Introduction
Autophagy is a conserved catabolic process in which
cytosolic macromolecules, lipid droplets, labeled proteins, and
damaged organelles are recycled via a lysosome-mediated digestion
process. This mechanism is normally important for cellular survival
and maintenance of energy homeostasis (1). It also establishes cellular
adaptations in response to stressful conditions (2). Autophagy is a highly regulated process
and can be influenced by aging, oxygen, nutrients and diseases,
especially cancers (3).
Promoting autophagy requires the activation of the
ULK initiation complex, which is mainly composed of UNC-51-like
kinase 1 (ULK1), autophagy-related factor 13 (ATG13), and focal
adhesion kinase family interacting protein of 200 kD (FIP200). Once
activated, an autophagosome forms to initiate the autophagy process
(4,5). ULK complex is under the direct
inhibition of the mechanistic target of rapamycin complex 1
(mTORC1), which is highly activated under nutrient-rich conditions
(6). On the other hand, the
inhibition of mTORC1 over the ULK complex is released under
starvation and stressful conditions, which in turn activates
autophagy as a survival mechanism (6). Cancer malignant traits, such as
increased proliferation and residency in oxygen and
nutrient-deprived environments, require alterations in their
metabolic activities, enabling cancerous cells to adapt to
stressful conditions (7). Among
several factors involved in metabolic reprogramming, autophagy has
been found to play a crucial role in cancer initiation, progression
and chemoresistance (8). The
interplay between autophagy and cancer is complicated and
contradictory, as it has been previously reported that autophagy
plays a bipolar role in cancer progression and operates differently
at different stages of cancer progression (9,10).
Seemingly, the opposing roles of autophagy are observed in the
prevention of early tumor development vs. the maintenance and
metabolic adaptation of established and metastasizing tumors to
promote cellular survival (11).
Recently, researchers have focused on enhancing
tumor sensitivity to chemotherapy by targeting autophagy. However,
this approach is complicated by the biphasic role of autophagy in
cancer progression; inhibiting it may suppress tumor survival but
could also eliminate its tumor-suppressive effects in early stages
(2). Therefore, the timing and
tumor context are critical in choosing the appropriate autophagy
intervention. In advanced cancers, inhibiting autophagy may be more
effective, whereas inducing it in apoptosis-resistant tumors could
promote cancer cell death (12).
These findings underscore that autophagy-targeted therapies should
be personalized, depending on cancer type and stage (2,12).
Combination therapies that use autophagy inhibitors, such as
hydroxychloroquine or chloroquine, alongside standard anticancer
medications, are being assessed in numerous clinical trials
(13-15).
However, targeting specific molecular nodes within the autophagy
pathway, such as ULK1, rather than using nonspecific
pharmacological agents, may help minimize adverse effects on normal
cells and provide more precise regulation (16).
A growing theory on the variety of ULK1 functions
pinpoints its critical role in several non-canonical processes,
primarily cellular fate determination, metabolic reprogramming,
stress response, and disease development, particularly cancers
(17,18). The mitotic delay and increased cell
death observed after ULK1 knockdown likely result from both
impaired autophagy and ULK1's autophagy-independent roles in
mitosis, including regulation of centrosome dynamics, spindle
assembly, and proper chromosome segregation, suggesting that
defects in cell division may substantially contribute to the
phenotype. Based on a previous study, cells are exposed to extreme
oxidative stress when ULK1 is inhibited. This is because of the
metabolic shifting from glycolysis to the utilization of
mitochondrial oxidative phosphorylation as an alternative mechanism
(19). Accordingly, LKB1 mutant
lung cancer cells are then re-sensitized to overcome
chemoresistance by the oxidative phosphorylation associated with
ULK1 inhibition (20). On the other
hand, Deng et al (20)
suggested that trametinib, a MAPK1/3 kinase inhibitor, prevents
bone metastases in MDA-MB-231 breast cancer (BC) by reestablishing
mitophagy function through upregulating ULK1 activity. Away from
its role in autophagy induction, ULK1 has also been demonstrated to
influence chromosomal segregation and mitotic spindle organization
during cell division. Thereafter, inhibition of ULK1 resulted in
defective chromosome alignment and delayed mitosis. This suggests
that ULK1 controls mitotic integrity, which is essential for the
advancement of the cell cycle and the proliferation of cancer cells
(21).
The ability of cancer cells to orchestrate autophagy
flux is extended beyond the genetic expression of autophagy-related
genes via several epigenetic and posttranslational modification
mechanisms, including ubiquitination and de-ubiquitination
processes (7). Several
autophagy-related factors were found to be either ubiquitinated or
de-ubiquitinated in favor of cancer development and spread.
Interestingly, distinct cancer types express distinct
ubiquitination/de-ubiquitination enzymes, and different tissue
types have varied roles for these enzymes. Accordingly, it has been
shown that activated ULK1 (or ULK-1) is further stabilized, and the
basal level of autophagy is maintained when the de-ubiquitination
peptidase USP20 removes ubiquitin from it, thereby preventing its
degradation. Moreover, USP20 upregulation was found to be
associated with various cellular biological processes such as cell
cycle progression, proliferation, migration and invasion, playing a
pivotal role in tumorigenesis (22). It has been hypothesized that under
prolonged starvation, the interaction between USP20 and ULK1 is
reduced to terminate autophagy, a protective response that drives
the cell away from apoptosis (22).
These data suggest that modulating proteolytic ubiquitination of
ULK1 via USP20 may be effective to bypass cancerous chemoresistance
and may play a role in deciding cellular fate, based on the fact
that persistent enhancement of autophagy could be a successful
strategy that drives cellular death and apoptosis in different
cancer types and sensitizes tumors to chemotherapeutic drugs.
As a mechanism that may be directly or indirectly
involved in deciding the fate of the cell, either survival or death
(3), it is necessary to ascertain
where and when autophagy contributes to or inhibits tumor
development and progression in different cancer types. This is
fundamental to establish autophagy as a potential therapeutic
target for personalized medication in different cancer types.
Moreover, understanding the dynamic conversion between
ubiquitination and de-ubiquitination status in different cancer
types, especially those related to the autophagy process, may
provide novel insights and new treatment candidates for cancer
therapy strategies. In the current study, it was aimed to perform
this task by measuring levels of basal autophagy flux in different
human cancer cell lines. Furthermore, a new system for selective
inhibition of autophagy was established using siRNA gene silencing
technology for both ULK1 and USP20, to evaluate the potential role
of autophagy inhibition on the fate of different cancer types when
combined with conventional therapeutic agents. This system will
also be evaluated for its efficiency in sensitizing different
multi-drug-resistant cancer types to their selective commercially
available chemotherapeutic drugs.
Materials and methods
Cell culture and maintenance
The following cell lines were involved in the
present study and provided from The American Type Culture
Collection, human pancreatic cancer cell line PANC-1
(CRL-1469™), Human lung carcinoma-A549
(CRM-CCL-185™), human glioblastoma of unknown origin
U-87 MG (HTB-14™), human liver cancer HepG2
(HB-8065™), human breast carcinoma MCF7
(HTB-22™), human breast adenocarcinoma MDA-MB-231
(CRM-HTB-26™), and human normal primary non-immortalized
dermal fibroblasts-HDFa (PCS-201-012, https://www.atcc.org/products/pcs-201-012).
As instructed by the supplier, PANC-1, A549,
MDA-MB-231, U-87, Fibroblasts and HepG2 cells were cultured in
Dulbecco's Modified Eagle Medium (cat. no. ECB7501L; Euroclone
SpA), while the MCF-7 cell line was cultured in RPMI-1640 culture
medium (cat. no. ECB9006L; Euroclone SpA). To prepare a complete
medium for the aforementioned cells, 10% heat-inactivated fetal
bovine serum (FBS; cat. no. 26170043; Gibco; Thermo Fisher
Scientific, Inc.), 1% of 200 mM L-Glutamine (cat. no. ECB3000D;
Euroclone SpA), 1% of 1M HEPES buffer (cat. no. ECM0180; Euroclone
SpA), and 1% of 100X penicillin-streptomycin (cat. no. ECB3001;
Euroclone SpA) were added to the media. Cultured cells were
passaged in a humidified incubator at 37˚C with 5% CO2
until being used for subsequent experiments.
Autophagy basal level in different
cell lines using the reverse transcription-quantitative (RT-q) PCR
technique
To investigate the baseline level of autophagy flux
in different cancer cell lines, the expression levels of ULK1 and
USP20 de-ubiquitinase enzyme were precisely measured. A total of
5x105 of each cell type was subjected to RNA extraction
using the extraction buffers provided by the RNeasy Plus Mini Kit
(Qiagen GmbH). The amount of 1,000 ng of RNA was converted to
complementary DNA (cDNA) using the PrimeScript 1st strand cDNA
Synthesis Kit (Takara Bio, Inc.). Then, qPCR was performed using
Applied Biosystems™ PowerTrack™ SYBR Green
Master Mix (Thermo Fisher Scientific, Inc.). Thermocycling
conditions were started with the initial activation at 95˚C for 2
min, followed by 40 cycles of denaturation at 95˚C for 15 sec and
annealing/extension at 60˚C for 1 min. The sequence of forward and
reverse primers used in this experiment is listed in Table I. The 18S rRNA was used as an
internal control of the experiment. The baseline expression of ULK1
and USP20 of each cell line was calculated relative to their
expression in normal fibroblast cells. Relative ULK1 and USP20
expression levels were quantified using 2-ΔΔCq method
(23), and further statistical
analysis was performed using GraphPad Prism software 9
(Dotmatics).
 | Table IList of primers used for quantitative
PCR. |
Table I
List of primers used for quantitative
PCR.
| Gene name | Primer sequence
(5'-3' direction) |
|---|
| ULK1 | F:
CCATCCCAGTCCCCACGCAG |
| | R:
GCGGATGGCAGAGGACCGAG |
| USP20 | F:
CAGTTGCGAGTGCAGGCTC |
| | R:
TGACACGAAGCCCACAGGAA |
| P62 | F:
CGCACTACCGCGATGAGGAC |
| | R:
TGTCATCCTTCACGTAGGACATGG |
| Bcl-2 | F:
GGACAACATCGCCCTGTGGA |
| | R:
TCCACAAAGGCATCCCAGCC |
| 18S rRNA | F:
AGAAACGGCTACCACATCCA |
| | R:
TACAGGGCCTCGAAAGAGTC |
| Bax | F:
GGTCCGGGGAGCAGC |
| | R:
GATCCTGGATGAAACCCTGAAG |
Knockdown of ULK1 and USP20 genes
using siRNA gene silencing therapy
To evaluate the differential response of different
cancer cells to autophagy inhibition, cells were transfected with
siRNAs to knockdown both ULK1 and USP20 mRNAs. Lipoplexes were
prepared by mixing each siRNA type: siULK1 (cat no. 5185429;
Microsynth AG), siUSP20 (cat no. 5185431; Microsynth AG), and siSCR
(scrambled negative control siRNA) (cat no. 5185433; Microsynth
AG), with Lipofectamine™ RNAiMAX Transfection Reagent
(cat no. 18324012, Gibco; Thermo Fisher Scientific, Inc.) in a
serum-free medium. Custom siRNAs were synthesized by Microsynth AG
(https://www.microsynth.com/home-ch.html), and the
sequence of each siRNA is indicated in Table II. Four different concentrations of
each siRNA lipoplex were prepared: 200, 100, 50 and 25 nM. A
transfection experiment was performed by plating 200x103
of each cell type into a 12-well plate and then kept overnight in
the incubator at 37˚C to firmly adhere. The day after, cells were
transfected with each concentration of lipoplexes in triplets and
transfected cells were starved for 6 h, then the medium was
adjusted to contain 10% FBS to end the starvation. Control
untreated cells were running alongside the experiment and subjected
only to starvation, like the transfected cells. After a total of
24-h of incubation with lipoplexes, cells were harvested for RNA
extraction using the RNeasy Plus Mini Kit (Qiagen). knockdown
efficiency of siULK1 and siUSP20 for both ULK1 and USP20 mRNAs was
confirmed via the qPCR technique. ULK1 and USP20 primers were
previously mentioned in the table and 18SrRNA was used as an
internal control. ΔΔCT values were calculated using Microsoft Excel
software, and subsequent significant inhibition was analyzed using
one-way ANOVA followed by Bonferroni's multiple comparisons post
hoc test, GraphPad Prism software 9 (Dotmatics). All siRNA
knockdown experiments included a well-validated scrambled siRNA
control to account for potential off-target effects or innate
immune activation. Experimental outcomes, including IC50
measurements, were analyzed relative to siSCR-treated cells to
ensure that observed effects reflect specific gene silencing.
| Table IIList of siRNA sequences used to
knockdown ULK1 and USP20 mRNAs. |
Table II
List of siRNA sequences used to
knockdown ULK1 and USP20 mRNAs.
| Target gene | siRNA sequences
(5'-3' direction) |
|---|
| ULK1 | sense:
GCUGGGGAAGGAAAUCAAA |
| | antisense:
UUUGAUUUCCUUCCCCAGCag |
| USP20 | sense:
GGACAAUGAUGCUCACCUA |
| | antisense:
UAGGUGAGCAUCAUUGUCCgg |
| Negative control
(scramble) | sense:
UUCUCCGAACGUGUCACGUTT |
| | antisense:
AAACGUGACACGUUCGGAGAA |
Effect of ULK1 and USP20 knockdown on
chemoresistance to several chemotherapeutic drugs
After determining the efficient knockdown
concentration of siULK1, siUSP20 and siSCR, different cancer types
were treated with a selective chemotherapeutic drug along with the
aforementioned siRNAs mentioned above. The chosen chemotherapeutic
agents were as follows: Gemcitabine for the treatment of PANC-1,
while HepG2, U-87, MCF-7 and MDA-MB-231 were treated with
Doxorubicin. A549 lung cancer cells were treated with Cisplatin.
Normal fibroblasts were treated with the three chemotherapeutic
agents, gemcitabine, doxorubicin and cisplatin. Briefly, different
cell types were seeded into a 96-well plate with a seeding density
of 7x103. Next, lipoplexes at a certain concentration of
each siRNA type (Table III) were
prepared as previously indicated and applied to cells. After 6 h of
incubation at 37˚C with lipoplexes, a serial dilution of each
chemotherapeutic agent was then applied over the cells. The serial
dilution of each chemotherapy started from 100 µM down to 15
concentrations. After 72 h, MTT assay was performed following the
manufacturer's protocol of CellTiter 96® Non-Radioactive
Cell Proliferation Assay (Promega Corporation) to measure the
cytotoxicity. After 3 h of incubation with the provided MTT
reagent, purple formazan crystals were dissolved by the provided
stop solution containing dimethyl sulfoxide, and the optical
density values were measured at 570 nm. Cells treated with
chemotherapy only, siRNA only, or untreated at all were running
alongside the experiment as experimental controls. GraphPad Prism 9
software (Dotmatics) was used to calculate the half-maximal
inhibitory concentration-IC50 values for each treatment
group using the logarithmic trend line of cytotoxicity graphs [log
(concentration vs. inhibition)].
| Table IIISelected safe and effective
concentrations of siRNAs over each cell type. |
Table III
Selected safe and effective
concentrations of siRNAs over each cell type.
| Cell type | siULK1 | siUSP20 | siSCR |
|---|
| Fibroblast | 25 nM | 25 nM | 25 nM |
| HepG2 | 200 nM | 100 nM | 25 nM |
| A549 | 25 nM | 25 nM | 25 nM |
| PanC1 | 50 nM | 200 nM | 25 nM |
| U87 | 200 nM | 100 nM | 25 nM |
| MDA-MD-231 | 200 nM | 100 nM | 25 nM |
| MCF-7 | 25 nM | 200 nM | 25 nM |
Effect of chemotherapy alone/siRNAs
alone on autophagy flux and apoptosis via qPCR
To evaluate the efficiency of combination therapy
over cell fate, it was mandatory to evaluate the effect of each
chemotherapy alone on autophagy flux, LC3B and P62, and
apoptosis-related genes, Bax and Bcl-2. Moreover, the effect of
knocking down ULK1 and USP20 on apoptosis-related genes was
important to be studied. For this purpose, 100,000 cells of each
cancer cell type were seeded in a 12-well plate, then they were
treated with the corresponding chemotherapy, siULK1, siUSP20, or
siSCR as mentioned in the previous sections. Three concentrations
of each chemotherapy were selected for this experiment, based on
the IC50 values of each chemotherapy for each cell line.
The concentrations of each siRNA that have been applied in this
experiment are listed in Table
III. Treated cells were cultured for 24 h at 37˚C, then total
RNA was extracted as aforementioned. qPCR was performed to test the
changes in the expression of autophagy flux genes, LC3B and P62,
and apoptosis-related genes, Bax and Bcl-2. The 18S rRNA gene was
used as a reference gene in this experiment. Primer sequences that
have been used are listed in Table
I.
Assessment for cellular fate after
combination therapy via flow cytometry
The ability of combination therapy to induce
apoptosis in treated cancerous cells was investigated, and this was
performed by treating 1x105 of each cancer type with the
selected siRNA concentration (Table
III) and two concentrations of each selected chemotherapy. A
period of 24-h treatment was ended by harvesting the cells,
followed by staining with TACS® Annexin V-FITC Apoptosis
Detection Kit (R&D Systems, Inc.). Apoptosis was detected and
analyzed using BD FACSCantoTM II Flow Cytometer, coupled
with BD FACS Diva software (BD Biosciences). Statistical analysis
was done using one-way ANOVA followed by Bonferroni's multiple
comparisons post hoc test (Dotmatics).
Statistical analysis
The IC50 values were determined using the
logarithmic trend line of cytotoxicity graphs [log (concentration
vs. inhibition)] in GraphPad Prism 8 software (Dotmatics). All the
experiments were performed in triplicate, and the mean ± SEM was
used to display the data. Data were analyzed using one-way ANOVA
followed by Bonferroni's multiple comparisons post hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Baseline expression of autophagy
markers via qPCR
ULK1, an important component of the autophagy
initiation complex, showed different expression levels in various
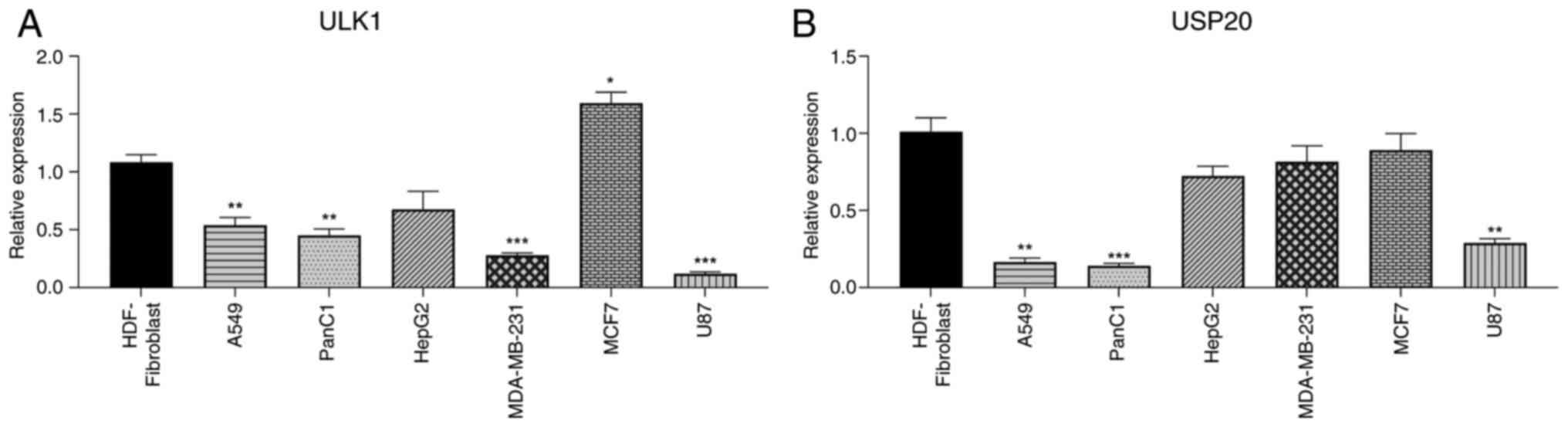
cancer types based on qPCR data (Fig.
1A). The triple-negative MDA-MB-231 cell line had a low level
of ULK1 compared with normal fibroblasts. By contrast, MCF7 BC
cells had the highest ULK1 expression. Additionally, U87 glioma
cells exhibited the lowest ULK1 expression among all the examined
cells when compared with normal fibroblasts (Fig. 1A). A549 and Panc1 also showed low
levels of ULK1 expression, at nearly half the level found in
fibroblasts. HepG2 cells demonstrated a slight decrease in ULK1
expression compared with normal fibroblasts.
Different cell lines also varied in their levels of
USP20 expression (Fig. 1B). USP20
levels in BC cell lines were similar to those in normal
fibroblasts, though it was very low and nearly absent in lung and
pancreatic cancer cell lines. In U87 cells, there was a significant
decrease in USP20 expression compared with normal cells. Finally,
the USP20 levels in HepG2 cells were slightly lower than those in
normal cells.
Knockdown of ULK1and USP20 genes using
siRNA gene silencing therapy
Gene silencing technology was used on the tested
cancer cell lines to modify autophagy. For each type of cancer
cell, four different siULK1 and siUSP20 concentrations were tested.
As shown in Table III, the
concentrations that led to a significant and effective suppression
of both genes varied among the different cells. This suggests that
different cell types respond differently to these silencing
molecules and the level of lipoplex absorption also varies among
the cell types. The concentrations of both siRNAs listed in
Table III were considered for
each cell type in the following experiments. The lowest tested
concentration of scrambled siRNA (25 nM) was used for all
experiments. This concentration should not show any significant
effect compared with the others.
To verify the efficiency of gene silencing, ULK1 and
USP20 mRNA expression was assessed in cells transfected with their
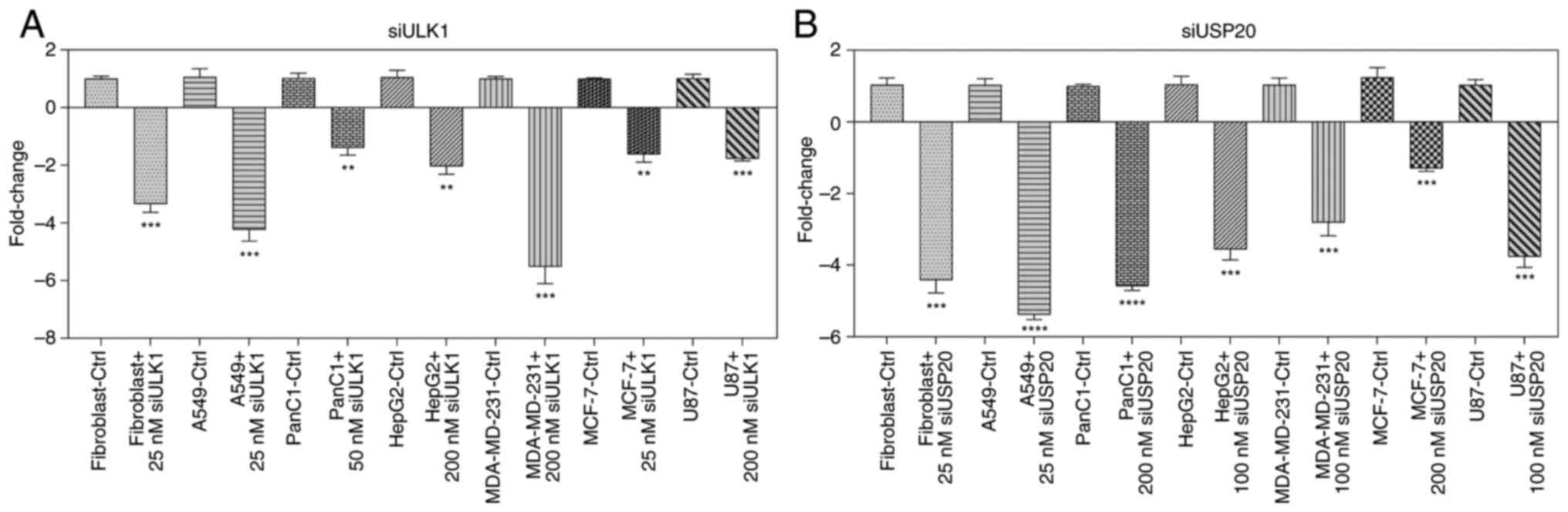
respective knockdown constructs using RT-qPCR. As shown in Fig. 2A and B, ULK1- and USP20-specific siRNA
significantly reduced transcript levels compared with cells
transfected with the non-targeting control. These results confirmed
successful and specific knockdown of ULK1 and USP20 under the
experimental conditions used.
The marked differences in siRNA potency across cell
lines are explained by intrinsic variations in transfection
efficiency, basal target gene expression, mRNA stability, RISC
loading capacity, endosomal escape and cell proliferation rates.
These biological and biophysical factors collectively determine the
concentration of siRNA required in each cell type to achieve
efficient ULK1 or USP20 silencing.
Effect of ULK1 and USP20 knockdown on
chemoresistance to several chemotherapeutic drugs
The possibility of shifting cancer cells'
sensitivity to conventional chemotherapy upon the knockdown of ULK1
and USP20 was tested using MTT assay. The results were interesting
and responses varied across different cell lines and even within
the same cell line when different genes were involved (Table IV). The table lists the
IC50 values for each chemotherapy used and those
obtained from combining chemotherapy with various siRNAs. Normal
fibroblast cells showed significant toxicity from cisplatin,
gemcitabine and doxorubicin. Adding siULK1 and siUSP20 to
cisplatin-treated fibroblasts offered slight protection from the
drug's toxic effects, but this was not significant. Notably, the
IC50 value for fibroblasts treated with siULK1 and
gemcitabine (24.85 µM) was nearly 4-fold higher than that for
fibroblasts treated with gemcitabine alone (6.38 µM). This
indicates that siULK1 protects normal fibroblasts from the drug's
harmful effects. Additionally, silencing USP20 led to a slight and
insignificant increase in the IC50 of
gemcitabine-treated fibroblasts. By contrast, adding siULK1 to
doxorubicin-treated fibroblasts had no significant effects, but
including siUSP20 increased doxorubicin toxicity compared with
fibroblasts treated with doxorubicin alone.
| Table IVIC50 values of various cancer cell
lines treated with chemotherapy alone or in combination with siULK1
or siUSP20. |
Table IV
IC50 values of various cancer cell
lines treated with chemotherapy alone or in combination with siULK1
or siUSP20.
| Cell type | Chemotherapy
type | IC50
(µM) of Chemotherapy only ± SD | IC50
(µM) of Chemotherapy and siULK1 ± SD | P-value | IC50
(µM) of Chemotherapy and siUSP20 ± SD | P-value | IC50
(µM) of Chemotherapy and siSCR ± SD | P-value |
|---|
| Fibroblast | Cisplatin | 54.02±2.46 | 57.94±5.34 | 0.31 | 63.11±11.76 | 0.26 | 79.35±12.68 | 0.09 |
| | Gemcitabine | 6.38±1.27 | 24.85±5.59 | 0.009b | 8.17±1.51 | 0.24 | 4.67±1.12 | 0.15 |
| | Doxorubicin | 0.30±0.076 | 0.28±0.01 | 0.69 ns | 0.29±0.011 | 0.81 | 0.23±0.02 | 0.23 |
| MCF-7 | Doxorubicin | 0.14±0.01 | 0.32±0.01 | 0.0001c | 0.04±0.008 | 0.0004c | 0.10±0.02 | 0.0373a |
| HepG2 | Doxorubicin | 0.28±0.02 | 0.09±0.02 | 0.0003c | 0.17±0.04 | 0.013a | 0.20±0.05 | 0.048a |
| A549 | Cisplatin | 11.23±0.69 | 18.71±1.95 | 0.003b | 8.63±1.57 | 0.057 | 10.64±0.20 | 0.22 |
| PanC1 | Gemcitabine | 2.70±0.28 | 2.10±0.25 | 0.0534 | 1.64±0.35 | 0.013a | 2.82±0.58 | 0.76 |
| U87 | Doxorubicin | 0.04±0.02 | 0.11±0.01 | 0.005b | 0.10±0.02 | 0.01a | 0.08±0.01 | 0.03 |
| MDA-MB-231 | Doxorubicin | 0.33±0.05 | 0.17±0.02 | 0.004b | 0.32±0.04 | 0.79 | 0.32±0.02 | 0.75 |
MCF-7 and MDA-MB-231 BC cell lines responded
differently to the knockdown of USP20 and ULK1. When siULK1 was
added to MCF-7 treated with doxorubicin, their resistance to the
drug increased by more than 2-fold (IC50=0.32 µM)
compared with chemotherapy-treated group (IC50 value
equals 0.14 µM). By contrast, siUSP20 significantly increased their
sensitivity to the drug by more than 3-fold (IC50=0.04
µM). Unlike MCF-7 cells, MDA-MB-231 cells became more sensitive to
doxorubicin with siULK1 addition (IC50=0.17 µM). USP20
knockdown had no noticeable effect on MDA-MB-231 cells treated with
doxorubicin, and the IC50 remained close to that of the
cells treated with the drug alone.
In HepG2 cells treated with doxorubicin, knocking
down ULK1 and USP20 significantly re-sensitized the cells to the
drug, with IC50 of 0.09 and 0.17 µM, respectively, when
compared with doxorubicin-treated HepG2 cells (IC50=0.28
µM). PanC1 cells treated with gemcitabine displayed a similar
pattern, becoming more sensitive to the drug; however, the effect
of ULK1 inhibition was minimal and statistically insignificant
(P=0.053). A549 cells treated with cisplatin revealed more
aggressive behavior when ULK1 was inhibited, but those cells were
slightly re-sensitized to the drug when USP20 was inhibited. The
response of glioblastoma U87 cells to siULK1 and siUSP20 was
similar; the cells became more aggressive and resistant to
doxorubicin with both knockdowns. Overall, the present findings
suggested that selectively inhibiting either USP20 or ULK1 could
help re-sensitize cancer cells to standard chemotherapy in
different cancer types. Thus, inhibiting USP20 or ULK1 may be
beneficial for some cancer types, but it should be avoided for
others to improve therapy effectiveness.
Effect of chemotherapeutic agents on
autophagy flux
To determine whether traditional chemotherapeutic
drugs involve autophagy or other mechanisms, the effect of these
agents on the basal levels of autophagy-related genes in different
cancer types was assessed. A total of 3 different concentrations of
each drug were selected in these experiments, based on the
IC50 values reported in Table IV. For this purpose, changes in the
expression of three autophagy-related genes: ULK1, USP20 and P62
were investigated. As demonstrated in Fig. 3, in HepG2 cells, doxorubicin reduced
ULK1 expression in a dose-dependent manner, while P62 expression
was increased in a dose-dependent manner. The drug did not
significantly affect USP20 expression in HepG2 cells. U87 exhibited
a similar response, but the changes were more pronounced. USP20 was
also decreased in these cells in a dose-dependent manner. These
results suggest that doxorubicin inhibits autophagy flow in these
cells to some extent.
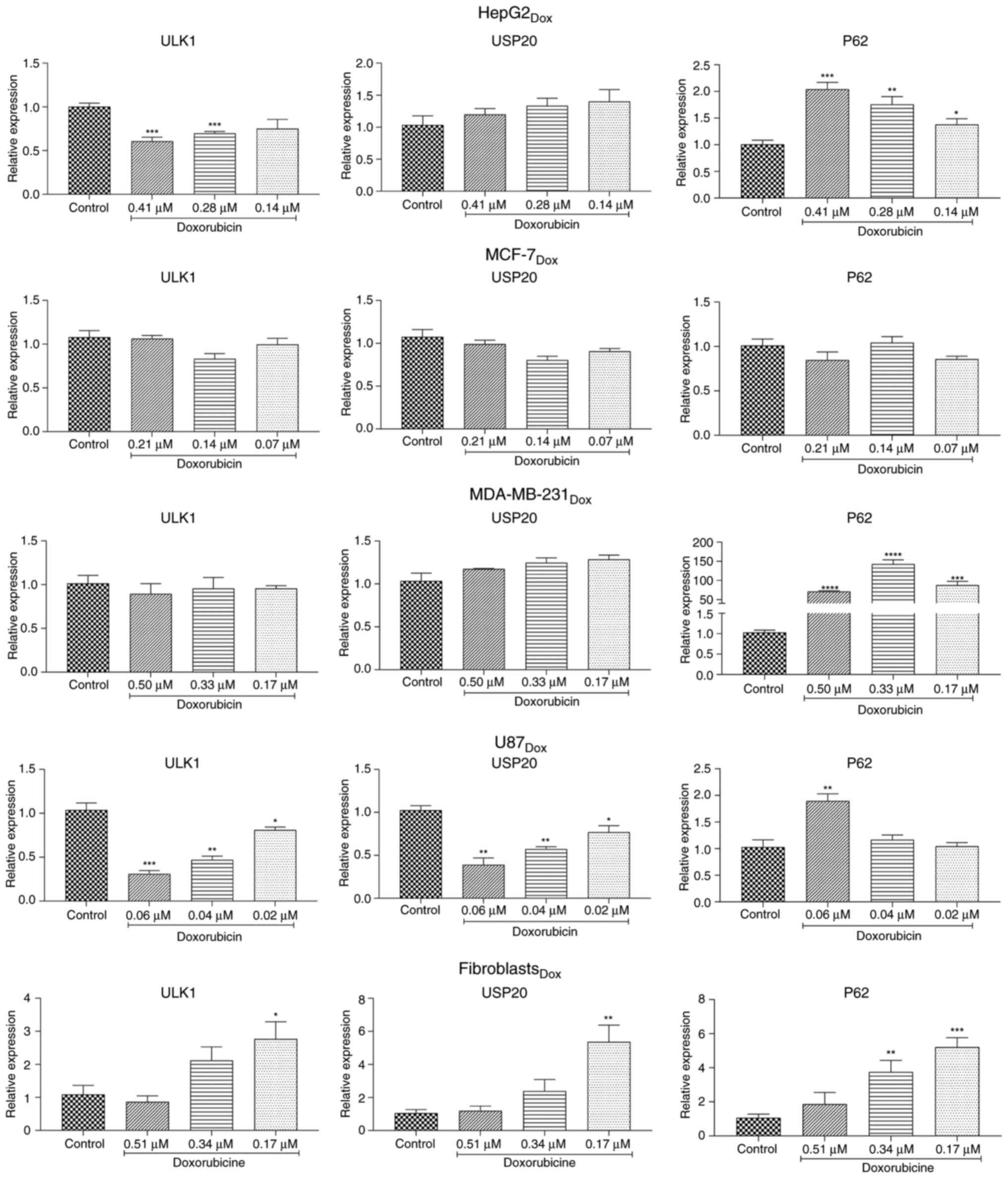 | Figure 3Changes in the basal autophagy flux
in different cancer cell lines in response to doxorubicin. HepG2,
MCF-7, MDA-MB-231, U87 and normal fibroblast cells were treated
with different concentrations of doxorubicin chemotherapy. The
level of expression of ULK1, USP20, and P62 was determined
via reverse transcription-quantitative PCR. (A-C) Relative
expression of ULK1, USP20 and P62 in HepG2 cells. (D-F) Relative
expression of ULK1, USP20 and P62 in MCF-7 breast cancer cells.
(G-I) Relative expression of ULK1, USP20 and P62 in MDA-MB-231
breast cancer cells. (J-L) Relative expression of ULK1, USP20 and
P62 in U87 cells. (M-O) Relative expression of ULK1, USP20 and P62
in normal fibroblast cells. Three repeats were considered for each
treatment group. Data are presented as the mean ± SEM. Statistical
significance was indicated as *P<0.05,
**P<0.01, ***P<0.001 and
****P<0.0001 (one-way ANOVA followed by Bonferroni's
multiple comparisons post hoc test). ULK1, Unc-51-like kinase
1. |
The BC cell lines MCF-7 and MDA-MB-231 reacted
differently to doxorubicin. Following doxorubicin treatment, ULK1
and USP20 expression changed very little and not significantly in
both cell lines. Additionally, P62 expression barely changed in
MCF-7 but rose sharply in the more aggressive MDA-MB-231 cell line
(Fig. 3). Interestingly, the
highest concentration of doxorubicin did not noticeably affect
ULK1, USP20, or P62 expression in normal fibroblast cells, but
lower doses significantly boosted their expression.
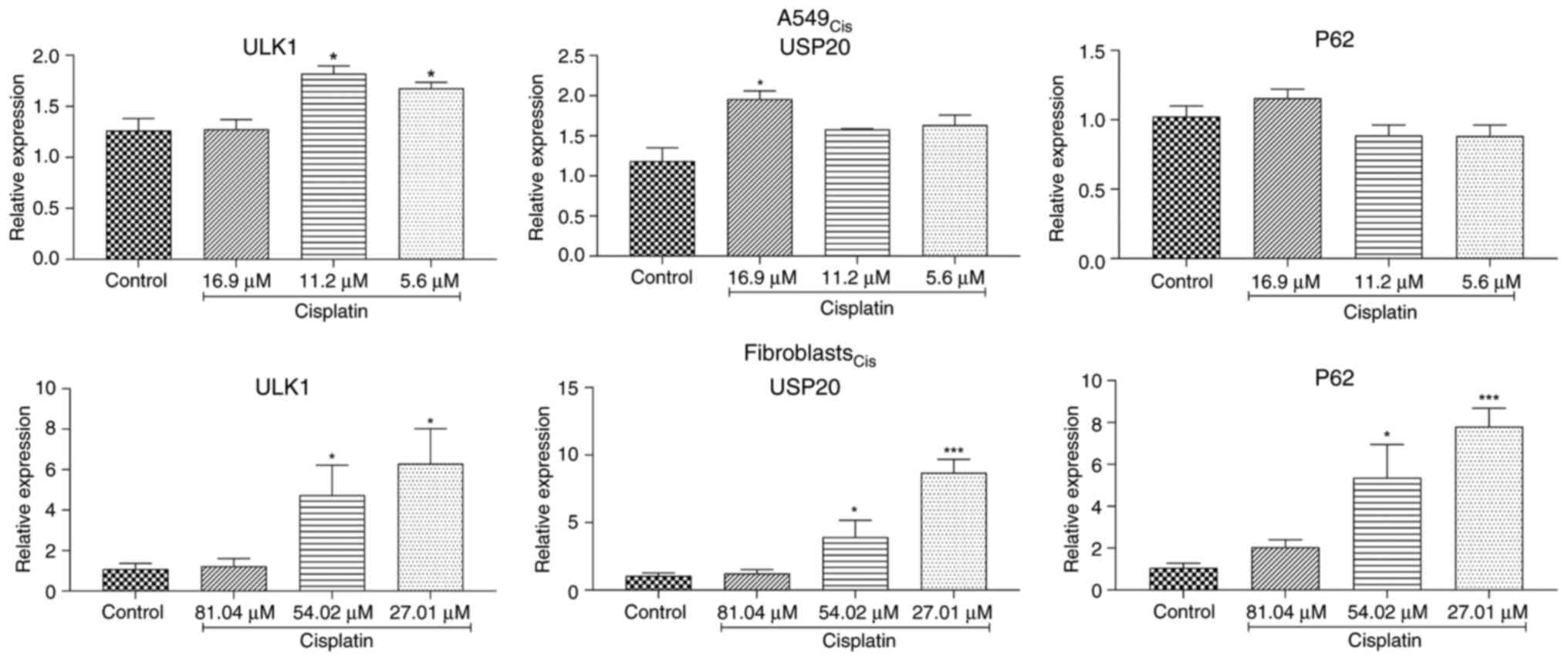
In A549 cells, the highest cisplatin concentration
significantly raised USP20 mRNA levels without affecting the
autophagy-related genes ULK1 and P62 (Fig. 4A-C). Lower concentrations of
cisplatin increased ULK1 mRNA levels but did not have a significant
impact on USP20 and P62 mRNA levels. The maximum concentration of
cisplatin did not significantly change mRNA levels in fibroblast
cells (Fig. 4D-F). However,
fibroblasts showed an increase in mRNA levels of all three examined
genes with lower concentrations of cisplatin.
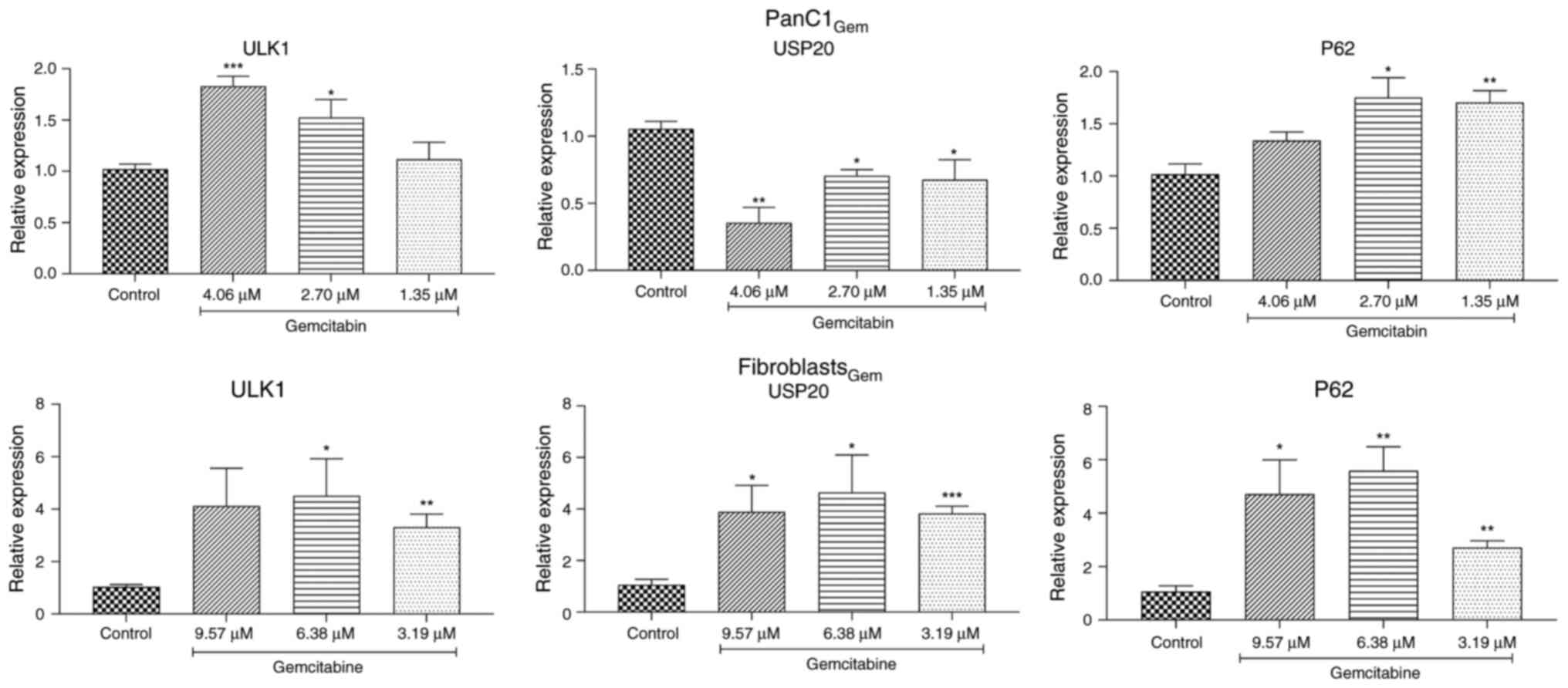
Similarly, gemcitabine treatment increased mRNA
levels of ULK1 and P62 in both PanC1 and normal fibroblast cells
(Fig. 5). Notably, PanC1 malignant
cells showed reduced USP20 expression after exposure to
gemcitabine, while normal fibroblast cells exhibited a
significantly higher level of it (Fig.
5B and E).
Effect of chemotherapeutic agents and
siRNA treatments on apoptosis via RT-qPCR
By tracking changes in the expression of
apoptosis-related genes, primarily Bax and Bcl-2, it was possible
to identify whether certain chemotherapeutic drugs and siRNAs may
lead to apoptosis (Tables V and
VI). To observe if a cell will
trigger or resist apoptosis in response to chemotherapy or siRNA
treatment, the ratio of Bax to Bcl-2 expression was precisely
measured. A ratio greater than 1 indicates a higher expression of
the pro-apoptotic factor Bax, which promotes apoptosis. By
contrast, a ratio less than one indicates a greater expression of
the anti-apoptotic factor Bcl-2, making cells resistant to
treatment. Among all the tested cells, cisplatin and gemcitabine
caused significant induction of apoptosis in A549 and PanC1 cells,
scoring ratios of 36.9 and 30.2, respectively, for the highest
concentration of the drugs, and this effect was dose dependent
(Table V). Notably, cisplatin works
on A549 by reducing the expression of the anti-apoptotic Bcl-2 gene
and slightly increasing the pro-apoptotic gene Bax. Gemcitabine
acts on PanC1 by promoting the expression of the pro-apoptotic gene
Bax. In both instances, the ratio of Bax to Bcl-2 favors the
induction of apoptosis in these cells.
| Table VBax: Bcl-2 ratio in different cell
types after treatment with different doses of chemotherapeutic
agents. |
Table V
Bax: Bcl-2 ratio in different cell
types after treatment with different doses of chemotherapeutic
agents.
| Cell line | Chemotherapy
type | Chemotherapy
concentration | ∆∆CtBax
± SD | ∆∆CtBcL2
± SD | ∆∆CtBax:
∆∆CtBcL2 Ratio |
|---|
| Fibroblast | Cisplatin | 81.04 | 2.09±0.86 | 0.54±0.85 | 3.9 |
| | | 54.02 | 2.6±1.2 | 0.30±1.2 | 8.7 |
| | | 27.01 | 6.9±2.4 | 0.23±2.3 | 30.5 |
| Fibroblast | Gemcitabine | 9.57 | 6.9±2.9 | 2.6±2.5 | 2.6 |
| | | 6.38 | 10.6±4.8 | 5.04±5.6 | 2.1 |
| | | 3.79 | 4.6±1.4 | 3.2±3.06 | 1.5 |
| Fibroblast | Doxorubicin | 0.51 | 0.99±0.13 | 0.58±0.61 | 1.7 |
| | | 0.34 | 0.68±0.36 | 4.3±5.3 | 0.16 |
| | | 0.17 | 2.09±0.73 | 0.67±0.63 | 3.1 |
| MCF-7 | Doxorubicin | 0.21 | 3.2±1.1 | 1.1±1.1 | 2.9 |
| | | 0.14 | 1.9±0.31 | 1.3±1.3 | 1.5 |
| | | 0.07 | 1.3±0.6 | 1.4±1.4 | 0.92 |
| HepG2 | Doxorubicin | 0.41 | 0.76±0.19 | 1.35±1.5 | 0.56 |
| | | 0.28 | 0.62±0.23 | 2.01±2.07 | 0.31 |
| | | 0.14 | 0.43±0.25 | 2.2±2.06 | 0.19 |
| A549 | Cisplatin | 16.9 | 2.9±0.31 | 0.08±0.06 | 36.9 |
| | | 11.2 | 2.7±0.95 | 0.07±0.08 | 37.5 |
| | | 5.6 | 2.7±1.2 | 0.41±0.44 | 6.6 |
| PanC1 | Gemcitabine | 4.06 | 31.9±0.35 | 1.05±1.07 | 30.2 |
| | | 2.7 | 39.4±1.1 | 1.3±1.26 | 31.03 |
| | | 1.35 | 32.3±2.9 | 1.4±1.37 | 23.04 |
| U87 | Doxorubicin | 0.06 | 1.5±0.46 | 1.2±1.05 | 1.2 |
| | | 0.04 | 1.3±0.27 | 0.82±0.85 | 1.6 |
| | | 0.02 | 1.08±0.26 | 0.92±0.91 | 1.18 |
| MDA-MB-231 | Doxorubicin | 0.5 | 1.03±0.01 | 0.52±0.51 | 1.99 |
| | | 0.33 | 1.5±0.59 | 0.91±0.91 | 1.64 |
| | | 0.17 | 1.9±0.2 | 1.19±1.22 | 1.58 |
| Table VIBax: Bcl-2 ratio in different cell
types after the treatment with siULK1 or siUSP20. |
Table VI
Bax: Bcl-2 ratio in different cell
types after the treatment with siULK1 or siUSP20.
| Cell type | siRNA Conc.
(nM) | ∆∆CtBax
± SD |
∆∆CtBcl-2 ± SD | ∆∆CtBax:
∆∆CtBcl-2 Ratio |
|---|
| Fibroblast | 25-siULK1 | 21.7±6.6 | 1.08±0.05 | 19.9 |
| | 25-siUSP20 | 83.4±1.8 | 3.03±0.003 | 27.5 |
| MCF-7 | 25-siULK1 | 0.8±0.4 | 1.1±0.5 | 0.68 |
| | 200-siUSP20 | 0.8±0.07 | 1.2±0.7 | 0.67 |
| HepG2 | 200-siULK1 | 7.7±4.3 | 1.2±1.2 | 6.3 |
| | 100-siUSP20 | 1.2±0.3 | 0.41±0.5 | 2.9 |
| A549 | 25-siULK1 | 0.25±0.07 | 1.1±0.2 | 0.22 |
| | 25-siUSP20 | 0.18±0.03 | 0.55±0.3 | 0.33 |
| PanC1 | 50-siULK1 | 16.5±2.05 | 3.7±3.9 | 4.5 |
| | 200-siUSP20 | 0.9±0.2 | 0.41±0.5 | 2.2 |
| U87 | 200-siULK1 | 0.21±0.2 | 0.42±0.2 | 0.5 |
| | 100-siUSP20 | 17.9±1.1 | 5.8±3.2 | 3.1 |
| MDA-MB-231 | 200-siULK1 | 0.2±0.01 | 0.07±0.05 | 2.2 |
| | 100-siUSP20 | 0.05±0.06 | 0.18±0.1 | 0.29 |
HepG2 cells appeared resistant to apoptosis when
exposed to doxorubicin, scoring a 0.56 ratio with the highest
tested concentration, and the ratio decreased with lower
concentrations. This may indicate that the cellular sensitivity
toward doxorubicin is adapted by pathways other than Bax-driven
apoptosis. Similar to A549 and PanC1, other cell types, including
U87, MDA-MB-231, MCF-7 and normal fibroblasts, induced apoptosis
when treated with the appropriate chemotherapy, but to a much
lesser degree (Table V).
Silencing ULK1 led to the induction of apoptosis in
HepG2, PanC1, MDA-MB-231 and normal fibroblast cells. However,
MCF-7, A549 and U87 showed the opposite response; ULK1 knockdown
made them more resistant to apoptosis (Table VI). Furthermore, USP20 silencing
changed cellular fate, as HepG2, PanC1 and U87 cancer cells
underwent apoptosis following USP20 knockdown (Table VI). It is noteworthy that normal
fibroblast cells had a strong response to USP20 knockdown and
underwent apoptosis when treated with siUSP20. Other cell types,
specifically MCF-7, A549 and MDA-MB-231, were protected from
apoptosis (Table VI), which may
lead these cells to develop aggressive and resistant traits when
lacking USP20 activity.
Notably, the high standard deviations in Bax: Bcl-2
ratios, such as in PanC1 cells treated with gemcitabine, likely
reflect biological heterogeneity and transient fluctuations in mRNA
expression during apoptosis, as well as technical variability in
mRNA quantification.
Assessment for cellular fate after
combination therapy via flow cytometry
The initial findings about how each chemotherapy or
siRNA affects apoptosis are controversial. For example, HepG2 cells
became resistant to apoptosis when treated with chemotherapy.
However, they induced apoptosis when ULK1 was suppressed with
siULK1. This raises questions about the fate of the cells, if they
would survive or die after combining the two treatments. To explore
how cells respond to combination therapy, an apoptosis assay was
performed. Two concentrations of chemotherapy were applied to each
cell type, either alone or with siULK1 or siUSP20. This assay
focused on four populations: healthy cells, necrotic cells, early
apoptotic cells and late apoptotic cells. The distribution of each
population for each treatment group is displayed as a percentage in
Table VII and Fig. S1, Fig.
S2, Fig. S3, Fig. S4, Fig.
S5, Fig. S6, Fig. S7, Fig.
S8 and Fig. S9. For data
analysis, each concentration of chemotherapy with siRNA was
compared with the same concentration of chemotherapy alone.
| Table VIIEffect of combination therapy on the
induction or inhibition of apoptosis in different cancer cell
lines. |
Table VII
Effect of combination therapy on the
induction or inhibition of apoptosis in different cancer cell
lines.
| Normal
Fibroblast-Treated with Doxorubicin |
|---|
| Treatment
groups | Control | 1.5 IC50 of
Dox | P-value | IC50 of Dox | P-value | 1.5 IC50 of Dox +
siULK1 | P-value | IC50 of Dox +
siULK1 | P-value | 1.5 IC50 of Dox +
siUSP20 | P-value | IC50 of Dox +
siUSP20 | P-value | 1.5 IC50 of Dox +
siSCR | P-value | IC50 of Dox +
siSCR | P-value |
|---|
| Q1 (Necrosis) | 2.15± 0.07 | 30.25± 2.19 | 0.003b | 11.2± 0.99 | 0.006 b | 16.1± 1.41 | 0.01a | 7.95± 1.76 | 0.15 | 5.15± 1.34 | 0.005b | 4±0.28 | 0.01a | 14.75± 0.07 | 0.009b | 9.6± 1.13 | 0.27 |
| Q2 (Late
apoptosis) | 4.05± 0.21 | 8.45± 1.06 | 0.02a | 10.6± 1.4 | 0.02a | 9.4± 0.56 | 0.38 | 9.4± 0.99 | 0.42 | 8.0± 0.14 | 0.61 | 7.85± 1.06 | 0.15 ns | 7.5± 1.13 | 0.47 | 8.95± 1.06 | 0.31 |
| Q3 (Healthy) | 90± 0.71 | 58.15± 0.21 | 0.0003c | 59.3± 0.71 | 0.0005c | 68.65± 0.49 | 0.001b | 76.95± 1.2 | 0.003b | 77.1± 1.83 | 0.004b | 83.4± 1.41 | 0.002b | 73.3± 1.27 | 0.003b | 74.95± 2.75 | 0.016a |
| Q4 (Early
apoptosis) | 3.8± 0.42 | 3.2± 0.85 | 0.46 | 18.9± 3.11 | 0.02a | 5.7± 1.13 | 0.12 | 5.7± 1.97 | 0.03 a | 9.75± 0.35 | 0.009 b | 4.75± 0.63 | 0.02 | 4.35± 0.21 | 0.20 | 6.5± 2.82 | 0.053 |
| Normal
Fibroblast-Treated with Cisplatin |
| Treatment
groups | Control | 1.5 IC50 of
Cis | P-value | IC50 of Cis | P-value | 1.5 IC50 of Cis +
siULK1 | P-value | IC50 of Cis +
siULK1 | P-value | 1.5 IC50 of Cis +
siUSP20 | P-value | IC50 of Cis +
siUSP20 | P-value | 1.5 IC50 of Cis +
siSCR | P-value | IC50 of Cis +
siSCR | P-value |
| Q1 (Necrosis) | 2.15± 0.07 | 0.7± 0.14 | 0.005b | 0.9± 0.63 | 0.02a | 1.95± 0.63 | 0.11 | 2.05± 0.54 | 0.18 | 4.05± 0.21 | 0.002b | 1.85± 0.07 | 0.04a | 1.35± 0.07 | 0.397 | 1.4± 0.0 | 0.12 |
| Q2 (Late
apoptosis) | 4.05± 0.21 | 11.2± 0.98 | 0.006b | 8.05± 1.06 | 0.04a | 8.85± 1.06 | 0.051 | 7.5± 0.73 | 0.77 | 5.65± 0.63 | 0.01a | 6.55± 0.49 | 0.24 | 8.9± 0.70 | 0.03a | 8.35± 0.35 | 0.76 |
| Q3 (Healthy) | 90± 0.71 | 65.15± 2.61 | 0.002b | 71.4± 0.39 | 0.007b | 77.6± 0.39 | 0.01a | 79.2± 1.26 | 0.057 | 84.85± 0.49 | 0.002b | 87.25± 1.76 | 0.009b | 82.25± 1.62 | 0.004b | 84.55± 1.34 | 0.01a |
| Q4 (Early
apoptosis) | 3.8± 0.42 | 22.95± 1.34 | 0.002b | 19.6± 1.3 | 0.01a | 11.65± 1.3 | 0.02a | 11.25± 2.08 | 0.076 | 5.45± 0.35 | 0.0009c | 4.35± 1.34 | 0.014a | 7.55± 0.77 | 0.001b | 5.7± 0.98 | 0.01a |
| Normal
Fibroblast-Treated with Gemcitabine |
| Treatment
groups | Control | 1.5 IC50 of
Gem | P-value | IC50 of Gem | P-value | 1.5 IC50 of Gem +
siULK1 | P-value | IC50 of Gem +
siULK1 | P-value | 1.5 IC50 of Gem +
siUSP20 | P-value | IC50 of Gem +
siUSP20 | P-value | 1.5 IC50 of Gem +
siSCR | P-value | IC50 of Gem +
siSCR | P-value |
| Q1 (Necrosis) | 2.15± 0.07 | 0.5± 0.16 | 0.03a | 0.8± 0.26 | 0.04a | 1.0± 0.31 | 0.06ns | 1.2± 0.54 | 0.07 | 1.55± 0.97 | 0.02a | 1.2± 0.06 | 0.0524 | 1.9± 0.14 | 0.07 | 2.05± 1.2 | 0.55 |
| Q2 (Late
apoptosis) | 4.05± 0.21 | 4.3± 1.31 | 0.51 | 6.6± 0.61 | 0.06ns | 8.7± 0.84 | 0.001b | 7.1± 0.13 | 0.097 | 5.85± 0.15 | 0.239 | 7.0± 0.14 | 0.53ns | 6.65± 1.34 | 0.38ns | 5.65± 0.63 | 0.08 |
| Q3 (Healthy) | 90± 0.71 | 64± 0.97 |
<0.0001d | 75.8± 2.1 | 0.0004c | 82.2± 1.3 |
<0.0001d | 83.9± 1.26 | 0.0001c | 77.4± 0.19 |
<0.0001d | 83.1± 0.27 | 0.0002c | 85.85± 1.76 | 0.0004c | 87.15± 0.78 | 0.0008c |
| Q4 (Early
apoptosis) | 3.8± 0.42 | 31.2± 1.01 |
<0.0001d | 16.8± 0.99 | 0.0003c | 8.2± 2.06 |
<0.0001d | 7.8± 1.05 |
<0.0001d | 15.2± 0.98 |
<0.0001d | 8.7± 0.04 |
<0.0001d | 5.65± 0.63 |
<0.0001d | 5.1± 1.13 | 0.0006c |
| U87-Treated with
Doxorubicin |
| | Control | 1.5 IC50 of
Dox | P-value | IC50 of Dox | P-value | 1.5 IC50 of Dox +
siULK1 | P-value | IC50 of Dox +
siULK1 | P-value | 1.5 IC50 of Dox +
siUSP20 | P-value | IC50 of Dox +
siUSP20 | P-value | 1.5 IC50 of Dox +
siSCR | P-value | IC50 of Dox +
siSCR | P-value |
| Q1 (Necrosis) | 3.3± 0.28 | 8.53± 4.19 | 0.1431 | 4.5± 0.99 | 0.2411 | 12.25± 0.92 | 0.5760 | 5.75± 0.78 | 0.2954 | 8.9± 0.99 | 0.6997 | 4.2± 0.42 | 0.7317 | 5.55± 2.33 | 0.2989 | 2.65± 2.61 | 0.4484 |
| Q2 (Late
apoptosis) | 11.4± 2.82 | 35.83± 2.67 | 0.018a | 30.2± 0.57 | 0.012a | 31.85± 2.05 | 0.33 | 31.2± 1.56 | 0.48 | 25.95± 1.2 | 0.035a | 23.7± 1.56 | 0.03a | 37.75± 0.07 | 0.51 | 27.3± 11.45 | 0.75 |
| Q3 (Healthy) | 70± 2.97 | 45.6± 2.27 | 0.013a | 53.75± 0.35 | 0.01a | 50.85± 1.06 | 0.117 | 55.35± 1.91 | 0.364 | 57.2± 0.14 | 0.027a | 61.3± 2.12 | 0.038a | 45.6± 3.25 | 0.874 | 55.1± 12.02 | 0.88 |
| Q4 (Early
apoptosis) | 15.3± 0.14 | 10.03± 1.78 | 0.007b | 11.55± 0.07 | 0.0009c | 5.1± 1.84 | 0.10 ns | 7.65± 0.35 | 0.004b | 7.95± 0.35 | 0.21 | 10.75± 1.06 | 0.39 | 11.15± 0.78 | 0.11 | 14.95± 2.19 | 0.15 |
| HepG2-Treated with
Doxorubicin |
| | Control | 1.5 IC50
Dox | P-value | IC50
Dox | P-value | 1.5 IC50 of Dox +
siULK1 | P-value | IC50 of Dox +
siULK1 | P-value | 1.5 IC50 of Dox+
siUSP20 | P-value | IC50 of Dox +
siUSP20 | P-value | 1.5 IC50 of Dox +
siSCR | P-value | IC50 of Dox +
siSCR | P-value |
| Q1 (Necrosis) | 4.55± 1.62 | 15.15± 0.35 | 0.0004c | 14.95± 0.07 | 0.0004c | 18.15± 0.21 | 0.01b | 12.02± 1.06 | 0.061 | 9.53± 2.28 | 0.022a | 7.4± 0.45 | 0.002b | 5.44± 1.42 | 0.002b | 5.4± 2.17 | 0.007b |
| Q2 (Late
apoptosis) | 7.1± 1.27 | 5.65± 0.63 | 0.24 | 4.6± 0.28 | 0.062 | 10.35± 0.07 | 0.009b | 15.55± 0.63 | 0.002b | 17.23± 3.86 | 0.028 | 13.83± 0.4 | 0.0001c | 11.46± 0.55 | 0.092 | 12.2± 1.05 | 0.002b |
| Q3 (Healthy) | 82.55± 3.32 | 77.65± 0.21 | 0.066 | 79.45± 0.49 | 0.173 | 68.4± 0.28 | 0.018a | 55.1± 0.28 | 0.0003c | 57.9± 3.84 | 0.006b | 65.56± 0.62 | 0.0001c | 75.8± 0.56 | 0.999 | 71.86± 1.01 | 0.002 b |
| Q4 (Early
apoptosis) | 5.8± 0.42 | 1.55± 0.49 | 0.0011b | 1± 0.28 | 0.0004c | 3.1± 0.14 | 0.051 | 17.3± 0.14 | 0.0002c | 15.4± 0.26 |
<0.0001d | 13.16± 0.37 |
<0.0001d | 7.2± 1.06 | 0.006b | 10.53 0.32 |
<0.0001d |
| A549-Treated with
Cisplatin |
| | Control | 1.5 IC50
Dox | P-value | IC50
Dox | P-value | 1.5 IC50 of Dox +
siULK1 | P-value | IC50 of Dox +
siULK1 | P-value | 1.5 IC50 of Dox+
siUSP20 | P-value | IC50 of Dox +
siUSP20 | P-value | 1.5 IC50 of Dox +
siSCR | P-value | IC50 of Dox +
siSCR | P-value |
| Q1 (Necrosis) | 0.9± 0.14 | 3.3± 0.2 | 0.0083b | 11.73± 0.83 |
<0.001c | 4.5± 0.42 | 0.0321a | 5.2± 0.14 | 0.032a | 11.53± 0.55 |
<0.0001d | 7.45± 0.49 | 0.018a | 7.9± 2.5 | 0.0143 | 9.4± 4.42 | 0.9456 |
| Q2 (Late
apoptosis) | 3.1± 0.07 | 24.57± 0.25 |
<0.0001d | 26.6± 0.95 |
<0.0001d | 14.8± 0.77 | 0.0002c | 12.05± 0.21 | 0.0001c | 30.56± 0.60 | 0.0009c | 33.1± 0.28 | 0.0009c | 23.63± 1.91 | 0.44 | 21.9± 1.34 | 0.003b |
| Q3 (Healthy) | 82.55± 3.32 | 56.03± 0.31 |
<0.0001d | 55.13± 0.8 |
<0.0001d | 59.1± 0.57 | 0.039a | 67.55± 0.49 | 0.007b | 51.26± 0.47 | 0.029a | 49.8± 0.26 | 0.033a | 58.46± 3.02 | 0.347 | 58.36± 4.67 | 0.5986 |
| Q4 (Early
apoptosis) | 5.8± 0.42 | 16.1± 0.26 |
<0.0001d | 6.53± 1.3 | 0.013a | 21.7± 0.21 | 0.0002c | 15.2± 0.14 | 0.031a | 6.63± 1.01 |
<0.0001d | 9.65± 0.07 | 0.0489a | 10± 0.9 | 0.0002c | 10.33± 4.5 | 0.298 |
| MCF-7-Treated with
Doxorubicin |
| | Control | 1.5 IC50 of
Dox | P-value | IC50 of Dox | P-value | 1.5 IC50 of Dox +
siULK1 | P-value | IC50 of Dox +
siULK1 | P-value | 1.5 IC50 of Dox +
siUSP20 | P-value | IC50 of Dox +
siUSP20 | P-value | 1.5 IC50 of Dox +
siSCR | P-value | IC50 of Dox +
siSCR | P-value |
| Q1 (Necrosis) | 4.16± 0.6 | 22.6± 3.4 | 0.0004c | 19.16± 3.5 | 0.0013 | 10.05± 4.1 | 0.006b | 5.2± 0.14 | 0.043a | 49.93± 1.05 | 0.0001c | 39.23± 2.77 | 0.0003c | 10± 0.28 | 0.006b | 10.5± 1.83 | 0.058 |
| Q2 (Late
apoptosis) | 2.53± 1.1 | 40.9± 5.03 | 0.0001c | 30.06± 5.9 | 0.0006c | 16.5± 7.4 | 0.002b | 12.05± 0.21 | 0.121 | 35.16± 1.51 | 0.462 | 26.26± 2.01 | 0.754 | 46.55± 0.91 | 0.602 | 38.4± 1.13 | 0.141 |
| Q3 (Healthy) | 77.06± 2.7 | 33.56± 5.62 | 0.0001c | 47.4± 4.2 | 0.0003c | 66.5± 5.7 | 0.0004c | 67.55± 0.49 | 0.004b | 14.7± 1.21 | 0.004b | 33.8± 2.17 | 0.003b | 37.8± 1.27 | 0.907 | 46.7± 1.83 | 0.84 |
| Q4 (Early
apoptosis) | 16.2± 1.4 | 2.9± 5.6 | 0.0001c | 3.4± 1.5 | 0.0001c | 2.6± 2.16 | 0.051ns | 15.2± 0.14 | 0.12 | 0.121± 0.003 | 0.012a | 0.7± 0.1 | 0.042a | 5.7± 0.56 | 0.182 | 4.45± 1.20 | 0.906 |
| PanC1-Treated with
Gemcitabine |
| | Control | 1.5 IC50 of
Gem | P-value | IC50 of Gem | P-value | 1.5 IC50 of Gem +
siULK1 | P-value | IC50 of Gem+
siULK1 | P-value | 1.5 IC50 of Gem+
siUSP20 | P-value | IC50 of Gem +
siUSP20 | P-value | 1.5 IC50 of Gem+
siSCR | P-value | IC50 of Gem +
siSCR | P-value |
| Q1 (Necrosis) | 3.05± 0.91 | 4.6± 0.84 | 0.652 | 6.16± 1.75 | 0.139 | 14.6± 2.96 | 0.005b | 7.36± 0.28 | 0.991 | 4.75± 1.06 | 0.99 | 10.6± 0.2 | 0.044a | 7.03± 1.15 | 0.490 | 4.4± 1.27 | 0.681 |
| Q2 (Late
apoptosis) | 19± 1.13 | 32.8± 0.42 | 0.004b | 28.8± 2.61 | 0.0106a | 36.1± 0.84 | 0.03a | 36.1± 5.16 | 0.043a | 31.75± 2.05 | 0.98 | 33± 0.6 | 0.391ns | 32± 2.26 | 0.998 | 34.9± 0.42 | 0.137 |
| Q3 (Healthy) | 71.6± 1.27 | 55.7± 0.28 | 0.006b | 56.43± 3.64 | 0.005b | 46.15± 1.34 | 0.006b | 49.76± 2.75 | 0.044 | 55.85± 0.07 | 0.999 | 52.85± 0.25 | 0.519ns | 55.06± 2.41 | 0.999 | 55.65± 2.05 | 0.80 |
| Q4 (Early
apoptosis) | 6.35± 0.77 | 6.9± 1.55 | 0.699ns | 7.9± 1.75 | 0.651 | 3.15± 0.77 | 0.039a | 6.76± 2.68 | 0.51 | 7.75± 1.06 | 0.99 | 3.55± 0.15 | 0.045a | 5.9± 0.65 | 0.99 | 5.05± 0.35 | 0.29 |
| MDA-MB-231-T |
| | Control | 1.5 IC50 of
Dox | P-value | IC50 of Dox | P-value | 1.5 IC50 of Dox +
siULK1 | P-value | IC50 of Dox +
siULK1 | P-value | 1.5 IC50 of Dox +
siUSP20 | P-value | IC50 of Dox +
siUSP20 | P-value | 1.5 IC50 of Dox +
siSCR | P-value | IC50 of Dox +
siSCR | P-value |
| Q1 (Necrosis) | 5.25± 0.35 | 16.72± 2.29 | 0.013a | 13.8± 1.83 | 0.030a | 10.3± 1.27 | 0.083 | 17± 0.42 | 0.13 | 14.6± 1.55 | 0.998 | 17.1± 1.13 | 0.12 | 22.5± 3.53 | 0.086 | 12.6± 0.42 | 0.99 |
| Q2 (Late
apoptosis) | 3.8± 0.28 | 38.07± 0.17 |
<0.0001c | 35± 0.84 | 0.0001c | 46.4± 0.14 | 0.001b | 40.15± 1.34 | 0.028a | 38.65± 1.62 | 0.66 | 35.05± 0.91 | 0.96 | 32.73± 2.75 | 0.053 | 26.55± 1.2 | 0.014a |
| Q3 (Healthy) | 85.8± 0.42 | 36.62± 1.37 | 0.0004b | 34.45± 1.06 | 0.0002c | 24.1± 1.55 | 0.013a | 30± 0.28 | 0.02a | 38.8± 0.70 | 0.18 | 36.9± 1.9 | 0.26 | 37± 1.62 | 0.76 | 44.5± 3.39 | 0.057 |
| Q4 (Early
apoptosis) | 5.15± 0.49 | 8.57± 0.74 | 0.032a | 16.75± 2.05 | 0.006b | 19.65± 0.63 | 0.003b | 12.85± 1.48 | 0.18 | 7.95± 0.63 | 0.94 | 10.9± 0.07 | 0.057 | 7.73± 2.4 | 0.85 | 16.4± 1.69 | 0.86 |
The results revealed that doxorubicin causes cell
death by activating necrosis (accounting for ~30.25%) instead of
apoptosis (accounting for only ~8.45%), leading to a decrease in
the healthy population (~58.15%) in normal fibroblasts. Adding
siULK1 offered some protection against doxorubicin's effects on
fibroblasts by reducing the number of necrotic cells nearly in half
(~16.1%) and significantly increasing the proportion of healthy
cells to 68.65% (P~0.001). Knocking down USP20 provided even
greater protection, significantly increasing the number of healthy
cells up to 77.1% (with P~0.005) (Fig.
S1).
Different cancer cell lines reacted differently to
doxorubicin therapy. For example, it caused glioblastoma U87 cells
to undergo apoptosis (late apoptotic population ~35.83%), and the
healthy population significantly decreased to 45.6% (P~0.013).
However, when siULK1 and siUSP20 were added, these cells developed
some resistance to doxorubicin, and the number of healthy cells
rose, reaching up to 50.9% (Fig.
S4). Notably, HepG2 cells responded to doxorubicin by
activating necrosis (the necrotic population accounting for 15.15%)
and suppressing apoptosis, with the apoptotic population accounting
for only 5.6% among all populations. Combining the drug with siULK1
or siUSP20 made HepG2 cells more vulnerable to it and significantly
increased the number of apoptotic cells (Fig. S5).
The responses of BC cell lines MCF-7 and MDA-MB-231
to doxorubicin also varied. The highest concentration of
doxorubicin given to MCF-7 cells significantly promoted both
apoptotic and necrotic pathways, resulting in a survival rate of
only 33.6%. Interestingly, knocking down USP20 enhanced the effects
of chemotherapy and increased the cell necrosis population up to
49.9% compared with the chemotherapy-only group (~22.6%), while
knocking down ULK1 allowed the cells to resist the effects of
doxorubicin and become more resistant to apoptosis and necrosis,
with population sizes reduced to nearly ~16.5 and 10.1% (Fig. S7), respectively. On the other hand,
combining siULK1 with doxorubicin made MDA-MB-231 cells markedly
more sensitive to the drug, with the apoptotic population increased
up to 46.4% compared with the apoptotic population of doxorubicin
only (~38.07%). However, USP20 knockdown had no noticeable effect
on these cells, and doxorubicin's impact remained unchanged
(Fig. S9).
Cisplatin treatment of A549 cells activated the
apoptotic cell death pathway dramatically. The healthy population
decreased significantly (P<0.0001), while the early and late
apoptotic populations increased, with values equal to 16.1 and
24.57%, respectively. Silencing ULK1, along with cisplatin, changed
how these cells behaved. They became more sensitive to cisplatin,
which decreased the healthy population up to 38.7%, while the
healthy population of cisplatin-treated cells reached up to 56.03%.
It also increased the late apoptotic population significantly up to
35.1%, with a P<0.0001 when compared with the cisplatin-treated
group. Treating A549 cells with siUSP20 and cisplatin also made
them vulnerable to the drug and significantly increased the
population of late apoptosis up to 30.56% with a P<0.0001,
compared with the cisplatin-treated group (Fig. S6). Normal fibroblasts were also
affected by cisplatin treatment, as the apoptotic pathway was
activated in these cells as well, raising the early apoptotic
population significantly up to 22.95% with a P=0.0008. Adding
siULK1 to cisplatin-treated fibroblasts protected them from the
drug's effects and significantly reduced early cellular apoptosis
to a value reaches 11.65% (Fig.
S2). Interestingly, normal fibroblasts became more resistant to
cisplatin when USP20 was silenced, and their population values were
very close to those in the control group.
Gemcitabine also had a mild effect on normal cells
and triggered the early apoptotic pathway. However, when
gemcitabine treatment was combined with siULK1 or siUSP20,
fibroblasts became resistant to gemcitabine. This was shown by an
increase in healthy populations (up to 82.2 and 77.4% for both
treatment groups, respectively) and a decrease in early and late
apoptotic cells (Fig. S3). When
gemcitabine was added to PanC1 cancer cells, it significantly
activated the apoptotic pathway and increased the late apoptotic
population up to 32.8%, with a P~0.003. Yet, when gemcitabine was
combined with siULK1, PanC1 cells became more sensitive, leading to
a higher number of cells undergoing late apoptosis (accounting for
36.1%, with P~0.03). Silencing USP20 did not significantly affect
the outcome for gemcitabine-treated PanC1 cells, and the cell
responses matched those seen in the gemcitabine-treated group
(Fig. S8).
Discussion
Autophagy is a homeostasis maintenance mechanism
that aims to prevent the accumulation of damaged cellular
components or compartments and maintain cellular balance. Besides
its canonical functions in the cell, it is well known to have
several noncanonical functions that affect numerous vital cellular
responses (24). In cancers, it has
been postulated that autophagy plays an important role in
reprogramming the cell, modulating cellular responses to
chemotherapy and determining cellular fate, either survival or
death. However, the available data were controversial and the
effect of autophagy on cancer progression and therapy effectiveness
was not the same for different cancer types and remained unclearly
defined. At present, to the best of our knowledge, the data that
precisely dissect the functional classification of autophagy in
different cancer types are still lacking and need to be further
clarified. The aim of the present study was to uncover the
functional role of autophagy in a number of cancer cell lines by
specifically evaluating its role in cancer cell fate and its
responses to the conventional chemotherapeutic agents. Direct
evidence that autophagy is differentially modulated in different
cancer cell lines for the sake of cancer survival was provided.
Powerful evidence was also presented that autophagy inhibition is
directly correlated with cancer cellular fates by being a
cytoprotective or cytotoxic mechanism. Furthermore, the precise
correlation between autophagy inhibition and the desensitization or
resensitization of different investigated cancer cells toward
conventional therapies was determined. Furthermore, it has been
successfully validated that the autophagy-related initiation
factor, ULK1, and its de-ubiquitinase stabilizing enzyme, USP20,
are powerful potential targets for the treatment of cancers. ULK1
was selected as the central autophagy reference molecule because of
its established, indispensable role in initiating the autophagy
cascade. USP20 was chosen as a parallel target because of its
potential but understudied role as a de-ubiquitinase that may
regulate ULK1 turnover and activity, thereby revealing a novel
post-translational mechanism that could reshape the understanding
of autophagy regulation.
The data of the present study demonstrated that ULK1
and USP20 expression varies among cancer cells, with either low or
high expression levels. Among the tested cells, ULK1 was mostly
expressed in the MCF-7 BC cell line, while the minimum expression
was detected in U87 glioblastoma cells. This variability in the
expression reflects the different dependence of cancer cells on the
autophagy pathway and may relate to chemoresistance, metastasis and
the fate of the cells. The data of the present study are in line
with other studies, where it has been previously postulated that
ULK1 is overexpressed in MCF-7 cells, while lowly expressed in
MDA-MB-231 cells (25). It has been
indicated that overexpression of ULK1 is conversely correlated with
the prognosis in a number of cancer types, including BC (26). Interestingly, low expression of ULK1
in MDA-MB231 has been correlated with more metastatic behavior,
while overexpression of ULK1 appears to be protective against
metastasis. This is because ULK1 is required to phosphorylate and
inhibit Exo70, an essential component of the exocyst complex, which
is necessary for invasion and metastasis (25). However, this differential expression
of ULK1 appears to affect the sensitivity of cancer cells to
doxorubicin. In the MCF-7 cell line, the high expression level of
ULK1 confers greater sensitivity to doxorubicin compared with the
MDA-MB-231 cell line. However, when ULK1 was suppressed, MCF-7
cells exhibited more aggressive behavior and became desensitized to
the drug. On the other hand, MDA-MB-231 is more resistant to
doxorubicin when compared with MCF-7 but becomes more vulnerable to
the drug when ULK1 is depressed. This can be explained as the
minimum level of ULK1 in MDA-MB-231 cells is essential to maintain
the aggressive behavior, but when it is suppressed, the cells lose
these aggressive traits.
Interestingly, the opposing effects of ULK1 on
doxorubicin sensitivity in MCF-7 vs. MDA-MB-231 cells likely
reflect its context-dependent roles in autophagy and apoptosis. In
MCF-7 cells, high basal ULK1 supports autophagy that facilitates
stress-induced apoptosis; therefore knockdown reduces doxorubicin
sensitivity. Conversely, in MDA-MB-231 cells, basal autophagy may
be primarily cytoprotective, and ULK1 knockdown disrupts this
survival mechanism, sensitizing cells to treatment. These
differences are influenced by cell-type-specific factors, including
p53 status, baseline autophagic flux, and apoptotic wiring,
highlighting ULK1 as a versatile regulator whose impact on
chemotherapy response is highly context-dependent. Furthermore, a
context-dependent approach to ULK1 modulation may be more effective
than uniform inhibition. In highly aggressive or metastatic
cancers, such as MDA-MB-231, ULK1 and USP20 appear to restrain
pro-metastatic programs, suggesting that ULK1 activation could
suppress invasion and metastasis. Conversely, in tumors where ULK1
supports cytoprotective autophagy, inhibition may enhance
chemosensitivity. Therefore, therapeutic strategies should be
guided by cell-type-specific autophagy dependency, basal ULK1/USP20
expression and metastatic potential, with activation or inhibition
tailored to exploit the tumor's particular vulnerabilities while
minimizing harm to normal tissues.
Tang et al (27) previously reported that ULK1 is
overexpressed in non-small cell lung cancer cells (NSCLC), compared
with normal lung cells and this is correlated with poor prognosis
and patients' survival rate. Inhibition of ULK1 with SBI0206965, a
selective ULK1 inhibitor, reversed the aggressive trait of these
cells and sensitized cells to cisplatin. The data of the present
study showed different outcomes, as ULK1 appears to prevent the
progression of the disease, and when it is inhibited, A549 cells
become more aggressive and resistant to cisplatin. However, the
correlation between ULK1 and drug-resistant genes must be further
investigated to understand the mechanism underlying these results.
The apparent contradiction between the present study's findings in
A549 cells and those reported by Tang et al (27) likely reflects differences in both
inhibition strategy and cellular context. Tang et al
(27) employed the small-molecule
ULK1 inhibitor SB10206965, which may affect ULK1 kinase activity
without fully depleting protein levels or disrupting its
scaffolding functions, whereas siRNA-mediated knockdown was used in
the present study, leading to a more complete loss of ULK1 protein
and potentially broader effects on autophagy-independent roles such
as mitotic regulation. Additionally, NSCLC encompasses
heterogeneous subtypes with distinct basal autophagy flux,
stress-response pathways and genetic backgrounds, all of which can
influence the outcome of ULK1 inhibition on cisplatin sensitivity.
Taken together, these methodological and biological differences
likely account for the divergent results and underscore the
context-dependent nature of ULK1 function in chemotherapy
response.
USP20 has been positively correlated to the
development and progression of cancers. It has been reported that
USP20 is differentially expressed between cancer and normal cells,
being highly expressed in most cancers compared with their
corresponding normal cells but found to be lowly expressed in a few
cancer types, including pancreatic cancers (28). Inconsistent with the previous
findings, the results of the present study are similar in the
context of USP20 expression level. Moreover, it has been reported
that USP20 is responsible for the stabilization of the labile
SNAI2/SLUG, which is a transcription factor that drives invasion
and metastasis in cancers (29).
When USP20 was inhibited, SNAI2/SLUG was subjected to degradation
after being ubiquitinated, which in turn inhibited tumor
progression (29). Consistently,
the results of the present study indicate that USP20 inhibition
re-sensitized numerous cancer types to their corresponding
chemotherapies, including MCF-7, HepG2, A549 and PanC1, and
enhanced the effectiveness of the chemotherapy. By contrast, the
triple-negative MDA-MB-231 BC and U87 glioblastoma cell lines
behaved differently, as they remained unchanged or became highly
resistant to doxorubicin, respectively. Overall, USP20 knockdown
sensitizes MCF-7 cells but not MDA-MB-231 cells, likely reflecting
cell-type-specific dependencies. The highly metastatic,
triple-negative MDA-MB-231 cells may rely on USP20-independent
pathways to maintain survival and invasion, using alternative
de-ubiquitinases or signaling networks to stabilize pro-metastatic
factors like SNAI2/SLUG. This highlights the context-dependent role
of USP20 and the plasticity of survival mechanisms in aggressive
BC. A comprehensive proteomic, metabolomic, and transcriptomic
study must be performed to widen the understanding of the
mechanisms underlying ULK1 and USP20 modulation and the
chemotherapeutic sensitivity of different cancer cell lines.
The present study determined whether
chemotherapeutic medications alter autophagy-related genes
including P62, USP20 and ULK1. In liver cancer HepG2 cells and
glioblastoma U87 cells, doxorubicin treatment leads to
downregulation of ULK1 and USP20 expression levels and the
overexpression of P62, suggesting the inhibition of autophagy.
Although autophagy induction is a well-described response to
chemotherapeutic stress, the present findings indicated that
doxorubicin can also suppress autophagy in a cell-type-dependent
manner, as reflected by ULK1 downregulation and p62 accumulation in
HepG2 and U87 cells. These seemingly opposing outcomes can be
reconciled by recognizing that chemotherapy produces biphasic and
context-specific autophagy responses. At early or lower doses,
genotoxic stress typically activates p53-, NF-κB-, and MAPK-driven
transcriptional programs that stimulate autophagy as a
cytoprotective mechanism. However, prolonged or high-dose exposure
can overwhelm cellular proteostasis, impair lysosomal function, or
promote ULK1 degradation, resulting in blocked autophagic flux
despite transcriptional activation of stress-responsive genes.
Thus, doxorubicin may either induce or inhibit autophagy depending
on dose, timing, and the intrinsic stress-response architecture of
each cell line, explaining why HepG2 and U87 exhibit autophagy
suppression while other models demonstrate induction.
Interestingly, the observations made in the present study suggested
that in U87 glioblastoma cells, basal autophagy maintained by ULK1
and USP20 may be pro-death rather than cytoprotective. Inhibition
of these autophagy regulators reduces stress-induced autophagic
activity, which in this context appeared to facilitate
doxorubicin-induced apoptosis. Consequently, knockdown of ULK1 or
USP20 diminishes pro-death autophagy, allowing cells to survive
chemotherapy, consistent with the notion that the functional
outcome of autophagy is highly context- and cell-type-dependent.
These findings highlight that in aggressive cancer types such as
glioblastoma, basal autophagy can act as a tumor-suppressive,
pro-apoptotic mechanism.
According to Sha et al (30), an elevated P62 level indicates a
compromised proteasomal function in cells. Moreover, doxorubicin
treatment markedly increased the level of P62 transcripts in
MDA-MB-231, indicating that autophagy is being inhibited in the
cells, but it did not affect the expression of ULK1 or USP20. These
findings could be explained by the fact that autophagy occurs
through a variety of routes inside the cell and that
autophagy-mediating factors other than ULK1 and USP20, including
ATG12 and ATG7, may be downregulated in response to the drug.
Another research group demonstrated that chemotherapy treatment of
liver cancer HepG2 cells increases autophagy flux, dictated by an
increase in LC3II level (31).
Their outcome contradicts the findings of the present study;
however, the chemotherapeutic agents evaluated in their study were
5-fluorouracil and cisplatin, but not doxorubicin, which was used
in the present study, suggesting that different chemotherapies
operate with different mechanisms.
Chemotherapeutic agents such as doxorubicin and
cisplatin appear to transcriptionally upregulate ULK1, USP20, and
P62 through activation of stress-responsive signaling pathways.
Both drugs induce substantial DNA damage, oxidative stress and ER
stress, which activate transcription factors including p53, NF-κB,
ATF4/CHOP and Nrf2. These regulators are well known to drive the
expression of autophagy-related genes as part of a compensatory
cytoprotective response. In parallel, activation of the DNA
damage-response kinases (ATM/ATR) and MAPK pathways further
enhances transcription of autophagy initiation factors and stress
adaptors. As a result, doxorubicin and cisplatin promote a
coordinated upregulation of ULK1, USP20, and P62 to enhance
autophagy and maintain cellular homeostasis under chemotherapeutic
stress.
Cisplatin was confirmed to induce ULK1 expression in
lung cancer A549 cells, and this elevation was correlated with poor
prognosis and the development of the cisplatin-resistant trait.
When ULK1 was knocked down, A549 became sensitized to cisplatin and
underwent apoptosis (27).
Conversely, the highest tested concentration of cisplatin in the
present research did not affect ULK1 expression, accompanied by an
elevation of P62 expression, indicating an impaired autophagy
process. When ULK1 was knocked down, resistance to cisplatin was
developed. Suggesting that the presence of ULK1 and a functional
autophagy process is important for effective chemotherapy
treatment. Interestingly, USP20 becomes elevated upon cisplatin
employment in A549 cells. It has been indicated that USP20 high
levels promote cell proliferation, migration and invasion, and are
directly correlated with poor prognosis and survival rates
(22). These data are in line with
our data, as USP20 knockdown leads to higher sensitivity of A549 to
cisplatin.
Regarding the pancreatic PanC1 cells, gemcitabine
chemotherapy massively induced ULK1 expression. However, the
induction of autophagy in this case appears to be important for
PanC1 survival after their exposure to gemcitabine, evidenced by
the enhanced sensitivity of PanC1 to the drug after siULK1
administration. Accordingly, one research group indicated that
gemcitabine acts by inhibiting the replication of DNA and inducing
its damage, and as a survival response, PanC1 cells induced
autophagy and the lysosomal functions upon drug exposure (32). The therapeutic efficiency of this
drug is highly limited due to the development of drug resistance by
the growing pancreatic cancers (33). Seemingly, PanC1 develops this
resistance by inducing autophagy as a survival mechanism to recycle
gemcitabine-induced damage in the cellular components and enhance
cell growth (34). Thus, inhibiting
autophagy along with gemcitabine administration may enhance
therapeutic effectiveness against the disease.
Although the present study focused on single-agent
chemotherapy, ULK1/USP20 modulation could influence combination
therapies in a context-dependent manner. Inhibiting ULK1/USP20 may
sensitize tumor cells to targeted therapies by blocking
cytoprotective autophagy, while activating ULK1 in pro-death
contexts could enhance immunotherapy by promoting immunogenic cell
death. Careful consideration of tumor subtype, basal autophagic
flux, and immune status will be essential to optimize such
combination strategies.
The fate of cancer cells when they are exposed to
chemotherapeutic treatment is governed by the complicated
relationship between apoptosis and autophagy, processes that may
operate synergistically or antagonize each other (35). Apoptosis is initiated by extrinsic
ligands or intrinsic cellular stresses, such as reactive oxygen
species, DNA damage and mitochondrial stress (36). The balance between the BCL-2 family
proteins, mainly the proapoptotic protein Bax and the antiapoptotic
protein BCL-2, dictates the fate of the cell, either survival or
death (37). However, studies that
investigate the correlation between ULK1, USP20, and apoptosis in
cancers are contradictory and minimal, especially for USP20. For
this aim, the dynamic relationship between the autophagy-related
gene, ULK1, the de-ubiquitinating enzyme USP20, and apoptosis is
further clarified in the present study. Although autophagy is
widely known to antagonize apoptotic cell death, it can operate
hand in hand with apoptosis, specifically after persistent exposure
to stressful conditions (35). This
contradictory function of autophagy over apoptotic proteins can be
observed in the present study. When ULK1 was inhibited in the
investigated cancer cell lines, the latter showed variable
responses regarding apoptosis induction. Chemotherapeutic agents
also show different patterns and magnitudes in inducing apoptosis
and exhibit different mechanisms that drive cellular death.
Accordingly, knockdown of ULK1 or USP20 likely
disrupts autophagic homeostasis, leading to the accumulation of
damaged organelles and cellular stress, which can trigger the
intrinsic apoptotic pathway. Specifically, autophagy inhibition may
promote mitochondrial dysfunction, resulting in the release of
cytochrome c and subsequent activation of pro-apoptotic proteins
such as Bax, while suppressing anti-apoptotic Bcl-2, thereby
shifting the Bax/Bcl-2 ratio in favor of apoptosis. This
mechanistic link is consistent with prior reports demonstrating
that impaired autophagy sensitizes cells to mitochondrial mediated
apoptosis. Furthermore, doxorubicin induces cellular apoptosis in
BC cell lines, MCF-7 and MDA-MB-231, and the glioblastoma U87 cell
line. On the contrary, doxorubicin treatment of liver cancer HepG2
cells inhibited the apoptotic pathway but enhanced the necrotic
pathway. Necrosis is another uncontrolled cell death mechanism,
which involves molecular mechanisms other than Bax and
Bcl-2(38).
Knockdown of USP20 also shows variable effects on
cancerous cells' fate. In a previous study, the knockdown of USP20
in the HeLa cervical cancer cell line promoted cellular survival
and growth by stabilizing P62, and activating the TNFα/NF-κB
survival pathway (39). When USP20
was depleted, apoptosis was induced in HeLa cells (39). Furthermore, USP20 prevents the
lysosomal degradation of ULK1 and thus enhances the operation of
autophagy under starvation conditions (40), and its depletion reduces autophagy
and drives apoptosis (40). These
data reflect the complex relationship between these factors and
apoptosis, as they may intersect at numerous points within
different cellular pathways. Altogether, cancer cell fate after
combining chemotherapy with ULK1 or USP20 silencing depends on the
dynamic balance between Bax and Bcl-2 triggered by both treatments.
Thus, the effect of ULK1 and USP20 silencing on the apoptotic
pathway may antagonize and reduce the effectiveness of the
conventional chemotherapy in some cancer types but synergize and
augment the apoptotic effect of chemotherapies in other cancer
cells.
Primary fibroblasts proliferate reliably, maintain a
stable karyotype, and are easier to culture than many primary
epithelial cells. This ensures reproducible comparisons across
experiments. In addition, HDFa cells lack the constitutive stress,
metabolic rewiring, and proliferative signaling that often
complicate the interpretation of cancer cell lines (41). This makes them a useful baseline for
identifying cancer-associated upregulation of proteins such as ULK1
and USP20. ULK1 and USP20 expression are highly tissue-dependent.
Some tissues naturally express higher or lower levels of autophagy
regulators (42,43). Tissue-matched controls might show
that certain cancer cell lines do not overexpress ULK1 or USP20
when compared with their native tissue.
Regarding the therapeutic implications, the
context-dependent effects of ULK1 inhibition indicate that careful
patient stratification is necessary to achieve a therapeutic
window. Tumors with high basal ULK1 expression or elevated
autophagic flux may be more dependent on ULK1 for survival, whereas
normal tissues with lower flux, such as fibroblasts, may be spared.
Additional factors, including p53 status, apoptotic priming, and
expression of co-regulators such as USP20, could further refine
predictions of ULK1 dependency. Assessing tumor subtype, metastatic
potential, and markers of oxidative stress or metabolic rewiring
may also help identify cancers that are particularly sensitive to
ULK1 inhibition, allowing selective targeting of tumor cells while
minimizing toxicity to normal tissue.
While the findings of the present study provide
valuable results, some limitations can be addressed. Nevertheless,
the involvement of more cell line types would broaden our
understanding of the multifaceted role of autophagy. Furthermore,
the present study compared the changes in the expression of ULK1
and USP20 in cancer cell lines relative to normal skin fibroblast
cells only. However, it would be more accurate to correlate the
changes in their expression with the normal cells of the same type
as the cancer cells. Another limitation of the present study is
that the autophagic flux at the protein level was not directly
assessed, as the antibodies for LC3 and p62 tested were
non-specific. Therefore, while the mRNA measurements provided
insight into transcriptional regulation of autophagy-related genes,
they may not fully reflect functional autophagic activity.
Additionally, a potential limitation was that siRNA reagents can
produce off-target or immune-stimulatory effects. While all
knockdowns were compared with a scrambled siRNA control, some
residual non-specific effects, such as modest IC50
shifts in fibroblasts, cannot be fully excluded and should be
considered when interpreting the data.
In conclusion, the present study provided a detailed
dataset that reflects the biphasic role of autophagy among
different cancer types. ULK1 and USP20 are differentially expressed
among different cancer types, and their knockdown is directly
correlated with chemoresistance of cancer cells toward the
conventional chemotherapeutic drugs. The presented data indicate
that different cancer cells react in various ways when ULK1 and
USP20 are silenced in the context of cellular death. The available
data help us determine where the inhibition of these genes can be
incorporated with conventional therapy to improve its effectiveness
and where it must be avoided to prevent the development of more
aggressive tumor characteristics. Further studies should be
conducted to uncover the molecular mechanisms underlying the
differential role of autophagy in these cancers, involving a
comprehensive metabolomics, transcriptomics, and proteomics
analysis. Also, key in vivo studies should use xenograft or
patient-derived models to test genetic or pharmacologic inhibition
of ULK1 and USP20 alongside chemotherapy, assessing tumor growth,
apoptosis, and drug response. Metastatic models can determine
effects on invasion and metastasis, while toxicity studies ensure
safety. Linking ULK1/USP20 expression and autophagy or apoptosis
markers with treatment outcomes will confirm target engagement and
support their therapeutic potential.
Supplementary Material
Flow cytometric analysis of apoptosis
in normal fibroblasts treated with Dox. Normal fibroblast cells
were treated with two different concentrations of Dox
chemotherapeutic drug, alone, or in combination with siULK1 or
siUSP20, or siSCR. A total of 24 h post-treatment, cells were
stained with Annexin V-FITC and PI, then analyzed by flow
cytometry. Representative dot plots show viable (lower left
quarter), early apoptotic (lower right quarter), late apoptotic
(Upper right quarter), and necrotic (Upper left quarter) cell
populations. Three sample repeats were used in this experiment.
Data were further analyzed using the one-way ANOVA statistical test
and presented in Table VII. Dox,
doxorubicin; si-, small interfering.
Flow cytometric analysis of apoptosis
in normal fibroblasts treated with Cisplatin. Normal fibroblast
cells were treated with two different concentrations of Cisplatin
chemotherapeutic drug, alone, or in combination with siULK1 or
siUSP20, or siSCR. 24 h post-treatment, cells were stained with
Annexin V-FITC and PI, then analyzed by flow cytometry.
Representative dot plots show the percentage of viable (lower left
quarter), early apoptotic (lower right quarter), late apoptotic
(Upper right quarter), and necrotic (Upper left quarter) cell
populations. Three sample repeats were used in this experiment.
Data was further analyzed using the One-Way ANOVA statistical test
and presented in Table VII.
Flow cytometric analysis of apoptosis
in normal fibroblasts treated with Gemcitabine. Normal fibroblast
cells were treated with two different concentrations of Gemcitabine
chemotherapeutic drug, alone, or in combination with siULK1 or
siUSP20, or siSCR. 24 h post-treatment, cells were stained with
Annexin V-FITC and PI, then analyzed by flow cytometry.
Representative dot plots show the percentage of viable (lower left
quarter), early apoptotic (lower right quarter), late apoptotic
(Upper right quarter), and necrotic (Upper left quarter) cell
populations. Three sample repeats were used in this experiment.
Data was further analyzed using the One-Way ANOVA statistical test
and presented in Table VII.
Flow cytometric analysis of apoptosis
in U87-Glioblastoma treated with Doxorubicin. U87-Glioblastoma
cells were treated with two different concentrations of Gemcitabine
chemotherapeutic drug, alone, or in combination with siULK1 or
siUSP20, or siSCR. A total of 24 h post-treatment, cells were
stained with Annexin V-FITC and PI, then analyzed by flow
cytometry. Representative dot plots show the percentage of viable
(lower left quarter), early apoptotic (lower right quarter), late
apoptotic (Upper right quarter), and necrotic (Upper left quarter)
cell populations. Three sample repeats were used in this
experiment. Data was further analyzed using the one-way ANOVA
statistical test and presented in Table VII.
Flow cytometric analysis of apoptosis
in HepG2 liver cancer treated with Doxorubicin. HepG2 liver cancer
cells were treated with two different concentrations of Gemcitabine
chemotherapeutic drug, alone, or in combination with siULK1 or
siUSP20, or siSCR. A total of 24 h post-treatment, cells were
stained with Annexin V-FITC and PI, then analyzed by flow
cytometry. Representative dot plots show the percentage of viable
(lower left quarter), early apoptotic (lower right quarter), late
apoptotic (Upper right quarter), and necrotic (Upper left quarter)
cell populations. Three sample repeats were used in this
experiment. Data was further analyzed using the one-way ANOVA
statistical test and presented in Table VII.
Flow cytometric analysis of apoptosis
in A549 lung cancer treated with Cisplatin. A549 lung cancer cells
were treated with two different concentrations of Cisplatin
chemotherapeutic drug, alone, or in combination with siULK1 or
siUSP20, or siSCR. A total of 24 h post-treatment, cells were
stained with Annexin V-FITC and PI, then analyzed by flow
cytometry. Representative dot plots show the percentage of viable
(lower left quarter), early apoptotic (lower right quarter), late
apoptotic (Upper right quarter), and necrotic (Upper left quarter)
cell populations. Three sample repeats were used in this
experiment. Data was further analyzed using the one-way ANOVA
statistical test and presented in Table VII.
Flow cytometric analysis of apoptosis
in MCF-7 breast cancer treated with Doxorubicin. MCF-7 breast
cancer cells were treated with two different concentrations of
Gemcitabine chemotherapeutic drug, alone, or in combination with
siULK1 or siUSP20, or siSCR. A total of 24 h post-treatment, cells
were stained with Annexin V-FITC and PI, then analyzed by flow
cytometry. Representative dot plots show the percentage of viable
(lower left quarter), early apoptotic (lower right quarter), late
apoptotic (Upper right quarter), and necrotic (Upper left quarter)
cell populations. Three sample repeats were used in this
experiment. Data was further analyzed using the one-way ANOVA
statistical test and presented in Table VII.
Flow cytometric analysis of apoptosis
in PanC1 pancreatic cancer treated with Gemcitabine. PanC1
pancreatic cancer cells were treated with two different
concentrations of Gemcitabine chemotherapeutic drug, alone, or in
combination with siULK1 or siUSP20, or siSCR. A total of 24 h
post-treatment, cells were stained with Annexin V-FITC and PI, then
analyzed by flow cytometry. Representative dot plots show the
percentage of viable (lower left quarter), early apoptotic (lower
right quarter), late apoptotic (Upper right quarter) and necrotic
(Upper left quarter) cell populations. Three sample repeats were
used in this experiment. Data was further analyzed using the
one-way ANOVA statistical test and presented in Table VII.
Flow cytometric analysis of apoptosis
in MDA-MB-231 breast cancer treated with Doxorubicin. MDA-MB-231
breast cancer cells were treated with two different concentrations
of Gemcitabine chemotherapeutic drug, alone, or in combination with
siULK1 or siUSP20, or siSCR. A total of 24 h post-treatment, cells
were stained with Annexin V-FITC and PI, then analyzed by flow
cytometry. Representative dot plots show the percentage of viable
(lower left quarter), early apoptotic (lower right quarter), late
apoptotic (Upper right quarter) and necrotic (Upper left quarter)
cell populations. Three sample repeats were used in this
experiment. Data was further analyzed using the One-Way ANOVA
statistical test and presented in Table VII.
Acknowledgements
The authors would like to thank Mrs Zain Hindash
(Department of Biology, University of Jordan, Amman, Jordan) for
technical support.
Funding
Funding: The present study was supported by the Deanship of
Scientific Research in The University of Jordan (grant no.
2024-172/2023).
Availability of data and materials
The data generated in the present study are included
in the figures and/or tables of this article.
Authors' contributions
AI, TA, DA and MZ conseptualized the present study
and performed the experimental design. AI and TA performed
statistical analysis and drafted the manuscript. All authors read
and approved the final version of the manuscript. AI and TA confirm
the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kitada M and Koya D: Autophagy in
metabolic disease and ageing. Nat Rev Endocrinol. 17:647–661.
2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Ahmadi-Dehlaghi F, Mohammadi P, Valipour
E, Pournaghi P, Kiani S and Mansouri K: Autophagy: A challengeable
paradox in cancer treatment. Cancer Med. 12:11542–11569.
2023.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Rakesh R, PriyaDharshini LC, Sakthivel KM
and Rasmi RR: Role and regulation of autophagy in cancer. Biochim
Biophys Acta Mol Basis Dis. 1868(166400)2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Yu L, Chen Y and Tooze SA: Autophagy
pathway: Cellular and molecular mechanisms. Autophagy. 14:207–215.
2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Hama Y, Fujioka Y, Yamamoto H, Mizushima N
and Noda NN: The triad interaction of ULK1, ATG13, and FIP200 is
required for ULK complex formation and autophagy. Elife.
13(RP101531)2025.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Follo C, Vidoni C, Morani F, Ferraresi A,
Seca C and Isidoro C: Amino acid response by halofuginone in cancer
cells triggers autophagy through proteasome degradation of mTOR.
Cell Commun Signal. 17(39)2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Sun T, Liu Z and Yang Q: The role of
ubiquitination and deubiquitination in cancer metabolism. Mol
Cancer. 19(146)2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Chakraborty P: Contrasting role of
autophagy in different types of cancer: A review toward biomarkers
and therapeutic improvement. Biomed Biotechnol Res J. 5:260–266.
2021.
|
|
9
|
Russell RC and Guan KL: The multifaceted
role of autophagy in cancer. EMBO J. 41(e110031)2022.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Yun CW, Jeon J, Go G, Lee JH and Lee SH:
The dual role of autophagy in cancer development and a therapeutic
strategy for cancer by targeting autophagy. Int J Mol Sci.
22(179)2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Debnath J, Gammoh N and Ryan KM: Autophagy
and autophagy-related pathways in cancer. Nat Rev Mol Cell Biol.
24:560–575. 2023.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Marinković M, Šprung M, Buljubašić M and
Novak I: Autophagy modulation in cancer: Current knowledge on
action and therapy. Oxid Med Cell Longev.
2018(8023821)2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ganzleben I, Neurath MF and Becker C:
Autophagy in cancer therapy-molecular mechanisms and current
clinical advances. Cancers (Basel). 13(5575)2021.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Mulcahy Levy JM and Thorburn A: Autophagy
in cancer: Moving from understanding mechanism to improving therapy
responses in patients. Cell Death Differ. 27:843–857.
2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Mahri S, Villa R, Shiau YP, Tang M,
Racacho KJ, Zong Q, Chowdhury SI, Hua T, Godinez F, Birkeland A, et
al: Nanomedicine approaches for autophagy modulation in cancer
therapy. Small Sci. 5(2400607)2025.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Walweel N and Aydin O: Enhancing
therapeutic efficacy in cancer treatment: Integrating nanomedicine
with autophagy inhibition strategies. ACS Omega. 9:27832–27852.
2024.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Zhu L, Li Z, Wang H, Cheng Z and Zhang L:
Unraveling the mysterious veil of ULK1: From non-canonical
functions to therapeutic applications. Int J Biol Macromol.
18(145177)2025.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Ma L, Li W, Zhang Y, Qi L, Zhao Q, Li N,
Lu Y, Zhang L, Zhou F, Wu Y, et al: FLT4/VEGFR3 activates AMPK to
coordinate glycometabolic reprogramming with autophagy and
inflammasome activation for bacterial elimination. Autophagy.
18:1385–1400. 2022.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zhang J, Tripathi DN, Jing J, Alexander A,
Kim J, Powell RT, Dere R, Tait-Mulder J, Lee JH, Paull TT, et al:
ATM functions at the peroxisome to induce pexophagy in response to
ROS. Nat Cell Biol. 17:1259–1269. 2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Deng R, Zhang HL, Huang JH, Cai RZ, Wang
Y, Chen YH, Hu BX, Ye ZP, Li ZL, Mai J, et al: MAPK1/3
kinase-dependent ULK1 degradation attenuates mitophagy and promotes
breast cancer bone metastasis. Autophagy. 17:3011–3029.
2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ji X, Zhang X and Li Z: ULK1 inhibitor
induces spindle microtubule disorganization and inhibits
phosphorylation of Ser10 of histone H3. FEBS Open Bio.
10:2452–2463. 2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Li Q, Ye C, Tian T, Jiang Q, Zhao P, Wang
X, Liu F, Shan J and Ruan J: The emerging role of
ubiquitin-specific protease 20 in tumorigenesis and cancer
therapeutics. Cell Death Dis. 13(434)2022.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wang Y, Wu L and Van Kaer L: Role of
canonical and noncanonical autophagy pathways in shaping the life
journey of B cells. Front Immunol. 15(1426204)2024.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Mao L, Zhan YY, Wu B, Yu Q, Xu L, Hong X,
Zhong L, Mi P, Xiao L, Wang X, et al: ULK1 phosphorylates Exo70 to
suppress breast cancer metastasis. Nat Commun.
11(117)2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Tang J, Deng R, Luo RZ, Shen GP, Cai MY,
Du ZM, Jiang S, Yang MT, Fu JH and Zhu XF: Low expression of ULK1
is associated with operable breast cancer progression and is an
adverse prognostic marker of survival for patients. Breast Cancer
Res Treat. 134:549–560. 2012.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Tang F, Hu P, Yang Z, Xue C, Gong J, Sun
S, Shi L, Zhang S, Li Z, Yang C, et al: SBI0206965, a novel
inhibitor of Ulk1, suppresses non-small cell lung cancer cell
growth by modulating both autophagy and apoptosis pathways. Oncol
Rep. 37:3449–3458. 2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Wang T, Bai Y, Dong Y, Qin J, Zhou X, Wang
A, Liu D, Li X, Ma Z and Hu Y: A comprehensive analysis of
deubiquitinase USP20 on prognosis and immunity in pan-cancer. FASEB
J. 39(e70499)2025.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Li W, Shen M, Jiang YZ, Zhang R, Zheng H,
Wei Y, Shao ZM and Kang Y: Deubiquitinase USP20 promotes breast
cancer metastasis by stabilizing SNAI2. Genes Dev. 34:1310–1315.
2020.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Sha Z, Schnell HM, Ruoff K and Goldberg A:
Rapid induction of p62 and GABARAPL1 upon proteasome inhibition
promotes survival before autophagy activation. J Cell Biol.
217:1757–1776. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Guo XL, Li D, Hu F, Song JR, Zhang SS,
Deng WJ, Sun K, Zhao QD, Xie XQ, Song YJ, et al: Targeting
autophagy potentiates chemotherapy-induced apoptosis and
proliferation inhibition in hepatocarcinoma cells. Cancer Lett.
320:171–179. 2012.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Marchand B, Poulin MA, Lawson C, Tai LH,
Jean S and Boucher MJ: Gemcitabine promotes autophagy and lysosomal
function through ERK-and TFEB-dependent mechanisms. Cell Death
Discov. 9(45)2023.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Gillson J, Abd El-Aziz YS, Leck LYW,
Jansson PJ, Pavlakis N, Samra JS, Mittal A and Sahni S: Autophagy:
A key player in pancreatic cancer progression and a potential drug
target. Cancers (Basel). 14(3528)2022.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Chavez-Dominguez R, Perez-Medina M,
Lopez-Gonzalez JS, Galicia-Velasco M and Aguilar-Cazares D: The
double-edge sword of autophagy in cancer: From tumor suppression to
pro-tumor activity. Front Oncol. 10(578418)2020.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Mariño G, Niso-Santano M, Baehrecke EH and
Kroemer G: Self-consumption: The interplay of autophagy and
apoptosis. Nat Rev Mol Cell Biol. 15:81–94. 2014.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Galluzzi L, Vitale I, Aaronson SA, Abrams
JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews
DW, et al: Molecular mechanisms of cell death: Recommendations of
the nomenclature committee on cell death 2018. Cell Death Differ.
25:486–541. 2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Tang D, Kang R, Berghe TV, Vandenabeele P
and Kroemer G: The molecular machinery of regulated cell death.
Cell Res. 29:364–374. 2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Kang S, Kim KR, Cho M, Hwang J, Yang JM,
Kim JK and Choi WJ: High-resolution imaging of morphological
changes associated with apoptosis and necrosis using single-cell
full-field optical coherence tomography. Biosensors (Basel).
15(522)2025.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Ha J, Kim M, Seo D, Park JS, Lee J, Lee J
and Park SH: The deubiquitinating enzyme USP20 regulates the
TNFα-induced NF-κB signaling pathway through stabilization of p62.
Int J Mol Sci. 21(3116)2020.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Kim JH, Seo D, Kim SJ, Choi DW, Park JS,
Ha J, Choi J, Lee JH, Jung SM, Seo KW, et al: The deubiquitinating
enzyme USP20 stabilizes ULK1 and promotes autophagy initiation.
EMBO Rep. 19(e44378)2018.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zhang F, Ma Y, Li D, Wei J, Chen K, Zhang
E, Liu G, Chu X, Liu X, Liu W, et al: Cancer associated fibroblasts
and metabolic reprogramming: Unraveling the intricate crosstalk in
tumor evolution. J Hematol Oncol. 17(80)2024.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Wang J, Zheng F, Wang D and Yang Q:
Regulation of ULK1 by WTAP/IGF2BP3 axis enhances mitophagy and
progression in epithelial ovarian cancer. Cell Death Dis.
15(97)2024.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Jin R, Luo Z, Jun-Li Tao Q, Wang P, Cai X,
Jiang L, Zeng C and Chen Y: USP20 is a predictor of poor prognosis
in colorectal cancer and associated with lymph node metastasis,
immune infiltration and chemotherapy resistance. Front Oncol.
13(1023292)2023.PubMed/NCBI View Article : Google Scholar
|