally matched across animals. Metastasis was defined as a
radiance value ≥3.0-fold above the background in the same animal,
together with a discrete focal bioluminescence signal exhibiting a
smooth intensity gradient.
Statistical analysis
Data are presented as mean ± SD with individual
values. Enrichment analyses were considered significant at adjusted
q<0.05 (Benjamini-Hochberg correction). A two-way mixed-effects
model (REML with Geisser-Greenhouse correction) was used to
evaluate the group, time, and interaction effects. Effect sizes
were quantified as partial eta squared (partial η2), and
statistical significance was defined as P<0.05. All analyses
were performed using GraphPad Prism 10.
Results
Suramin-induced transcriptomic
alterations revealed by RNA-sequencing
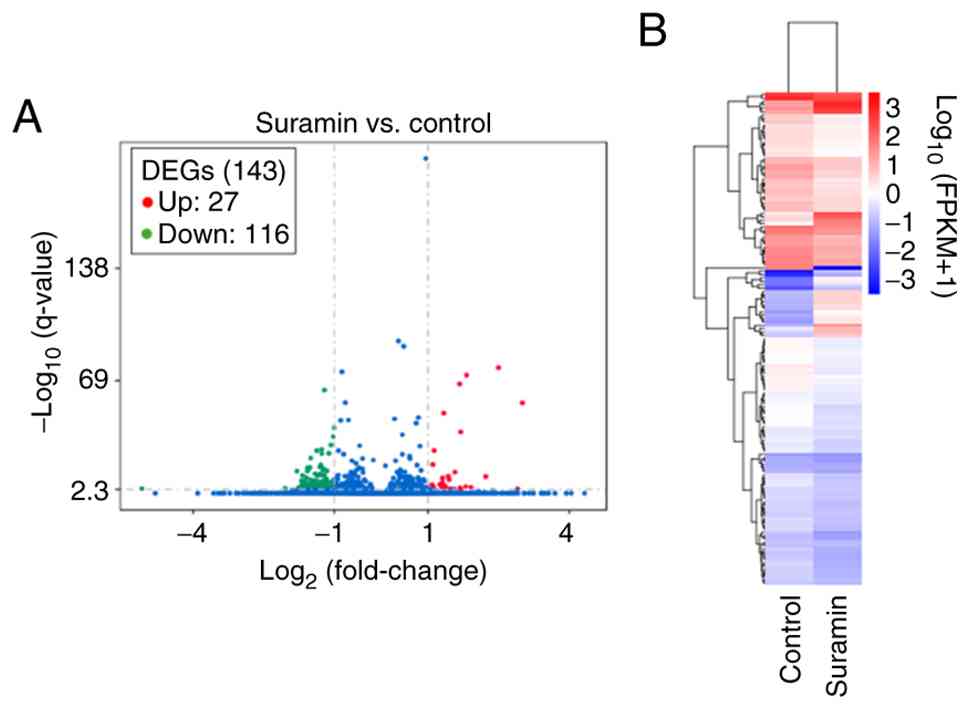
RNA-sequencing detected 60,448 genes, of which
12,114 were commonly expressed in both groups. Differential
expression analysis identified 116 downregulated and 27 upregulated
genes in suramin-treated cells (Fig.
1A), and cluster analysis showed distinct expression patterns
(Fig. 1B). Downregulated genes were
dominated by regulators of the G2/M transition and
mitotic progression (e.g., CCNB1, CDC20,
AURKA), kinetochore and spindle assembly (e.g.,
CENPF, TPX, TUBB), DNA metabolism (RRM2
and TK1), chromatin regulation (DNMT1), cytokinesis
(ANLN), chromosome segregation (MKI67 and
TOP2A), nuclear envelope integrity (LMNB1/2), DNA
damage response (HMGB1/2 and PCNA), and metabolic
regulators (LDHA and PSAT1) were broadly suppressed
(Table I).
 | Table ITop 30 downregulated genes identified
by mRNA-sequencing in suramin-treated HSC-3 cells. |
Table I
Top 30 downregulated genes identified
by mRNA-sequencing in suramin-treated HSC-3 cells.
| Rank | Gene symbol | Known function | Fold change | q-value |
|---|
| 1 | TUBB | Microtubule
component | 0.43 |
4.61x10-64 |
| 2 | LDHA | Glycolysis, lactate
production, Warburg effect | 0.50 |
5.32x10-41 |
| 3 | EIF5A | Translation
elongation, mRNA turnover | 0.49 |
1.85x10-35 |
| 4 | HMGB1 | Chromatin
remodeling, DAMPs | 0.48 |
2.30x10-30 |
| 5 | TUBB4B | Microtubule
assembly | 0.42 |
1.42x10-27 |
| 6 | LMNB2 | Nuclear lamina
component | 0.39 |
7.51x10-27 |
| 7 | TOP2A | DNA topology,
mitotic chromosome condensation | 0.46 |
3.31x10-25 |
| 8 | MKI67 | Chromatin
organization, cell proliferation | 0.42 |
4.51x10-25 |
| 9 | UBE2S | Ubiquitination,
mitotic progression | 0.35 |
2.29x10-22 |
| 10 | TUBA1C | Microtubule
assembly | 0.41 |
3.88x10-18 |
| 11 | KPNA2 | Nuclear import | 0.40 |
6.47x10-17 |
| 12 | CCNB1 | G2/M
transition | 0.35 |
1.43x10-16 |
| 13 | CDC20 | Anaphase-promoting
complex activator | 0.34 |
2.30x10-16 |
| 14 | HMGB2 | Chromatin
remodeling, | 0.42 |
6.15x10-16 |
| 15 | RRM2 | dNTP synthesis, DNA
replication | 0.34 |
1.59x10-15 |
| 16 | RANBP1 | Nucleocytoplasmic
transport | 0.42 |
5.75x10-15 |
| 17 | DNMT1 | DNA methylation
maintenance | 0.45 |
1.43x10-14 |
| 18 | TK1 | dTTP synthesis, DNA
replication | 0.34 |
1.96x10-14 |
| 19 | TUBA1B | Microtubule
assembly | 0.29 |
2.16x10-14 |
| 20 | ANLN | Cytokinesis, actin
cytoskeleton regulation | 0.38 |
1.03x10-12 |
| 21 | TCOF1 | rRNA
processing | 0.40 |
1.32x10-12 |
| 22 | LMNB1 | Nuclear lamina
structure | 0.40 |
6.85x10-12 |
| 23 | CENPF | Kinetochore
function, chromosome segregation | 0.43 |
6.95x10-12 |
| 24 | TPX2 | Spindle assembly,
microtubule nucleation | 0.42 |
8.74x10-12 |
| 25 | PSAT1 | Serine
biosynthesis | 0.31 |
1.19x10-11 |
| 26 | CKS1B | CDK regulation,
cell cycle | 0.39 |
1.96x10-11 |
| 27 | AURKA | Mitotic spindle,
centrosome maturation | 0.33 |
3.28x10-10 |
| 28 | PCNA | DNA replication and
repair | 0.45 |
1.65x10-9 |
| 29 | HNRNPAB | Pre-mRNA
processing, mRNA transport | 0.49 |
6.15x10-9 |
| 30 | CKS2 | CDK regulation,
cell cycle progression | 0.35 |
6.31x10-9 |
In contrast, the upregulated genes encoded several
matrix metalloproteinases (MMPs) with their inhibitor TIMP3,
stress-response genes (TXNIP and TP53INP1),
pro-apoptotic ligand TNFSF10 (TRAIL), and immune-related
regulators such as PIK3IP1 and FYB (Table II).
| Table IIThe 27 upregulated genes identified
by mRNA-sequencing in suramin-treated HSC-3 cells. |
Table II
The 27 upregulated genes identified
by mRNA-sequencing in suramin-treated HSC-3 cells.
| Rank | Gene symbol | Known function | Fold change | q-value |
|---|
| 1 | MMP10 | ECM
degradation | 5.63 |
1.16x10-77 |
| 2 | MMP1 | ECM degradation,
invasion | 3.52 |
3.18x10-73 |
| 3 | CLU | Apoptosis
modulation, extracellular chaperone | 3.17 |
9.02x10-68 |
| 4 | MMP13 | ECM degradation,
collagen degradation | 8.02 |
3.43x10-56 |
| 5 | TXNIP | Oxidative stress
response, redo regulation | 2.52 |
6.30x10-50 |
| 6 | CLDN1 | Tight junctions,
epithelial barrier | 3.22 |
2.25x10-38 |
| 7 | GLUL | Metabolic
adaptation, glutamine synthesis | 2.18 |
7.04x10-27 |
| 8 | MT2A | Metal ion
homeostasis, oxidative stress response | 2.14 |
2.62x10-18 |
| 9 | CYBRD1 | Iron metabolism,
ferric reductase activity | 2.96 |
9.73x10-14 |
| 10 | CCDC80 | Cell adhesion,
matrix assembly | 2.68 |
3.69x10-11 |
| 11 | MMP12 | ECM degradation,
elastin remodeling | 4.67 |
5.89x10-11 |
| 12 | TIMP3 | MMP inhibition, ECM
stabilization | 2.47 |
4.20x10-10 |
| 13 | FBXO32 | E3 ligase, protein
degradation | 2.71 |
3.74x10-9 |
| 14 | PTGES | PGE2
synthesis, inflammatory response | 2.47 |
2.96x10-7 |
| 15 | PIK3IP1 | Negative regulator
of PI3K | 2.37 |
3.38x10-6 |
| 16 | PLEKHA6 | PH
domain-containing protein | 2.12 |
5.33x10-6 |
| 17 | DHRS3 | Retinal to retinol
reduction | 2.57 |
4.47x10-5 |
| 18 | FYB | T-cell signaling
adaptor | 3.48 |
7.23x10-5 |
| 19 | TNFSF10 | Apoptosis
induction | 2.15 |
8.09x10-5 |
| 20 |
TP53INP1 | Stress-induced
apoptosis | 3.76 |
1.87x10-4 |
| 21 | KLHL24 | BCR E3 ligase
complex | 2.44 |
1.88x10-4 |
| 22 | NATD1 | Acetyltransferase
domain-containing protein | 2.58 |
2.14x10-4 |
| 23 | MMP3 | ECM
degradation | 3.19 |
3.75x10-4 |
| 24 | MT1E | Heavy metal
binding | 2.77 |
1.37x10-3 |
| 25 | TG | Substrate for
thyroid hormone synthesis | 3.22 |
1.66x10-3 |
| 26 | CTSF | Intracellular
degradation, turnover of proteins | 7.46 |
1.78x10-3 |
| 27 | ITGB8 | RGD
recognition | 2.94 |
2.32x10-3 |
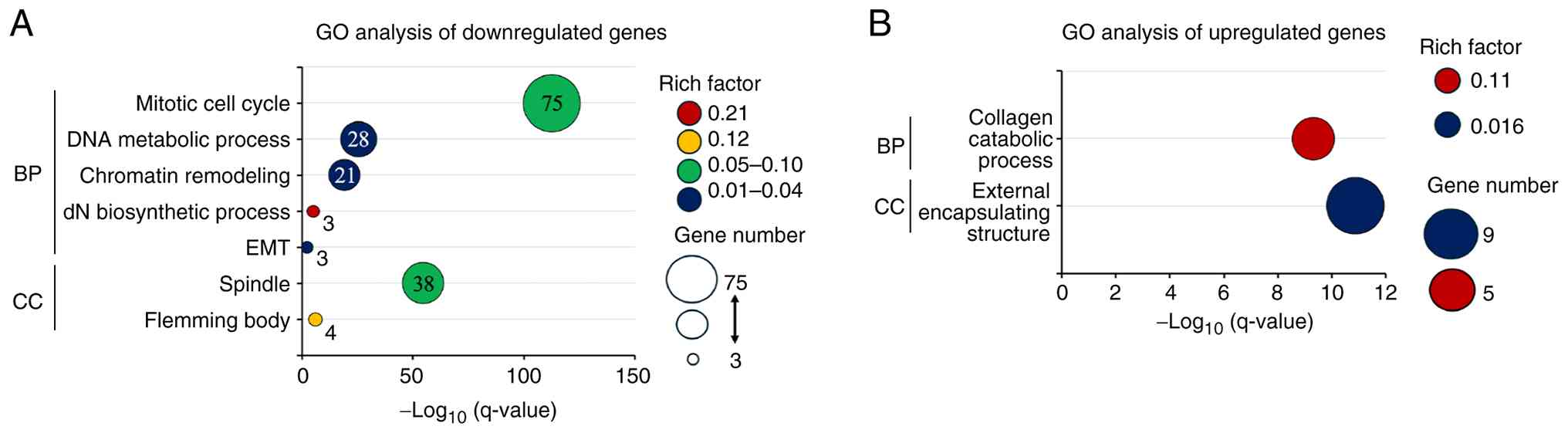
GO enrichment analysis
GO enrichment analysis of the downregulated gene set
demonstrated significant overrepresentation of mitosis-related
pathways, including the mitotic cell cycle, DNA metabolic process,
chromatin remodeling, and deoxyribonucleotide biosynthetic process,
indicating the coordinated downregulation of genes governing
G2/M progression, DNA replication/repair, and chromatin
regulation (Fig. 2A). In contrast,
the upregulated genes were enriched in extracellular matrix-related
processes, such as collagen catabolism, consistent with the
activation of ECM remodeling (Fig.
2B).
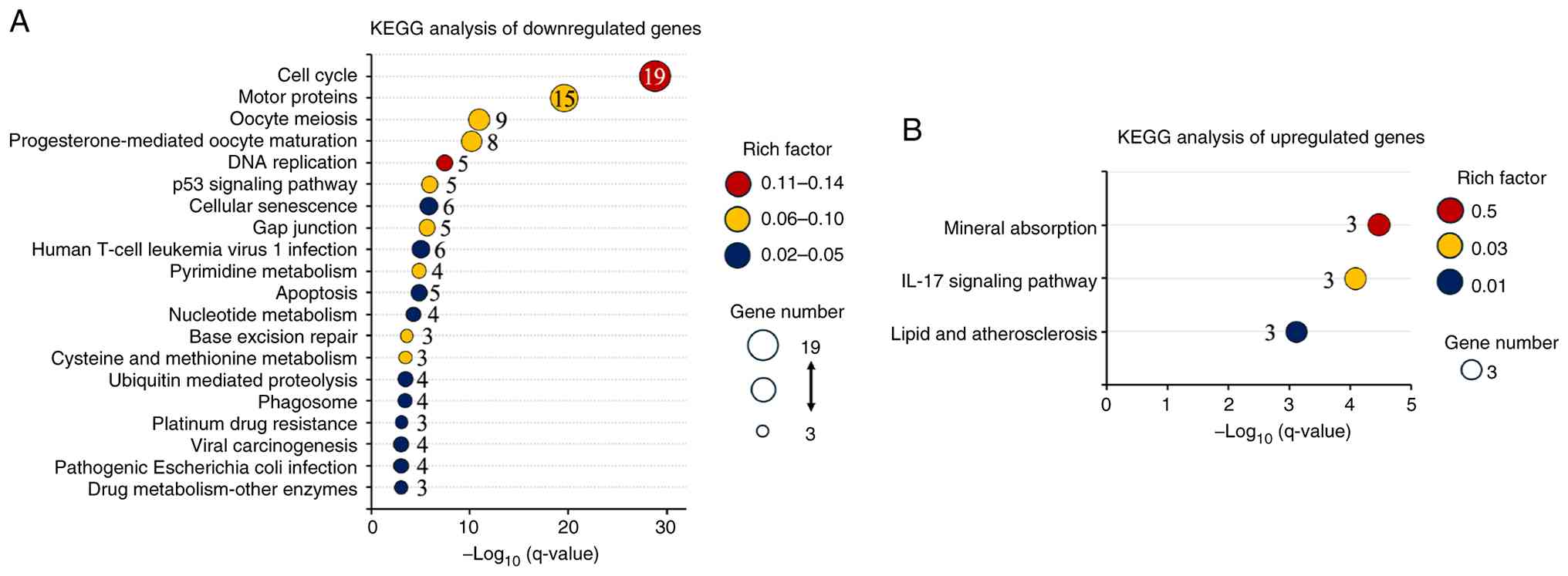
KEGG analysis of differentially
expressed genes
The KEGG pathway analysis demonstrated significant
enrichment of pathways central to cell proliferation and genome
stability in the downregulated gene set. The cell cycle was most
prominently enriched, with associated terms involving
G2/M transition, spindle assembly, and checkpoint
regulation. Additional enrichment in DNA replication, p53
signaling, and base excision repair supports the notion of impaired
genome maintenance. Enrichment of cellular senescence,
ubiquitin-mediated proteolysis, and apoptosis further indicated
perturbation of protein turnover and checkpoint surveillance
(Fig. 3A). In contrast, the
upregulated genes were enriched in only three pathways, mineral
absorption, IL-17 signaling, and lipid and atherosclerosis,
suggesting adaptive changes in metal ion homeostasis, immune
modulation, and lipid metabolism (Fig.
3B).
Protein-protein interaction network
analysis
The PPI network of downregulated genes formed a
densely interconnected module dominated by cell cycle and mitotic
regulators. Hub genes included CDK1, CCNA2,
CCNB1, TOP2A, CDC20, KIF11,
KIF20A, BUB1, PLK1, AURKB, TPX2,
and CENPF, all of which are central to the G2/M
transition, spindle function, and chromosome segregation (Table III).
| Table IIITop 30 downregulated and upregulated
genes based on degree centrality in the PPI network. |
Table III
Top 30 downregulated and upregulated
genes based on degree centrality in the PPI network.
| A, Downregulated
genes |
|---|
| Rank | Gene symbol | Degree |
|---|
| 1 | CDK1 | 77 |
| 2 | CCNA2 | 71 |
| 3 | CCNB1 | 70 |
| 4 | TOP2A | 69 |
| 5 | CDC20 | 67 |
| 6 | KIF11 | 67 |
| 7 | KIF20A | 66 |
| 8 | BUB1 | 66 |
| 9 | PLK1 | 65 |
| 10 | DLGAP5 | 64 |
| 11 | AURKB | 63 |
| 12 | BUB1B | 62 |
| 13 | TPX2 | 62 |
| 14 | CCNB2 | 61 |
| 15 | BIRC5 | 61 |
| 16 | KIF2C | 61 |
| 17 | AURKA | 60 |
| 18 | KIF4A | 60 |
| 19 | CDCA8 | 60 |
| 20 | CENPF | 59 |
| 21 | UBE2C | 59 |
| 22 | ASPM | 59 |
| 23 | KIF23 | 57 |
| 24 | NUSAP1 | 57 |
| 25 | CEP55 | 56 |
| 26 | MELK | 56 |
| 27 | CENPA | 55 |
| 28 | PBK | 55 |
| 29 | NCAPG | 54 |
| 30 | RRM2 | 54 |
| B, Upregulated
genes |
| Rank | Gene symbol | Degree |
| 1 | MMP3 | 4 |
| 2 | MMP1 | 3 |
| 3 | TIMP3 | 3 |
| 4 | MMP10 | 2 |
| 5 | MMP12 | 1 |
| 6 | MMP13 | 1 |
| 7 | MT1E | 1 |
| 8 | MT2A | 1 |
In contrast, the network of upregulated genes was
sparse, but revealed coherent submodules. Notably, MMPs, together
with their inhibitor TIMP3, highlighted extracellular matrix
remodeling, whereas MT1E and MT2A played roles in
metal ion homeostasis and oxidative stress responses (Table III).
Transcription factor analysis
TFs were identified among the differentially
expressed genes FOXM1, MYBL2, and TCF19. All
three were significantly downregulated (FOXM1,
FDR=5.01x10-6, log2FC=-1.27; MYBL2,
FDR=1.58x10-5, log2FC=-1.65; TCF19,
FDR=1.71x10-5, log2FC=-1.53). Binding motifs
for FOXM1 and MYBL2 were validated in JASPAR 2024,
and their binding activity was further supported by ENCODE ChIP-seq
datasets. In contrast, TCF19 lacked motif evidence and
demonstrated only limited ChIP-seq support. Therefore, it was
regarded as putative in this context.
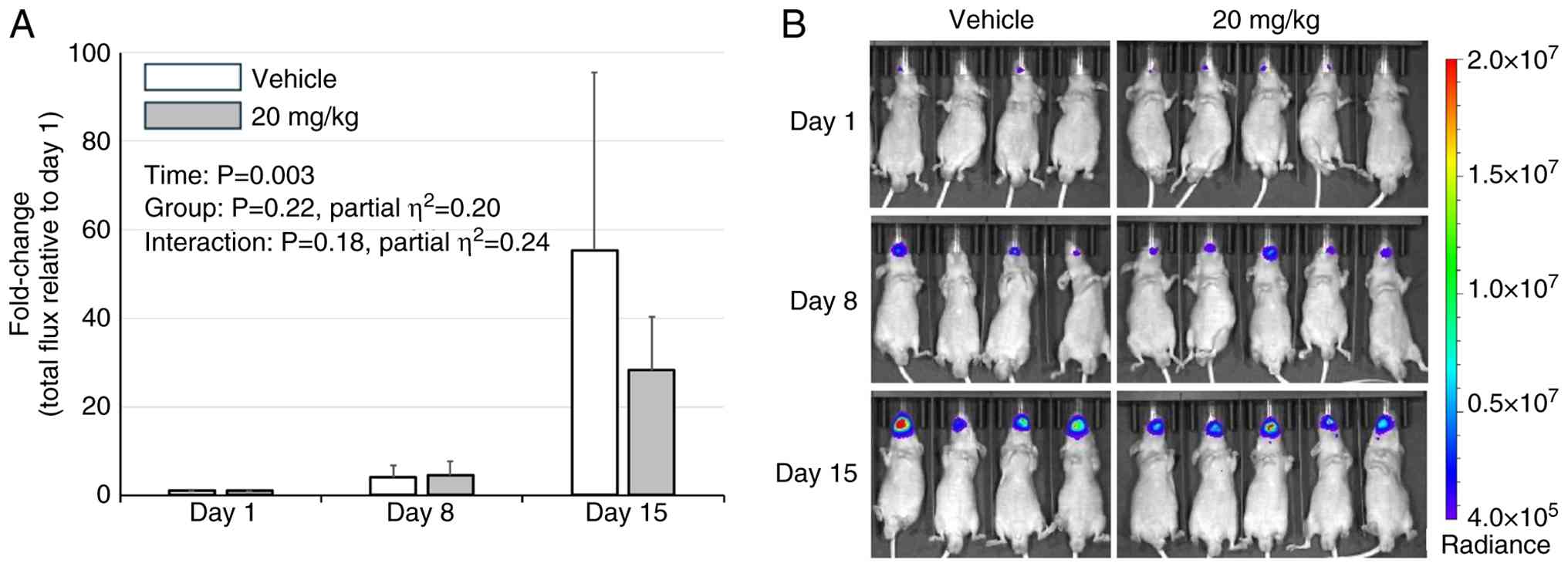
Effects of suramin treatment in an
orthotopic tongue cancer mouse model
Although five mice per group were initially
enrolled, early accidental deaths resulted in four mice in the
vehicle group and five in the 20 mg/kg group available for
longitudinal analyses. Body weights were measured during IVIS
imaging and suramin administration. Mice were regularly monitored,
and euthanasia was performed at humane endpoints (>20% body
weight loss with progressive debilitation due to tumor burden).
Accordingly, analyses were restricted to day 15, when all the mice
remained alive, and the BLI signals were within the linear
detection range. Although BLI values were lower in the suramin 20
mg/kg group than in the vehicle group at this time point, the
mixed-effects model did not detect a significant group effect
(P=0.22), or a significant time x group interaction (P=0.18).
Effect size estimates were relatively large for both the group
effect (partial η2=0.20) and the time x group
interaction (partial η2=0.24), but were insufficient to
establish a clear inhibitory effect of suramin (Fig. 4A and B). In contrast, tumor growth in the
suramin 60 mg/kg group was comparable to that in the vehicle group,
and no difference was detected (Fig.
S1A and B). Further studies
with larger cohorts are necessary to determine whether suramin
exerts a measurable influence on tumor progression. Data collected
after day 15 were excluded from the quantitative analysis owing to
signal saturation and attrition. Lymph node metastases were
detected in the 60 mg/kg (day 22), 20 mg/kg (day 25), and vehicle
groups (two cases on day 29). Pulmonary metastasis was detected in
one vehicle-treated group on day 29. Because necropsies were
performed at different time points, the timing and frequency of
metastases could not be statistically evaluated. The final
confirmation of metastasis was based on ex vivo BLI during
necropsy. The mixed-effects model up to day 22 demonstrated a
significant main effect of time (P=0.0006), whereas neither the
group effect (P=0.16) nor the time x group interaction (P=0.36) was
significant. Although statistical group differences were not
demonstrated, the 60 mg/kg group began to show a marked decline
from day 18, and one mouse died by day 21, supporting clear
systemic toxicity at a high dose (Fig.
S2). Ex vivo BLI confirmed the metastatic involvement of
the cervical lymph nodes and lungs. The corresponding radiance
values and images are provided in Table SI and Fig. S3. In the 20 mg/kg group, metastasis
was not counted in mouse #5 because the lung signal lacked a smooth
intensity gradient and was judged as background rather than a
discrete metastatic focus based on the predetermined imaging
criteria (Fig. S3).
Discussion
Transcriptomic reprogramming induced by suramin in
TSCC cells highlights a dual antitumor mechanism: suppression of
proliferative networks and modulation of the tumor
microenvironment. On the proliferative side, broad downregulation
of genes essential for the G2/M transition (e.g.,
CCNB1, CDC20, AURKA) and spindle assembly
(e.g., TPX2, TUBA1C, TUBB4B) indicates
impaired mitotic entry, chromosome segregation, and spindle
formation. This aligns with previous findings that suramin induces
G2/M arrest in breast cancer cells (12), suggesting a conserved mechanism of
cell cycle inhibition across different malignancies.
Our previous study demonstrated that suramin reduces
the HuR-mediated stabilization of CCNB1 and CCNA2
(7). Although CCNA2 was not
listed among the top 30 downregulated genes, it ranked 55th among
116 downregulated genes. In addition, other HuR-stabilized
transcripts, such as HMGB1, RRM2, and DNMT1, reported
in previous studies, were also downregulated in the present dataset
(13-15).
This concordance between known HuR-dependent transcripts and the
current RNA-seq results supports HuR inhibition as a contributing
mechanism.
In parallel, suramin reprogrammed transcriptional
pathways related to extracellular matrix remodeling. Upregulation
of several MMPs, together with TIMP3, may indicate a more
regulated ECM-associated transcriptional program, distinct from the
unchecked MMP/TIMP imbalance typical of aggressive tumors (16-18).
The co-upregulation of MMP9, MMP11, and MMP14
with TIMP3 has been regarded as a failed compensatory
response in advanced ovarian cancer (19), whereas in cartilage models, suramin
suppresses MMP13 while inducing TIMP3, exerting
protective ECM effects (20,21).
Taken together, these observations indicate that suramin is
consistent with a balanced ECM remodeling-related transcriptional
program in TSCC. However, the functional consequences of this
MMP/TIMP3 pattern were not evaluated in this exploratory
study, and this interpretation should, therefore, be regarded as
tentative.
Suramin also enhances the stress- and
immune-adaptive pathways. Upregulation of TXNIP,
MT1E, and MT2A indicates reinforcement of oxidative
stress responses; TXNIP promotes oxidative stress and
apoptosis (22), MT2A
protects against oxidative injury (23), and MT1E, which is often
silenced in cancers such as HCC (24,25),
was re-induced, consistent with tumor-suppressive reprogramming.
Pro-apoptotic mediators (TNFSF10 and TP53INP1)
(26,27), the PI3K pathway inhibitor
PIK3IP1 (28,29), and the immune adaptor FYB
(30) were also upregulated,
suggesting broad modulation of apoptosis and immune-related
processes.
Our PPI network analysis supported this dual
reprogramming: a densely connected mitotic module was suppressed,
whereas smaller yet coherent subnetworks related to ECM remodeling
and stress responses were activated, consistent with the modular
reorganization reported in cancer systems biology (31).
Importantly, transcription factor analysis revealed
the concurrent downregulation of MYBL2, which cooperates
with the DREAM complex to initiate late S/G2 transcription, and
FOXM1, the master regulator of G2/M progression
(32). TCF19, another
repressed factor, promotes β-cell stress survival partly through
FOXM1 regulation (33) and
promotes proliferation and tumor formation in lung carcinoma by
activating the RAF/MEK/ERK pathway (34). Thus, the suramin-mediated
suppression of TCF19 may synergize with
MYBL2/FOXM1 repression, reinforcing the collapse of
mitotic transcriptional programs and contributing to anti-invasive
reprogramming. Taken together, these findings support a dual
mechanism of action: profound suppression of mitotic progression
and regulation of the tumor microenvironment (35).
Multiple studies have reported diverse suramin
dosing regimens and antitumor effects, reflecting its dual role as
a direct cytotoxic agent at high doses and as a chemosensitizer at
low exposures. In preclinical models, high-dose monotherapy (~60
mg/kg per injection) suppressed tumor growth, including in human
osteosarcoma xenografts (36). In
contrast, lower-dose schedules (5-10 mg/kg, twice weekly) are
commonly employed to enhance the efficacy of paclitaxel, docetaxel,
doxorubicin, and cisplatin with minimal added toxicity (37,38).
In our orthotopic TSCC model, suramin administered
at 20 mg/kg twice weekly reduced tumor growth, although statistical
significance (P=0.18) was not achieved because of the limited
cohort size; the effect size was relatively large (partial
η2=0.24). This dosing range was selected based on
previous reports indicating that the intraperitoneal administration
of up to 60 mg/kg in mice yielded pharmacologically active systemic
exposure, although plasma suramin levels were not measured in this
study. This finding is broadly consistent with previous reports of
in vivo activity, including significant growth inhibition of
DU145 prostate cancer xenografts with suramin monotherapy at
effective doses (39) and enhanced
inhibition of ovarian cancer xenografts when combined with
cisplatin (38). Furthermore,
previous in vitro studies, including our own report
(7), have demonstrated that suramin
suppresses the proliferation and invasion of oral squamous cell
carcinoma cells, including our own report (7), supporting the notion that suramin
exerts multifaceted antitumor effects in oral cancer. When
contextualized using body surface-area scaling (40), the 20 and 60 mg/kg doses in mice
fell within the clinically explored dosing range; however, such a
conversion provides only a theoretical estimate and does not
predict plasma exposure or toxicity. However, in our model, the 60
mg/kg regimen was associated with more pronounced adverse effects
than the 20 mg/kg regimen. This was accompanied by marked body
weight loss, indicating high toxicity at this dose. At excessively
high doses, further increases in antitumor efficacy may not
necessarily occur, and several angiogenesis- and immune-related
anticancer agents have been reported to exhibit non-linear
dose-response relationships in which the therapeutic benefit
plateaus or declines beyond an optimal window (41). Although this phenomenon has not been
established for suramin, the substantial toxicity observed at 60
mg/kg in our model raises the possibility that excessively high
exposure may limit the therapeutic benefits. The assessment of
metastasis was limited because the mouse numbers declined as early
as day 21 in the 60 mg/kg group and from day 23 in the 20 mg/kg and
vehicle groups. Because lymph node metastases typically appeared
after day 25, a reliable evaluation would require a larger number
of mice to ensure sufficient survival in the later phase of tumor
progression. Ex vivo BLI findings of lymph nodes and lungs
are presented in the supplementary data for completeness; however,
differences in necropsy timing prevent quantitative comparison of
the metastatic burden.
Suramin dosing is clinically guided by a nomogram.
Regimens targeting non-cytotoxic plasma concentrations (10-50 µM
for ≥48 h) were primarily applied in combination chemotherapy
trials, where suramin enhanced the effects of agents such as
paclitaxel and carboplatin (42).
In contrast, a large randomized Phase III trial in HRPC employed
dosing schedules designed to maintain plasma concentrations within
the range of 100-300 µg/ml together with hydrocortisone (8). This approach produced a modest
symptomatic benefit but no survival advantage, and the overall
incidence of severe adverse events, including neurotoxicity, was
relatively low owing to careful pharmacokinetic control (8). Earlier studies, however, demonstrated
that when plasma concentrations exceeded 350 µg/ml, the risk of
dose-limiting neurotoxicity increased substantially (43). A more recent clinical
pharmacokinetic study reported that a single 20 mg/kg infusion in
humans produced peak plasma concentrations close to this level
(Cmax=328 µg/ml), without exceeding the upper boundary
of the recommended therapeutic window (44). Collectively, these results suggest
that suramin is unlikely to be effective as a single agent but may
have substantial value as a chemosensitizer in rational combination
regimens. This finding warrants further validation in larger
preclinical cohorts.
Suramin also exhibits anti-inflammatory activity in
addition to its antitumor effects. In a DNCB-induced atopic
dermatitis mouse model, intraperitoneal administration at 20 mg/kg
twice weekly for 3 weeks significantly reduced serum IL-6, IL-1β,
TNF-α, and IgE, improving dermatitis scores (45). These effects are plausibly linked to
the blockade of HMGB1/HMGB2 signaling, as suramin's broad
polyanionic interactions can neutralize cationic DAMPs, such as
HMGB1 and extracellular histones, providing a mechanistic rationale
for its dual anti-inflammatory and antitumor activities.
In conclusion, this study demonstrates that suramin
exerts a dual antitumor effect in TSCC by suppressing proliferative
regulators such as MYBL2, FOXM1, and TCF19,
thereby impairing the G2/M transition, while activating
pathways associated with ECM remodeling and stress responses. In
our orthotopic TSCC model, 20 mg/kg suramin reduced tumor growth
compared to controls without achieving statistical significance;
however, the relatively large effect size highlights suramin as a
promising candidate for further translational investigation.
This study is exploratory, relying on single-sample
transcriptomics and a small in vivo cohort. The in
vivo analysis was limited to day 15 due to signal saturation
and attrition, which precluded the evaluation of longer-term
effects. Functional validation of key findings-including
G2/M arrest, FOXM1/MYBL2 activity, and the
MMP/TIMP3 profile-was not performed. Accordingly, the
present findings should be interpreted as hypothesis-generating and
require validation in larger, adequately powered studies. In
addition, systematic toxicity scoring was not performed, limiting
quantitative evaluation of dose-related tolerability, and hub genes
identified by the PPI network were not validated at the protein
level by western blotting. Future studies using ChIP-qPCR to
directly assess FOXM1/MYBL2 binding to target gene promoters, as
well as validation in additional TSCC and HNSCC cell lines, will be
required to determine whether the observed transcriptomic changes
are not cell-line-specific.
Supplementary Material
In vivo effects of suramin on
tumor growth in an orthotopic tongue squamous cell carcinoma model.
(A) BLI signals in the vehicle-, 20 and 60 mg/kg-treated groups are
shown, but the difference was not statistically significant. (B)
In vivo bioluminescence images of orthotopic HSC-3-Luc
tumors in vehicle and suramin (20 and 60 mg/kg) groups.
Longitudinal body-weight changes
during treatment. Body weight (mean ± SD) is shown for the vehicle,
suramin 20 mg/kg, and suramin 60 mg/kg groups. Body weight was
plotted over the entire observation period, and the number of
surviving animals at each time point is shown below the graph. No
statistically significant differences in the mean body weight were
detected between the groups.
Quantitative ex vivo
bioluminescence imaging radiance of cervical lymph nodes and lungs.
The figure shows ex vivo images of dissected organs: left
and right cervical lymph nodes per mouse in the upper row, and the
whole lung specimen per mouse in the lower row (L/R-LN=left and
right cervical lymph nodes). Images from mice dissected on the same
necropsy day are grouped into a single figure. Mouse ID indicates
the individual identification number of each animal. One mouse in
the 60 mg/kg group was excluded from ex vivo imaging because
it died outside the scheduled necropsy time, and prolonged
post-mortem delay precluded reliable assessment. Background
radiance was measured in an adjacent tissue-free area within the
same dish and imaging session for each necropsy day, and was
therefore determined separately for each session. Arrows indicate
metastatic foci.
Quantitative ex vivo
bioluminescence radiance of cervical lymph nodes and lungs.
Acknowledgements
The authors would like to thank Dr Takashi Murakami
(Department of Microbiology, Saitama Medical University, Moroyama,
Japan) for kindly providing the luciferase-expressing HSC-3 cells
used in this study. This work was also supported by the
infrastructure and equipment at GI-CoRE GSQ, Hokkaido University
(Sapporo, Japan).
Funding
Funding: This work was supported by the Japan Society for the
Promotion of Science KAKENHI (grant nos. JP18K17022, JP22K09922,
and JP25K12969).
Availability of data and materials
The RNA-sequencing data generated in the present
study may be found in the NCBI BioProject database under accession
number PRJNA1347865 or at the following URL: https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA1347865.
All other data generated in the present study may be requested from
the corresponding author.
Authors' contributions
WK designed the study, performed RNA extraction,
animal experiments and bioinformatics analyses, interpreted the
data and drafted the manuscript. MM performed and analyzed the
animal experiments. NM and KH contributed to the design and
analysis of the animal experiments. KT contributed to the
acquisition of animal experimental data, including data collection.
TN, SO and KM contributed to the interpretation and discussion of
the suramin-related data. YO contributed to the analysis and
interpretation of data. WK and YO confirms the authenticity of all
the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
All procedures were approved by the Institutional
Animal Care and Use Committee of Hokkaido University (approval no.
18-0050; Sapporo, Japan) and conducted in accordance with the
institutional guidelines for animal welfare.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Use of artificial intelligence tools
During the preparation of this work, AI tools
(ChatGPT) were used to improve the readability and language of the
manuscript, and subsequently, the authors revised and edited the
content produced by the AI tools as necessary, taking full
responsibility for the ultimate content of the present
manuscript.
References
|
1
|
Coropciuc R, Moreno-Rabié C, De Vos W, Van
de Casteele E, Marks L, Lenaerts V, Coppejans E, Lenssen O, Coopman
R, Walschap J, et al: Navigating the complexities and controversies
of medication-related osteonecrosis of the jaw (MRONJ): A critical
update and consensus statement. Acta Chir Belg. 124:1–11.
2024.PubMed/NCBI View Article : Google Scholar
|
|
2
|
National Comprehensive Cancer Network
(NCCN): NCCN Clinical Practice Guidelines in Oncology: Head and
Neck Cancers. Version 1.2026. NCCN, Plymouth Meeting, PA, 2026.
https://www.nccn.org. Accessed January 8,
2026.
|
|
3
|
Steverding D and Troeberg L: 100 years
since the publication of the suramin formula. Parasitol Res.
123(11)2023.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Wiedemar N, Hauser DA and Mäser P: 100
years of suramin. Antimicrob Agents Chemother. 64:e01168–19.
2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Zaragoza-Huesca D, Rodenas MC,
Peñas-Martínez J, Pardo-Sánchez I, Peña-García J, Espín S, Ricote
G, Nieto A, García-Molina F, Vicente V, et al: Suramin, a drug for
the treatment of trypanosomiasis, reduces the prothrombotic and
metastatic phenotypes of colorectal cancer cells by inhibiting
hepsin. Biomed Pharmacother. 168(115814)2023.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Naviaux RK, Curtis B, Li K, Naviaux JC,
Bright AT, Reiner GE, Westerfield M, Goh S, Alaynick WA, Wang L, et
al: Low-dose suramin in autism spectrum disorder: A small, phase
I/II, randomized clinical trial. Ann Clin Transl Neurol. 4:491–505.
2017.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kakuguchi W, Nomura T, Kitamura T,
Otsuguro S, Matsushita K, Sakaitani M, Maenaka K and Tei K:
Suramin, screened from an approved drug library, inhibits HuR
functions and attenuates malignant phenotype of oral cancer cells.
Cancer Med. 7:6269–6280. 2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Small EJ, Meyer M, Marshall ME, Reyno LM,
Meyers FJ, Natale RB, Lenehan PF, Chen L, Slichenmyer WJ and
Eisenberger M: Suramin therapy for patients with symptomatic
hormone-refractory prostate cancer: Results of a randomized phase
III trial comparing suramin plus hydrocortisone to placebo plus
hydrocortisone. J Clin Oncol. 18:1440–1450. 2000.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Cheng B, Gao F, Maissy E and Xu P:
Repurposing suramin for the treatment of breast cancer lung
metastasis with glycol chitosan-based nanoparticles. Acta Biomater.
84:378–390. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Wang L, Feng Z, Wang X and Zhang X:
DEGseq: An R package for identifying differentially expressed genes
from RNA-seq data. Bioinformatics. 26:136–138. 2010.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Young TR, Yamamoto M, Kikuchi SS, Yoshida
AC, Abe T, Inoue K, Johansen JP, Benucci A, Yoshimura Y and
Shimogori T: Thalamocortical control of cell-type specificity
drives circuits for processing whisker-related information in mouse
barrel cortex. Nat Commun. 14(6077)2023.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Foekens JA, Sieuwerts AM, Stuurman-Smeets
EM, Peters HA and Klijn JG: Effects of suramin on cell-cycle
kinetics of MCF-7 human breast cancer cells in vitro. Br J Cancer.
67:232–236. 1993.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Al-Kharashi LA, Al-Mohanna FH, Tulbah A
and Aboussekhra A: The DNA methyl-transferase protein DNMT1
enhances tumor-promoting properties of breast stromal fibroblasts.
Oncotarget. 9:2329–2343. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Zhang J, Wu Q, Xie Y, Li F, Wei H, Jiang
Y, Qiao Y, Li Y, Sun Y, Huang H, et al: Ribonucleotide reductase
small subunit M2 promotes the proliferation of esophageal squamous
cell carcinoma cells carcinoma cells via HuR-mediated mRNA
stabilization. Acta Pharm Sin B. 14:4329–4344. 2024.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Wang J, Zhao L, Li Y, Feng S and Lv G: HuR
induces inflammatory responses in HUVECs and murine sepsis via
binding to HMGB1. Mol Med Rep. 17:1049–1056. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Stetler-Stevenson WG: The tumor
microenvironment: regulation by MMP-independent effects of tissue
inhibitor of metalloproteinases-2. Cancer Metastasis Rev. 27:57–66.
2008.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Brew K and Nagase H: The tissue inhibitors
of metalloproteinases (TIMPs): An ancient family with structural
and functional diversity. Biochim Biophys Acta. 1803:55–71.
2010.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Cabral-Pacheco GA, Garza-Veloz I,
Castruita-De la Rosa C, Ramirez-Acuña JM, Perez-Romero BA,
Guerrero-Rodriguez JF, Martinez-Avila N and Martinez-Fierro ML: The
roles of matrix metalloproteinases and their inhibitors in human
diseases. Int J Mol Sci. 21(9739)2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Escalona RM, Kannourakis G, Findlay JK and
Ahmed N: Expression of TIMPs and MMPs in ovarian tumors, ascites,
ascites-derived cells, and cancer cell lines: Characteristic
modulatory response before and after chemotherapy treatment. Front
Oncol. 11(796588)2022.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Guns LA, Monteagudo S, Kvasnytsia M,
Kerckhofs G, Vandooren J, Opdenakker G, Lories RJ and Cailotto F:
Suramin increases cartilage proteoglycan accumulation in vitro and
protects against joint damage triggered by papain injection in
mouse knees in vivo. RMD Open. 3(e000604)2017.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Chanalaris A, Doherty C, Marsden BD,
Bambridge G, Wren SP, Nagase H and Troeberg L: Suramin inhibits
osteoarthritic cartilage degradation by increasing extracellular
levels of chondroprotective tissue inhibitor of metalloproteinases
3. Mol Pharmacol. 92:459–468. 2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Deng J, Pan T, Liu Z, McCarthy C, Vicencio
JM, Cao L, Alfano G, Suwaidan AA, Yin M, Beatson R and Ng T: The
role of TXNIP in cancer: A fine balance between redox, metabolic,
and immunological tumor control. Br J Cancer. 129:1877–1892.
2023.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ling XB, Wei HW, Wang J, Kong YQ, Wu YY,
Guo JL, Li TF and Li JK: Mammalian metallothionein-2a and oxidative
stress. Int J Mol Sci. 17(1483)2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Si M and Lang J: The roles of
metallothioneins in carcinogenesis. J Hematol Oncol.
11(107)2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Liu Q, Lu F and Chen Z: Identification of
MT1E as a novel tumor suppressor in hepatocellular carcinoma.
Pathol Res Pract. 216(153213)2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Niroshika KKH, Weerakoon K, Molagoda IMN,
Samarakoon KW, Weerakoon HT and Jayasooriya RGPT: Exploring the
dynamic role of circulating soluble tumor necrosis factor-related
apoptosis-inducing ligand (TRAIL) as a diagnostic and prognostic
marker; a review. Biochem Biophys Res Commun.
751(151415)2025.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Deng Y, Li AM, Zhao XM, Song ZJ and Liu
SD: Downregulation of tumor protein 53-inducible nuclear protein 1
expression in hepatocellular carcinoma correlates with poor
prognosis. Oncol Lett. 13:1228–1234. 2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
He X, Zhu Z, Johnson C, Stoops J, Eaker
AE, Bowen W and DeFrances MC: PIK3IP1, a negative regulator of
PI3K, suppresses the development of hepatocellular carcinoma.
Cancer Res. 68:5591–5598. 2008.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Uche UU, Piccirillo AR, Kataoka S,
Grebinoski SJ, D'Cruz LM and Kane LP: PIK3IP1/TrIP restricts
activation of T cells through inhibition of PI3K/Akt. J Exp Med.
215:3165–3179. 2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Peterson EJ, Woods ML, Dmowski SA,
Derimanov G, Jordan MS, Wu JN, Myung PS, Liu QH, Pribila JT,
Freedman BD, et al: Coupling of the TCR to integrin activation by
Slap-130/Fyb. Science. 293:2263–2265. 2001.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Chuang HY, Lee E, Liu YT, Lee D and Ideker
T: Network-based classification of breast cancer metastasis. Mol
Syst Biol. 3(140)2007.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Sadasivam S and DeCaprio JA: The DREAM
complex: Master coordinator of cell cycle-dependent gene
expression. Nat Rev Cancer. 13:585–595. 2013.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Krautkramer KA, Linnemann AK, Fontaine DA,
Whillock AL, Harris TW, Schleis GJ, Truchan NA, Marty-Santos L,
Lavine JA, Cleaver O, et al: Tcf19 is a novel islet factor
necessary for proliferation and survival in the INS-1 β-cell line.
Am J Physiol Endocrinol Metab. 305:E600–E610. 2013.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Tian Y, Xin S, Wan Z, Dong H, Liu L, Fan
Z, Li T, Peng F, Xiong Y and Han Y: TCF19 promotes cell
proliferation and tumor formation in lung cancer by activating the
Raf/MEK/ERK signaling pathway. Transl Oncol.
45(101978)2024.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Egeblad M and Werb Z: New functions for
the matrix metalloproteinases in cancer progression. Nat Rev
Cancer. 2:161–174. 2002.PubMed/NCBI View
Article : Google Scholar
|
|
36
|
Walz TM, Abdiu A, Wingren S, Smeds S,
Larsson SE and Wasteson A: Suramin inhibits growth of human
osteosarcoma xenografts in nude mice. Cancer Res. 51:3585–3589.
1991.PubMed/NCBI
|
|
37
|
Zhang Y, Song S, Yang F, Au JL and
Wientjes MG: Nontoxic doses of suramin enhance activity of
doxorubicin in prostate tumors. J Pharmacol Exp Ther. 299:426–433.
2001.PubMed/NCBI
|
|
38
|
Kikuchi Y, Hirata J, Hisano A, Tode T,
Kita T and Nagata I: Complete inhibition of human ovarian cancer
xenografts in nude mice by suramin and
cis-diamminedichloroplatinum(II). Gynecol Oncol. 58:11–15.
1995.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Church D, Zhang Y, Rago R and Wilding G:
Efficacy of suramin against human prostate carcinoma DU145
xenografts in nude mice. Cancer Chemother Pharmacol. 43:198–204.
1999.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Nair AB and Jacob S: A simple practice
guide for dose conversion between animals and human. J Basic Clin
Pharm. 7:27–31. 2016.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Reynolds AR: Potential relevance of
bell-shaped and u-shaped dose-responses for the therapeutic
targeting of angiogenesis in cancer. Dose Response. 8:253–284.
2010.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Chen D, Song SH, Wientjes MG, Yeh TK, Zhao
L, Villalona-Calero M, Otterson GA, Jensen R, Grever M, Murgo AJ
and Au JL: Nontoxic suramin as a chemosensitizer in patients:
dosing nomogram development. Pharm Res. 23:1265–1274.
2006.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Chaudhry V, Eisenberger MA, Sinibaldi VJ,
Sheikh K, Griffin JW and Cornblath DR: A prospective study of
suramin-induced peripheral neuropathy. Brain. 119 (Pt 6):2039–2052.
1996.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Wu G, Zhou H, Lv D, Zheng R, Wu L, Yu S,
Kai J, Xu N, Gu L, Hong N and Shentu J: Phase I, single-dose study
to assess the pharmacokinetics and safety of suramin in healthy
Chinese volunteers. Drug Des Devel Ther. 17:2051–2061.
2023.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Alyoussef A: Suramin attenuated
inflammation and reversed skin tissue damage in experimentally
induced atopic dermatitis in mice. Inflamm Allergy Drug Targets.
13:406–410. 2015.PubMed/NCBI View Article : Google Scholar
|