Introduction
Neuroendocrine tumours represent a diverse and
complex group of neoplasms originating from neuroendocrine cells
found throughout the body, predominantly in the GI tract, pancreas,
and lungs (1). These tumours are
unique in their ability to produce hormones and other bioactive
substances, which can lead to various clinical syndromes depending
on the type and quantity of hormones secreted (2). Despite being relatively rare, the
incidence of NETs has steadily risen over the past few decades,
partly due to advancements in diagnostic techniques and heightened
clinical awareness (3).
NETs are heterogeneous, both in terms of their
biological behaviour and clinical presentation. The overall
incidence is estimated to be around 5.86 per 100,000 individuals
annually, and it has been increasing over the years (4). This rise in incidence can be
attributed to improved imaging techniques, more widespread use of
endoscopy, and increased screening practices that lead to the
detection of asymptomatic cases (5).
The World Health Organization classifies NETs based
on their histopathological characteristics (6). NETs are generally categorized into
well-differentiated and poorly differentiated tumours, further
classified into grades 1, 2, and 3 based on the Ki-67 index
(6). Well-differentiated NETs
typically have a low behaviour, whereas poorly differentiated
neuroendocrine carcinomas (NECs) are more aggressive and have
poorer prognoses (7).
The pathophysiology of NETs is closely linked to
their neuroendocrine origin, which endows them with the ability to
secrete a variety of peptides and amines. This leads to distinct
clinical syndromes (8). For
instance, carcinoid syndrome, often associated with
gastrointestinal NETs, results from the secretion of serotonin and
other vasoactive substances (9).
The genetic underpinnings of NETs are also diverse, with common
mutations identified in genes such as MEN1, DAXX, and ATRX,
particularly in pNETs (10,11). Mutations in the MEN1 gene, which
encodes the protein menin, are frequently observed in familial
cases of NETs and are also present in sporadic cases (12). The loss of function of menin leads
to unchecked cellular proliferation and tumour formation.
Similarly, mutations in DAXX and ATRX, which are involved in
chromatin remodelling, have been associated with more aggressive
forms of NETs and are considered poor prognostic markers (13).
The clinical presentation of NETs can vary widely
depending on the tumour's location, its size, the grade of the
tumour, and its functional status. For example, insulinomas, which
secrete insulin, can cause recurrent hypoglycaemia, while
gastrinomas, which secrete gastrin, lead to Zollinger-Ellison
syndrome, characterized by severe peptic ulcers (14). In contrast, non-functional NETs,
which do not secrete hormones, may remain asymptomatic for a long
time and are often discovered incidentally during imaging studies
performed for other reasons (15).
When symptoms do occur in these tumours, they are typically related
to the tumour's mass effect or metastasis. The liver is the most
common site of metastatic spread, particularly in gastrointestinal
and pNETs (16).
Therapeutic strategies for neuroendocrine tumours
have evolved rapidly alongside advances in pathway-targeted
treatments. A significant milestone was reached on 26 March 2025,
when the U.S. Food and Drug Administration authorized cabozantinib
for patients with unresectable or metastatic pancreatic and extra
pancreatic NETs who had progressed after prior systemic therapy
(17). In addition, belzutifan, a
selective hypoxia-inducible factor-2α inhibitor, was approved for
the management of malignant pheochromocytoma and paraganglioma,
representing a new metabolism-oriented therapeutic paradigm in
neuroendocrine oncology (18).
At the same time, peptide receptor radionuclide
therapy continues to advance beyond conventional
177Lu-DOTATATE, with ongoing development of somatostatin
receptor antagonists and alpha-particle-emitting radionuclides
aimed at improving tumour targeting and cytotoxic efficacy
(19). Beyond radionuclide-based
approaches, novel immunotherapeutic strategies are emerging as
promising avenues that may substantially reshape systemic treatment
options for NETs in the near future.
The diagnosis of NETs involves a combination of
clinical evaluation, biochemical testing of markers (17,18)
and imaging studies, such as CT, MDI and DORATATE PET/CT (19,20).
When it comes to histopathological confirmation, the assessment of
the Ki-67 index is important in determining the tumour grade and
guiding treatment decisions (21,22).
The management of NETs is complex and requires a
multidisciplinary approach. Surgical resection remains the
cornerstone of treatment for localized NETs and offers the best
chance for cure (23). Many
patients are admitted to the hospital with advanced or metastatic
disease at diagnosis, for which systemic therapies are the core of
treatment (24). SSAs such as
octreotide and lanreotide are used to control symptoms and slow
tumour progression in patients with functional NETs (25). Targeted therapies such as the
mammalian target of rapamycin (mTOR) inhibitor everolimus have
demonstrated efficacy in treating advanced pNETs, providing both
antiproliferative and symptom control effects (26). Similarly, the tyrosine kinase
inhibitor sunitinib is approved for treating advanced pNETs and has
been shown to improve progression-free survival in this patient
population (27).
Despite advances in the understanding and treatment
of NETs, several challenges remain. The heterogeneous nature of
these tumours poses significant challenges in diagnosis, treatment,
and prognosis (28). The rarity of
NETs has also limited the ability to conduct large-scale clinical
trials, resulting in a reliance on retrospective studies and small,
prospective cohorts for much of the available evidence (29). Moreover, while new therapies have
improved outcomes for some patients, others continue to experience
poor prognosis, particularly those with high-grade or poorly
differentiated NETs (30).
Accordingly, this systematic review aimed to
evaluate contemporary therapeutic strategies for adult patients
with neuroendocrine tumours. Using the PICO framework, the
population included adult patients with NETs; interventions
comprised surgery, systemic therapies, targeted agents,
immunotherapy, and peptide receptor radionuclide therapy;
comparators included alternative treatment strategies or standard
of care where applicable; and outcomes of interest were
progression-free survival, overall survival, and treatment-related
outcomes.
Materials and methods
Study design
This systematic review was conducted following the
Preferred Reporting Items for Systematic Reviews and Meta-Analyses
(PRISMA) guidelines, which ensure a rigorous and transparent
approach to synthesizing evidence from the literature (31). This review evaluated the efficacy
and safety of various treatment modalities for NETs, including
surgery, chemotherapy, targeted therapy, and PRRT. The review was
not prospectively registered in the PROSPERO database as the study
was designed as an exploratory synthesis of recently emerging
therapeutic strategies in neuroendocrine tumours, and the review
process was initiated prior to protocol registration. Nevertheless,
all methodological steps, including eligibility criteria, search
strategy, study selection, data extraction, and synthesis methods,
were predefined and consistently applied throughout the review.
Eligibility criteria
This review included randomized controlled trials
(RCTs), cohort studies, and observational studies. Only studies
published in peer-reviewed journals were considered, and
prospective and retrospective studies were included. Systematic
reviews and meta-analyses were used for reference and comparison
but were not part of the primary analysis.
PICO framework (32,33):
i) Participants: Eligible studies involved adult
patients (aged 18 years and older) diagnosed with neuroendocrine
tumours, regardless of tumour location (e.g., gastrointestinal,
pancreatic, pulmonary NETs) or grade (well-differentiated or poorly
differentiated). Studies involving paediatric populations or animal
models were excluded.
ii) Interventions: The review focused on therapeutic
interventions for NETs, including, but not limited to, surgical
interventions, chemotherapy (both single-agent and combination
regimens), targeted therapies (including mTOR inhibitors, e.g.,
everolimus and tyrosine kinase inhibitors, e.g., sunitinib), and
PRRT.
iii) Comparison: Evaluating different treatment
plans.
iv) Outcomes: The primary outcomes of interest were
overall survival (OS) and progression-free survival (PFS).
Only studies published in English were included in
this review. To ensure the relevance of findings to current
clinical practices, the search timeframe was January 2019 to July
2025.
Information sources
The literature search was conducted across multiple
databases, including PubMed (https://pubmed.ncbi.nlm.nih.gov/), Cochrane Library
(https://www.cochranelibrary.com/), and
ScienceDirect (https://www.sciencedirect.com/). The search strategy
was developed in collaboration with an experienced medical
librarian to ensure comprehensive coverage of the relevant
literature. Key words related to neuroendocrine tumours and their
treatments were used, including ‘neuroendocrine tumours’, ‘NETs’,
‘carcinoid’, ‘pancreatic neuroendocrine tumours’, ‘treatment’,
‘therapy’, ‘surgery’, ‘chemotherapy’, ‘targeted therapy’ and
‘radiotherapy’.
Search strategy
The search strategy was tailored for each database
but generally followed a similar structure. Boolean operators
(34) were used to combine key
words, and filters were applied to limit results to human studies
and publications in English. The initial search was conducted in
March 2024, and an update was performed in July 2024 to capture any
newly published studies.
PubMed database: ‘neuroendocrine tumors’ OR
‘neuroendocrine tumours’ OR ‘NETs’ OR ‘carcinoid’ OR ‘pancreatic
neuroendocrine tumor’ OR ‘gastroenteropancreatic neuroendocrine
tumor’ AND ‘treatment’ OR ‘therapy’ OR ‘surgery’ OR ‘chemotherapy’
OR ‘targeted therapy’ OR ‘immunotherapy’ OR ‘peptide receptor
radionuclide therapy’ OR ‘PRRT’. Filters applied: Humans, English
language, Adults (≥18 years), publication date January 2019-July
2024.
Cochrane Library: ‘neuroendocrine tumor*’ OR ‘NET*’)
in Title/Abstract/Keywords AND ‘treatment’ OR ‘therapy’ OR
‘chemotherapy’ OR ‘targeted therapy’ OR ‘PRRT’.
ScienceDirect: TITLE-ABSTR-KEY ‘neuroendocrine
tumor*’ OR ‘NET*’ AND (‘treatment’ OR ‘therapy’ OR ‘immunotherapy’
OR ‘targeted therapy’).
Selection process
Two independent reviewers screened the titles and
abstracts of all retrieved studies to assess their eligibility
based on the predefined inclusion and exclusion criteria. They then
retrieved and reviewed in detail full-text articles of potentially
eligible studies. Discrepancies between the reviewers regarding
study eligibility were resolved through discussion, and if
necessary, a third reviewer was consulted.
Data collection process
Data was extracted using a standardized extraction
form explicitly designed for this review. The form captured vital
study characteristics, including: i) Study details: Authors,
publication year, study design, and sample size. ii) Participant
characteristics: Age, sex, tumour type, and tumour grade. iii)
Intervention details: Treatment type, duration, and follow-up
period. iv) Outcomes: Primary and secondary outcomes as outlined
above, including any reported measures of effect and statistical
significance.
Data items
The following data items were extracted from each
included study, where available: First author and year of
publication, study design, sample size, and duration of follow-up.
Patient-related variables included median age, sex distribution,
tumour site, tumour grade, and disease stage. Treatment-related
variables comprised type of therapeutic intervention, treatment
regimen, dosing schedule, and duration of therapy. Outcome
variables included progression-free survival, overall survival,
objective response rate when reported, and treatment-related
adverse events. Only outcomes explicitly reported in the original
studies were extracted, and no assumptions or imputations were made
for missing data.
Risk of bias assessment
The risk of bias for included RCTs was assessed
using the Cochrane Risk of Bias Tool (35), which evaluates bias across several
domains, including selection, performance, detection, attrition,
and reporting. Sensitivity analyses were performed to assess the
robustness of the findings by excluding studies with a high risk of
bias or those with small sample sizes (36).
Synthesis methods
Due to substantial clinical and methodological
heterogeneity across included studies, including differences in
tumour sites, study designs, treatment modalities, and reported
outcomes, a quantitative meta-analysis was not conducted. Instead,
a structured narrative synthesis was performed. Studies were
grouped according to treatment modality, and outcomes were
summarized descriptively using reported median progression-free
survival and overall survival values.
Certainty assessment
The overall certainty of evidence was evaluated
qualitatively, taking into account study design, risk of bias,
consistency of results, and methodological heterogeneity. Given the
inclusion of randomized controlled trials alongside single-arm
phase II studies, retrospective analyses, and observational
studies, and the substantial heterogeneity in tumour types and
treatment strategies, a formal GRADE assessment was not performed.
The certainty of evidence was therefore considered moderate to low,
depending on treatment modality, with higher confidence attributed
to outcomes derived from randomized controlled trials.
Software and statistical analysis
Statistical analyses were conducted using SPSS
version 29(37). All statistical
tests were two-sided, and P<0.05 was considered to indicate a
statistically significant difference (38). The review results were reported per
PRISMA guidelines, ensuring transparency and completeness in
reporting (39,40). Key findings were summarized in text
and tables, and graphs were used to visually present the results.
No formal quantitative meta-analysis was conducted due to marked
heterogeneity in study design, tumour types, treatment modalities,
and reported outcomes. Statistical analyses were therefore limited
to descriptive comparisons and exploratory survival analyses based
on reported median progression-free and overall survival values.
Descriptive statistics, log-rank tests and χ2 test were
used to summarize reported median progression-free survival and
overall survival values across studies. The Φ (phi) effect size was
calculated from the χ2 statistic obtained in the
log-rank test, using the standard formula Φ=√(χ²/n), where χ²
represents the χ2 value from the survival distribution
comparison and n the total number of observations included in the
analysis. Exploratory survival analyses were performed using
Kaplan-Meier estimates based on extracted median survival times,
with censoring applied where appropriate, for illustrative purposes
only. Differences in survival distributions between treatment
categories were explored using the log-rank test, without
adjustment for covariates.
Results
Study selection and population
characteristics
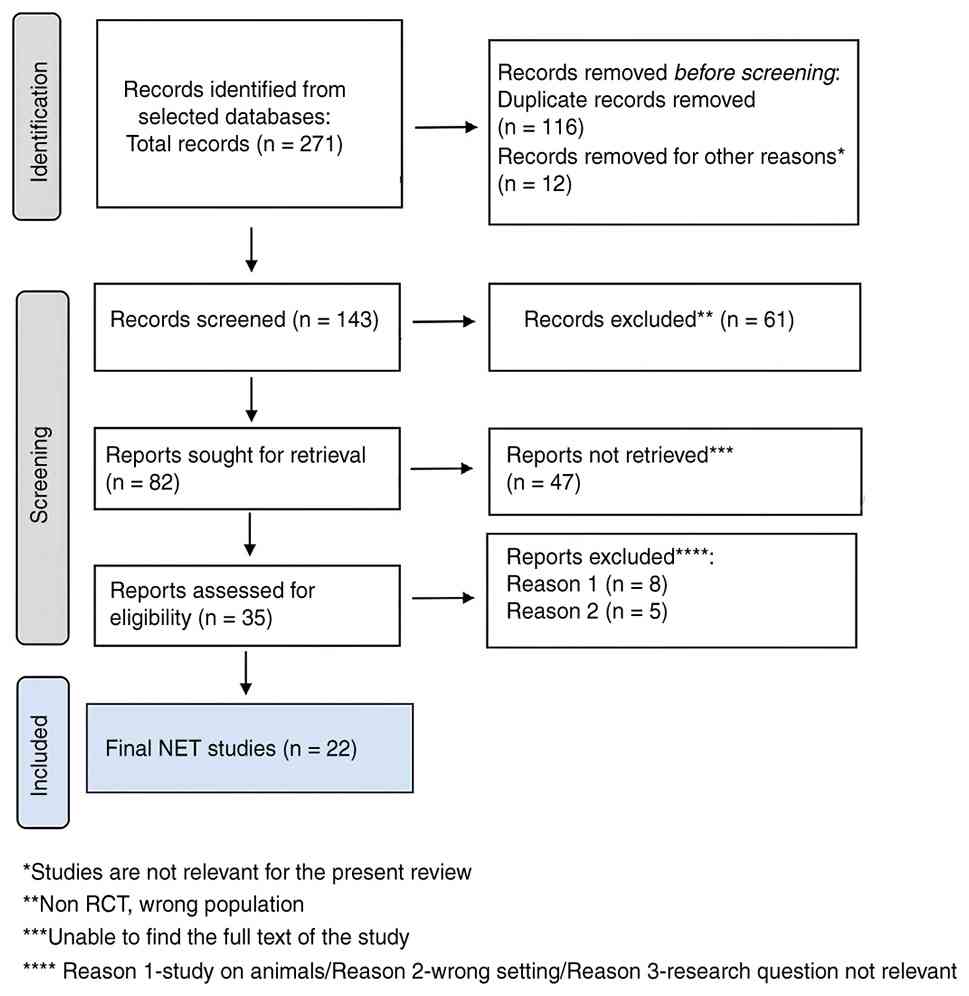
A total of 271 citations were retrieved after
scanning the databases mentioned above. After eliminating duplicate
entries and excluding 12 items that did not satisfy the search
parameters, the list was reduced to 143 remaining articles. Based
on the abstracts, 61 studies were excluded from this research as
they did not meet the criteria. Additionally, 47 papers were
eliminated because they needed the necessary data for extraction
and analysis. Another 13 studies were omitted because they were
commentary or editorial rather than original research, or they were
disregarded, as the full text was not available. Thus, the final
analysis was based on a total of 22 search results that met the
criteria for this investigation. The search yielded 120 citations
for ‘neuroendocrine tumours’ available on PubMed, three on Cochrane
Library, and six on ScienceDirect. The search for ‘pancreatic
neuroendocrine tumours’ led to 8 articles on PubMed, three on
Cochrane Library, and one on ScienceDirect.
Table SI (41-62)
presents a comprehensive summary of the 22 clinical studies
included in this systematic review (Fig. 1), encompassing a diverse range of
therapeutic modalities for neuroendocrine tumours and related
malignancies. Each study is detailed according to sample size,
patient demographics, tumour classification, treatment type and
duration, and principal clinical outcomes, including
progression-free survival and overall survival.
NETs
Table SI presents a
comprehensive overview of the 22 selected studies focused on
various treatment approaches for patients with advanced or
metastatic NETs and other related solid tumours. Key findings from
these studies include each treatment approach's potential benefits
and challenges. Therapies, such as nivolumab with ipilimumab and
newer agents like belzutifan, are targeted at improving PFS and
OS.
A majority of the studies, such as those by Patel
et al (41), Amaria et
al (48) and Eggermont et
al (52), have smaller sample
sizes, typically under 100 patients. Only a few studies, like
Larkin et al (45) and
Motzer et al (49), have
significantly larger sample sizes, reaching nearly 1000 patients.
The majority of studies, including Marabelle et al (42), Tawbi et al (43), and Dummer et al (51), report an average patient age centred
around the early 60s. Most studies, such as those by Olson et
al (54) and Taylor et
al (53), have a male
participation rate of 50 to 70%. However, a few studies, like Puca
et al (44), have a much
higher male representation.
Next, we performed a Kaplan-Meier survival analysis
(63), for which we prepared
Table I. This non-parametric
statistical technique is used in survival analysis to estimate the
survival function from lifetime data (64,65).
Table I includes the key data
points necessary to calculate and plot the survival curve. The
Status column was used to differentiate between the occurrence of
death (coded as 1) and censored data (coded as 0) (66). Censored data represents subjects who
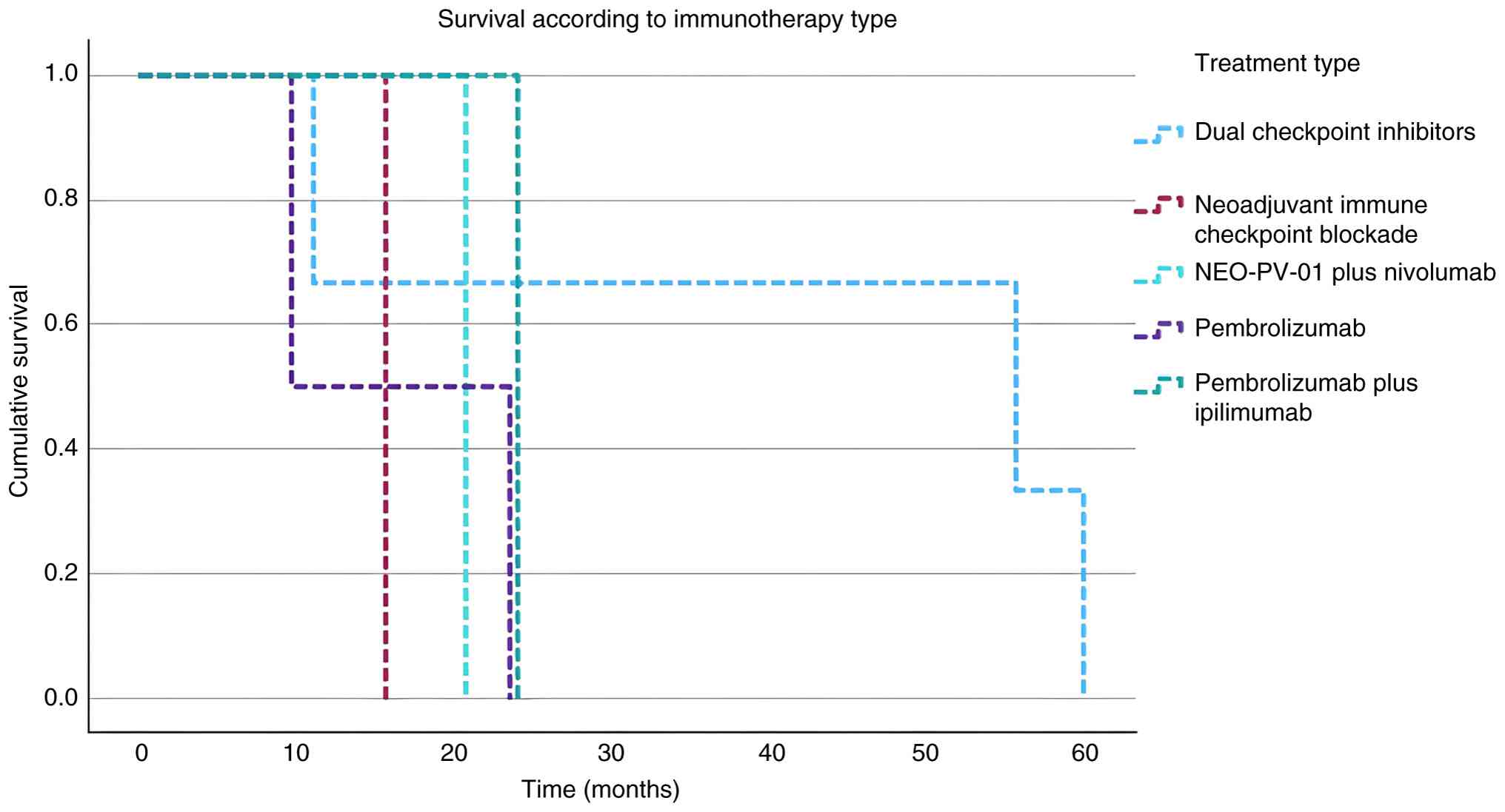
did not experience the event during the study period (67). The Kaplan-Meier survival curve
(Fig. 2) shows a steady decline in
survival probability over up to 60 months. The survival probability
begins at 1.0 (or 100%) and gradually decreases, with significant
drops occurring at various points, indicating times when multiple
events (such as deaths) likely occurred. The curve shows a gradual
decline in survival probability over time, indicating that events
(such as deaths or failures) occurred consistently throughout the
observed period. The curve reflects a steady decrease in survival
probability, with survival rates dropping significantly as time
progresses, particularly after around 20 to 30 months. Fig. 2 shows the Kaplan-Meier survival
curve based on treatment.
 | Table IKey points from NETs for Kaplan-Meier
analysis. Key variables extracted from selected studies for
Kaplan-Meier analysis, including survival time (months), event
status (death or censored), sex distribution, and treatment
category used for stratification. |
Table I
Key points from NETs for Kaplan-Meier
analysis. Key variables extracted from selected studies for
Kaplan-Meier analysis, including survival time (months), event
status (death or censored), sex distribution, and treatment
category used for stratification.
| First author
(year) | Time, months | Event status for OS
(0=censored; 1=death) | Male, % | Treatment type | Strata | (Refs.) |
|---|
| Patel, 2020 | 11 | 1 | 59 | Dual checkpoint
inhibitors | Immunotherapy | (41) |
| Marabelle,
2019 | 23.5 | 0 | 41.2 | Pembrolizumab | Immunotherapy | (42) |
| Puca, 2019 | 35 | 1 | 100 | Delta-like protein
3 | Targeted
therapy | (44) |
| Larkin, 2019 | 60 | 1 | 66 | Dual checkpoint
inhibitors | Immunotherapy | (45) |
| Morizane, 2022 | 12.5 | 0 | 68.8 | Etoposide and
Cisplatin | Chemotherapy | (47) |
| Amaria, 2018 | 15.6 | 1 | 75 | Neoadjuvant immune
checkpoint blockade | Immunotherapy | (48) |
| Motzer, 2022 | 55.7 | 0 | 73.7 | Dual checkpoint
inhibitor | Immunotherapy | (49) |
| Ott, 2020 | 20.7 | 1 | 68 | NEO-PV-01 plus
nivolumab | Immunotherapy +
vaccine | (50) |
| Eggermont,
2018 | 9.6 | 1 | 56 | Pembrolizumab | Immunotherapy | (52) |
| Olson, 2021 | 24 | 1 | 67 | Pembrolizumab plus
ipilimumab | Immunotherapy | (54) |
| Reidy-Lagunes,
2019 | 24 | 1 | 50 | Lu-satoreotide
tetraxetan | Radiolabelled
therapy | (59) |
| Iyer, 2020 | 32.7 | 1 | 53 | Nintedanib | Targeted
therapy | (60) |
Larkin et al (45) and Motzer et al (49) stand out with the highest OS, both
above 55 months. Larkin's study focused on skin melanoma and used
nivolumab and ipilimumab. Motzer's study, which focused on advanced
renal cell carcinoma, administered a combination of nivolumab and
ipilimumab, followed by sunitinib. Eggermont et al (52) show the lowest PFS and OS, both under
ten months, despite using pembrolizumab for treating small-cell
lung cancer. Marabelle et al (42) and Olson et al (54) have moderate OS (around 20 to 30
months) but relatively low PFS (under five months). Marabelle's
study involved pembrolizumab across multiple MSI-H/dMMR tumours,
while Olson's focused on skin melanoma with a combination of
pembrolizumab and low-dose Ipilimumab.
Morizane et al (47) and Patel et al (41) show a similar relationship between
PFS and OS, with PFS around ten months and OS around 20-25 months.
These studies involved Etoposide plus Cisplatin vs. Irinotecan plus
Cisplatin in multiple tumour types and ipilimumab plus nivolumab in
gastrointestinal and lung NETs, respectively. Iyer et al
(60) reported an OS of around 35
months, which is notable considering its moderate PFS of about 11
months. This study used nintedanib combined with octreotide LAR for
gastrointestinal and pancreatic NETs.
Table II presents
the log-rank test for NETs. For chemotherapy, the observed event
count is 1, with an expected frequency of 0.27, suggesting that
fewer events were anticipated based on the overall survival data.
Immunotherapy has an observed count of 7 events, closely matching
its expected frequency of 7.03, indicating that the survival
outcomes in this group align with expectations. In contrast, the
immunotherapy/vaccine group has 0 observed events against an
expected frequency of 0.39, suggesting that this treatment group is
outperforming expectations, with fewer patients experiencing the
event (death). Both radiotherapy and targeted therapy show 1
observed event each, compared to expected frequencies of 0.86 and
1.44, respectively.
| Table IILog-rank test for NETs. Observed and
expected event frequencies for each treatment category, used to
assess differences in survival distributions across groups using
the log-rank test. |
Table II
Log-rank test for NETs. Observed and
expected event frequencies for each treatment category, used to
assess differences in survival distributions across groups using
the log-rank test.
| Group | Observed
frequency | Expected
frequencies |
|---|
| Chemotherapy | 1 | 0.274242424 |
| Immunotherapy | 7 | 7.033982684 |
|
Immunotherapy/vaccine | 0 | 0.385353535 |
| Radiotherapy | 1 | 0.861544012 |
| Targeted
therapy | 1 | 1.444877345 |
The χ2 test statistic from Table III (χ2=2.47) falls
within the 95% region of acceptance, meaning that there is no
statistically significant difference in survival outcomes between
the five treatment groups. Any observed differences in survival may
be due to random variation rather than the effects of the different
treatments. The Φ effect size (Φ=0.80) indicates a moderate
relationship between the treatment groups and survival
outcomes.
| Table IIIχ² test for comparing the survival
distributions. χ² statistics, degrees of freedom, and effect size
(Φ coefficient) summarizing the comparison of survival
distributions among the different treatment modalities. |
Table III
χ² test for comparing the survival
distributions. χ² statistics, degrees of freedom, and effect size
(Φ coefficient) summarizing the comparison of survival
distributions among the different treatment modalities.
| Statistic | Value | Description |
|---|
| Number of groups
(k) | 5 | Number of
groups |
| Sample size
(n) | 10 | Sample size |
| χ² value | 2.465397 | χ² test
statistic |
| m | 0 | Estimated
parameters |
| DF | 4 |
DF=k-m-1=5-0-1=4 |
| Φ effect size | 0.796528 | Φ=√(χ2 /
n) |
Table IV as well as
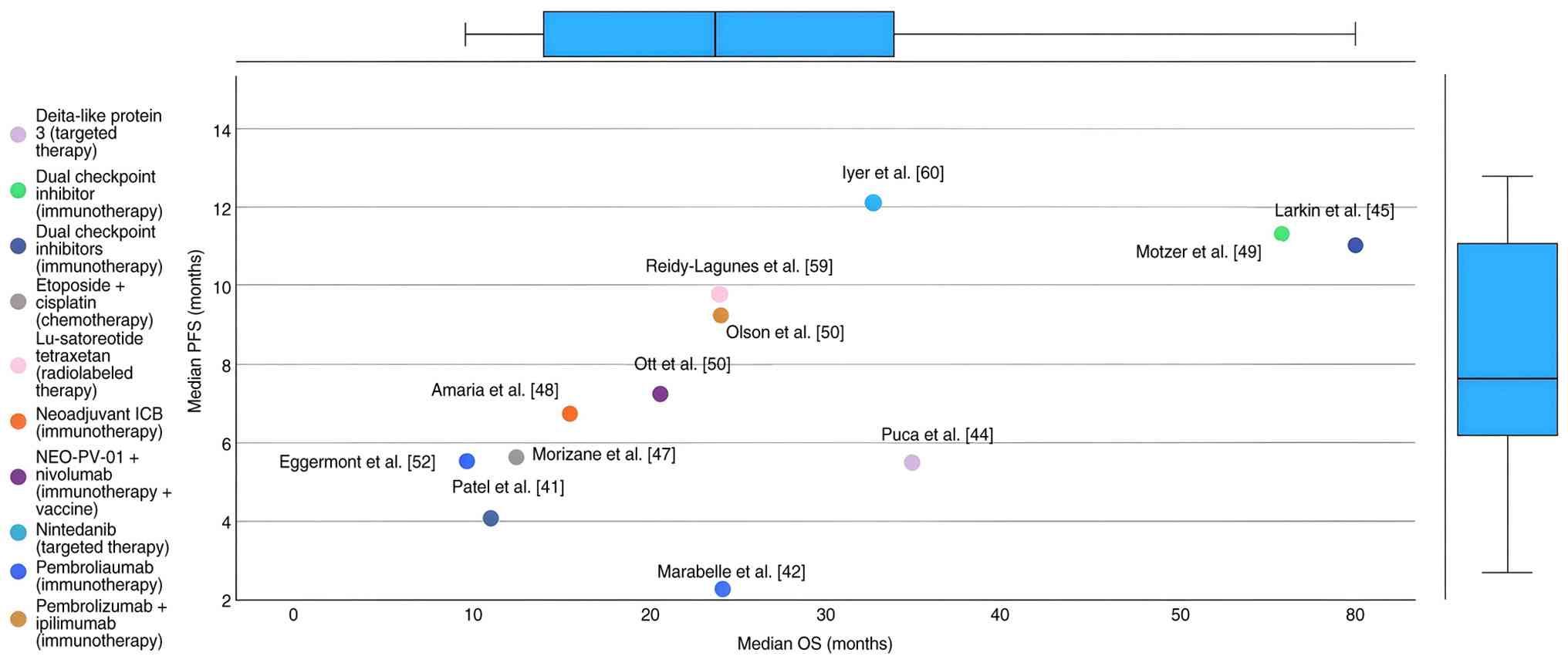
the scatter plot in Fig. 3
highlights significant variability in PFS and OS outcomes.
Treatments involving combinations of Nivolumab and Ipilimumab tend
to show higher OS, particularly in studies like Larkin et al
(45) and Motzer et al
(49). In contrast, Pembrolizumab
alone or in combination, shows more varied outcomes depending on
the tumour type and setting. The plot also reveals a pattern where
specific treatments provide long-term survival benefits even if
they don't immediately prolong PFS, as seen in studies like Olson
et al (54) and Iyer et
al (60).
| Table IVMedian PFS and OS according to
treatment type. Median PFS and OS values reported across studies,
stratified by treatment modality, illustrating variability in
survival outcomes among different therapeutic approaches. |
Table IV
Median PFS and OS according to
treatment type. Median PFS and OS values reported across studies,
stratified by treatment modality, illustrating variability in
survival outcomes among different therapeutic approaches.
| First author
(year) | Median PFS,
months | Median OS,
months | Strata/treatment
type | (Refs.) |
|---|
| Patel, 2020 | 4.1 | 11 | Dual checkpoint
inhibitors (immunotherapy) | (41) |
| Marabelle,
2019 | 2 | 23.5 | Pembrolizumab
(immunotherapy) | (42) |
| Puca, 2019 | 5.5 | 35 | Delta-like protein
3 (targeted therapy) | (44) |
| Larkin, 2019 | 11 | 60 | Dual checkpoint
inhibitors (immunotherapy) | (45) |
| Morizane, 2022 | 5.6 | 12.5 | Etoposide +
Cisplatin (chemotherapy) | (47) |
| Amaria, 2018 | 6.7 | 15.6 | Neoadjuvant ICB
(immunotherapy) | (48) |
| Motzer, 2022 | 11.2 | 55.7 | Dual checkpoint
inhibitor (immunotherapy) | (49) |
| Ott, 2020 | 7.2 | 20.7 | NEO-PV-01 +
Nivolumab (immunotherapy + vaccine) | (50) |
| Eggermont,
2018) | 5.5 | 9.6 | Pembrolizumab
(immunotherapy) | (52) |
| Olson, 2021 | 9.2 | 24 | Pembrolizumab +
Ipilimumab (immunotherapy) | (54) |
| Reidy-Lagunes,
2019 | 9.8 | 24 | Lu-satoreotide
tetraxetan (radiolabelled therapy) | (59) |
| Iyer, 2020 | 12.1 | 32.7 | Nintedanib
(targeted therapy) | (60) |
The middle blue dashed line at HR=1 indicates no
effect, serving as a reference point. Most studies have hazard
ratios that hover around the null value of 1, indicating that the
treatments under consideration have varying degrees of
effectiveness on PFS and OS. Studies like Tawbi et al
(43), Morizane et al
(47), and Rinke et al
(61) show hazard ratios greater
than 1, which could suggest a potential reduction in survival
compared to the baseline or control. Conversely, other studies like
Puca et al (44) and
Reidy-Lagunes et al (59)
have HRs below 1, indicating a possible survival benefit.
Risk of bias assessment using the Cochrane Risk of
Bias 2 (RoB 2) tool was performed only for randomized controlled
trials, in accordance with Cochrane recommendations. Observational
studies, retrospective analyses, single-arm phase I/II trials, and
basket trials without randomization were not suitable for RoB 2
assessment and were therefore excluded from this analysis. Of the
22 included studies, five met the criteria for randomized
controlled trials and were assessed for risk of bias (Tables V and VI). Two trials [Larkin et al
(45) and Motzer et al
(49)] were judged to have a low
overall risk of bias, with low risk across most bias domains. The
remaining three studies [Tawbi et al (43), Morizane et al (47) and Singh et al (62)] presented some concerns regarding
overall risk of bias, mainly related to deviations from intended
interventions or open-label study designs. No trial was classified
as having a high overall risk of bias.
| Table VStudy design of included clinical
studies. Distribution of the included studies according to study
design, including randomized controlled trials, single-arm phase II
trials, retrospective cohort studies, observational studies, and
exploratory basket trials. |
Table V
Study design of included clinical
studies. Distribution of the included studies according to study
design, including randomized controlled trials, single-arm phase II
trials, retrospective cohort studies, observational studies, and
exploratory basket trials.
| Study type | Number of
studies |
|---|
| Randomized
controlled trials | 5 |
| Single-arm phase II
trials | 9 |
| Retrospective
cohort studies | 4 |
| Observational
prospective studies | 3 |
| Basket/exploratory
trials | 1 |
| Table VIOverall risk of bias. Evaluation of
risk of bias across five domains: Randomization process, deviations
from intended interventions, missing outcome data, outcome
measurement, and selective reporting, for the five randomized
controlled trials included in the review, together with the overall
risk of bias judgment for each study. |
Table VI
Overall risk of bias. Evaluation of
risk of bias across five domains: Randomization process, deviations
from intended interventions, missing outcome data, outcome
measurement, and selective reporting, for the five randomized
controlled trials included in the review, together with the overall
risk of bias judgment for each study.
| First author
(year) | Bias arising from
the randomization process (Selection bias) | Bias due to
deviations from intended interventions (Performance bias) | Bias due to
missinge outcom data (Attrition bias) | Bias in measurement
of the outcome (Detection bias) | Bias in selection
of the reported (Reporting result bias) | Overall risk of
bias | (Refs.) |
|---|
| Larkin, 2019 | Low risk | Some concerns | Low risk | Low risk | Low risk | Low risk | (45) |
| Motzer, 2022 | Low risk | Low risk | Some concerns | Low risk | Low risk | Low risk | (49) |
| Tawbi, 2022 | Low risk | Some concerns | Low risk | Low risk | Low risk | Some concerns | (43) |
| Morizane, 2022 | Low risk | Some concerns | Low risk | Low risk | Low risk | Some concerns | (47) |
| Singh, 2024 | Low risk | High risk
(open-label design) | Some concerns | Low risk | Low risk | Some concerns | (62) |
Discussion
This systematic review provides an integrated
evaluation of therapeutic strategies across neuroendocrine tumours
and selected related malignancies, highlighting the diversity of
available treatments and their variable impact on survival
outcomes.
Across the included studies, immunotherapy emerged
as a central area of investigation. Dual checkpoint blockade with
nivolumab plus ipilimumab demonstrated durable benefits in several
cohorts, with overall survival exceeding 55 months in certain
populations, as shown by Larkin et al (45) and Motzer et al (49). These results confirm the potential
of immune-based regimens to extend long-term survival, even though
toxicity profiles and treatment costs remain limiting factors
(68,69). This finding is also stated by
Pánczél et al (70) who
concluded that dual checkpoint blockade with ipilimumab and
nivolumab achieved higher response and disease control rates,
albeit with increased toxicity, suggesting a potential benefit for
selected patients. In contrast, pembrolizumab monotherapy and
combination regimens yielded more heterogeneous outcomes. For
example, Eggermont et al (52) reported OS under 10 months in
small-cell lung cancer, whereas Marabelle et al (42) observed more favourable responses in
mismatch repair-deficient tumours. Similar to this, Hektoen et
al (71) noticed a doubling in
patients surviving more than 2 years when comparing improvement in
survival with pembrolizumab relative to previous platinum-based
chemotherapy. It continues to be used in the management of
high-grade or poorly differentiated NETs and related carcinomas.
The trial by Morizane et al (47) compared etoposide-cisplatin with
irinotecan-cisplatin in advanced digestive neuroendocrine
carcinoma, showing no significant difference in survival but
confirming both regimens as standard first-line options. Choucair
et al (72) investigated
Irinotecan in combination with cisplatin for treating advanced
poorly differentiated GEP-NETs, and in phase II of his trial, the
researchers found that the objective response rate was comparable
between IP and EP in small-cell NETs.
Targeted therapies also demonstrated clinically
relevant benefits. Nintedanib in non-pancreatic
gastroenteropancreatic NETs was associated with PFS of 11 months
and OS approaching 33 months. Similarly, rovalpituzumab tesirine,
directed at DLL3 in neuroendocrine prostate cancer, showed
promising efficacy.
Another notable therapeutic avenue is radiolabelled
peptide receptor therapy and other radiolabelled approaches. The
trial by Reidy-Lagunes et al (59) demonstrated that Lu-satoreotide
tetraxetan achieved a median PFS of 21 months and favourable
long-term OS rates in somatostatin receptor-positive NETs. This is
similar to what Wild et al (73) found. In their phase I/II study,
Lu-satoreotide tetraxetan, administered at a median cumulative
activity of 13.0 GBq over three cycles, has an acceptable safety
profile with a promising clinical response in patients with
progressive, SSTR-positive NETs.
From a clinical practice perspective, the findings
summarized support an individualized approach to neuroendocrine
tumour management. Peptide receptor radionuclide therapy
demonstrated the most durable disease control, with
177Lu-DOTATATE and Lu-satoreotide tetraxetan achieving
the longest PFS in somatostatin receptor-positive tumours (59,62).
In addition, dual immune checkpoint blockade with nivolumab and
ipilimumab was associated with the longest OS in their selected
cohorts (45,49). From a policy and future research
perspective, the heterogeneity of outcomes highlights the need for
equitable access to advanced therapies such as PRRT and combination
immunotherapy (59,62). Healthcare policies should facilitate
referral to specialized centres and support reimbursement for these
treatments. Future research should prioritize NET-specific
randomized trials and biomarker-driven approaches, building on
existing evidence for PRRT and immunotherapy (45,49,62),
to optimize patient selection, treatment sequencing, and long-term
outcomes.
Despite encouraging results, the variability of
outcomes across treatment modalities reflects the challenges of
managing NETs. Some regimens, such as checkpoint inhibitors, may
provide long-lasting benefit in a subset of patients but limited
responses in others. Moreover, the included studies varied
substantially in sample size, design, and endpoints. Thus, the
analysis indicates that immunotherapy, targeted agents, and
radiolabelled therapies offer meaningful clinical benefits for
selected patients with NETs outside the pancreas.
The overall certainty of evidence for the main
outcomes was considered moderate for PFS and OS in treatment
modalities supported by randomized controlled trials, particularly
for dual immune checkpoint inhibition and peptide receptor
radionuclide therapy. In contrast, the certainty of evidence was
low for outcomes derived primarily from single-arm phase II
studies, retrospective analyses, and exploratory trials, due to
limited sample sizes, lack of randomization, and heterogeneity in
tumour types and treatment regimens.
This study presents some limitations. First of all,
it includes a mix of RCTs, single-arm studies, and retrospective
analyses. This variability in study design introduces heterogeneity
that can affect the comparability of outcomes across studies.
Including studies with different methodologies, sample sizes, and
bias levels may have influenced the overall results. We included in
the analysis various cancer types, stages, and patient
demographics. While providing a broad overview of treatment
effects, this diversity also limits the ability to draw specific
conclusions about individual treatments' efficacy for particular
cancer subtypes. Differences in tumour biology, patient
characteristics, and previous treatments contribute to the
variability in survival outcomes. We also found many small sample
sizes and single-arm study designs, limiting the robustness and
generalizability of the results.
To sum up, immunotherapy, particularly dual
checkpoint blockade with nivolumab and ipilimumab, demonstrated the
most durable effects, with progression-free survival around 11
months and overall survival extending to 55-60 months in advanced
settings. Single-agent checkpoint inhibitors showed variable
results: pembrolizumab achieved PFS between 2 and 5 months, with OS
ranging from 9.6 months in small-cell lung cancer to 23.5 months in
mismatch repair-deficient tumours. Chemotherapy remained a mainstay
for high-grade carcinomas, with etoposide-cisplatin or
irinotecan-cisplatin achieving OS of approximately 11-12.5 months.
Targeted therapies, such as nintedanib, improved survival outcomes
(PFS 11 months; OS 32.7 months), indicating benefit in selected
cases. Radiolabelled therapies like Lu-satoreotide tetraxetan
further extended PFS to 21 months, with long-term survival rates of
85% at two years and 63% at three years. Collectively, results
emphasize tailored, multimodal strategies.
Supplementary Material
Selected studies representing NETs
(n=22). Summary of the included studies, detailing sample size,
patient demographics, tumour type, treatment regimen, treatment
duration, key findings, survival outcomes (PFS and OS), and
principal study limitations.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
ADC, SD, MC, LM, LSNC, ACIP, RAN, CAC and NC
contributed to the conceptualization and design of the study. ADC
and SD developed the methodology. MC and LM performed the formal
analysis. LSNC and ACIP were responsible for data curation. RAN and
CAC conducted the investigation and managed resources. NC performed
the statistical analysis and data validation. ADC drafted the
original manuscript. SD, MC and LM critically revised and edited
the manuscript. CAC generated figures and tables. NC supervised the
project. ADC and SD acquired funding. SD and ADC confirm the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Klöppel G: Neuroendocrine neoplasms:
Dichotomy, origin and classifications. Visc Med. 33:324–330.
2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Gheorghișan-Gălățeanu AA, Ilieșiu A,
Lambrescu IM and Țăpoi DA: The complex histopathological and
immunohistochemical spectrum of neuroendocrine tumors-an overview
of the latest classifications. Int J Mol Sci.
24(1418)2023.PubMed/NCBI View Article : Google Scholar
|
|
3
|
The International Agency for Research on
Cancer (IARC): Global Cancer Observatory. https://gco.iarc.who.int/. Accessed October 2,
2025.
|
|
4
|
Durma AD, Saracyn M, Kołodziej M,
Jóźwik-Plebanek K, Dmochowska B, Kapusta W, Żmudzki W, Mróz A,
Kos-Kudła B and Kamiński G: Epidemiology of neuroendocrine
neoplasms and results of their treatment with
[177Lu]Lu-DOTA-TATE or [177Lu]Lu-DOTA-TATE
and [90Y]Y-DOTA-TATE-A six-year experience in
high-reference polish neuroendocrine neoplasm center. Cancers
(Basel). 15(5466)2023.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Guccione L, Gough K, Drosdowsky A, Price
T, Pavlakis N, Wyld D, Ransom D, Michael M and Schofield P: The
unmet information needs, quality of life, and care experiences of
patients with neuroendocrine tumours (NETs) at follow-up: 6 months
from diagnosis. Support Care Cancer. 31(577)2023.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Helderman NC, Suerink M, Kilinç G, van den
Berg JG, Nielsen M and Tesselaar MET: Relation between WHO
classification and location- and functionality-based
classifications of neuroendocrine neoplasms of the digestive tract.
Neuroendocrinology. 114:120–133. 2024.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Park JH, Shin SJ, Jeon N and Lim BJ:
Clinicopathologic characteristics of neuroendocrine tumors with
assessment by digital image analysis for Ki-67 index with a focus
on the gastroenteropancreatic tract: A single-center study. Int J
Clin Exp Pathol. 16:225–234. 2023.PubMed/NCBI
|
|
8
|
Zandee WT, Kamp K, van Adrichem RC,
Feelders RA and de Herder WW: Effect of hormone secretory syndromes
on neuroendocrine tumor prognosis. Endocr Relat Cancer.
24:R261–R274. 2017.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Hofland J and de Herder WW:
Gastrointestinal neuroendocrine tumors and the carcinoid syndrome.
(Updated 2023 Aug 25). In: Endotext [Internet]. Feingold KR, Adler
RA, Ahmed SF, Anawalt B, Blackman MR, Chrousos G, Corpas E, de
Herder WW, Dhatariya K, Dungan K, et al (eds). MDText.com, Inc., South Dartmouth, MA, 2000.
|
|
10
|
Chan CS, Laddha SV, Lewis PW, Koletsky MS,
Robzyk K, Da Silva E, Torres PJ, Untch BR, Li J, Bose P, et al:
ATRX, DAXX or MEN1 mutant pancreatic neuroendocrine tumors are a
distinct alpha-cell signature subgroup. Nat Commun.
9(4158)2018.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Coman M, Hîncu M, Surlin P, Mateescu G,
Nechita A and Banu M: Comparative histomorphometric study of bone
tissue synthesized after electric and ultrasound stimulation. Rom J
Morphol Embryol. 52 (Suppl 1):S455–S458. 2011.PubMed/NCBI
|
|
12
|
Thakker RV: Multiple endocrine neoplasia
type 1 (MEN1) and type 4 (MEN4). Mol Cell Endocrinol. 386:2–15.
2014.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Feng Z, Wang L, Sun Y, Jiang Z, Domsic J,
An C, Xing B, Tian J, Liu X, Metz DC, et al: Menin and Daxx
interact to suppress neuroendocrine tumors through epigenetic
control of the membrane metallo-endopeptidase. Cancer Res.
77:401–411. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Metz DC and Jensen RT: Gastrointestinal
neuroendocrine tumors: Pancreatic endocrine tumors.
Gastroenterology. 135:1469–1492. 2008.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Sulciner ML and Clancy TE: Surgical
maknagement of pancreatic neuroendocrine tumors. Cancers (Basel).
15(2006)2023.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Lee DW, Kim MK and Kim HG: Diagnosis of
pancreatic neuroendocrine tumors. Clin Endosc. 50:537–545.
2017.
|
|
17
|
Neuroendocrine Tumor Research Foundation:
A new treatment option for NET patients. Retrieved from https://netrf.org/2025/03/26/a-new-treatment-option-for-net-patients/.
Accessed March 27, 2025.
|
|
18
|
Alkaissi H, Talvacchio S, Derkyi A, Gubbi
S, Pappo A, Gordon CM, Glod J, Zhuang Z and Pacak K: Belzutifan for
HIF2A-related pheochromocytoma and paraganglioma: A retrospective
study of real-world data. Endocr Pract. 32:201–205. 2026.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Prado-Wohlwend S, Bernal-Vergara JC,
Utrera-Costero A, Cañón-Sánchez JR, Agudelo-Cifuentes M and
Bello-Arques P: Endocrinology Working Group of the SEMNIM. Peptide
receptor radionuclide therapy with [177Lu]Lu-DOTA-TATE.
Rev Esp Med Nucl Imagen Mol (Engl Ed). 41:55–65. 2022.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Reddy RP, Ross Schmidtlein C, Giancipoli
RG, Mauguen A, LaFontaine D, Schoder H and Bodei L: The quest for
an accurate functional tumor volume with 68Ga-DOTATATE
PET/CT. J Nucl Med. 63:1027–1032. 2022.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Reubi JC and Schonbrunn A: Illuminating
somatostatin analog action at neuroendocrine tumor receptors.
Trends Pharmacol Sci. 34:676–688. 2013.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Soliman NA and Yussif SM: Ki-67 as a
prognostic marker according to breast cancer molecular subtype.
Cancer Biol Med. 13:496–504. 2016.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Souche R, Hobeika C, Hain E and Gaujoux S:
Surgical management of neuroendocrine tumours of the pancreas. J
Clin Med. 9(2993)2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
O'Neill CB, Atoria CL, O'Reilly EM, Henman
MC, Bach PB, Elkin EB, O'Neill CB, Atoria CL, O'Reilly EM, Henman
MC, et al: ReCAP: Hospitalizations in older adults with advanced
cancer: The role of chemotherapy. J Oncol Pract. 12:151–152,
e138-e148. 2016.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Siddiqui Z, Marginean H, Leung M, Asmis T,
Vickers M and Goodwin R: Real world use of lanreotide in
neuroendocrine tumors. J Gastrointest Oncol. 14:1488–1495.
2023.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Phan AT and Dave B: The pivotal role of
mammalian target of rapamycin inhibition in the treatment of
patients with neuroendocrine tumors. Cancer Med. 5:2953–2964.
2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Delbaldo C, Faivre S, Dreyer C and Raymond
E: Sunitinib in advanced pancreatic neuroendocrine tumors: Latest
evidence and clinical potential. Ther Adv Med Oncol. 4:9–18.
2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Deacu S, Neculai-Cândea L, Pricop S,
Aschie M, Mocanu L and Popa M: Vascular adhesive peptide-1 (VAP-1)
expression in wounds-a new vital reaction marker? Rom J Leg Med.
29:347–351. 2021.
|
|
29
|
Bundschuh RA, Habacha B, Lütje S and
Essler M: Therapy of patients with neuroendocrine
neoplasia-evidence-based approaches and new horizons. J Clin Med.
8(1474)2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Pan WX, Zhang XM, Hao SL and Han W:
Progress in immunotherapy for neuroendocrine neoplasm of the
digestive system. World J Gastroenterol. 29:4174–4185.
2023.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Page MJ, McKenzie JE, Bossuyt PM, Boutron
I, Hoffmann TC, Mulrow CD, Shamseer L, Tetzlaff JM, Akl EA, Brennan
SE, et al: The PRISMA 2020 statement: An updated guideline for
reporting systematic reviews. Syst Rev. 10(89)2021.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Covvey JR, McClendon C and Gionfriddo MR:
Back to the basics: Guidance for formulating good research
questions. Res Social Adm Pharm. 20:66–69. 2024.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Icahn School of Medicine at Mount Sinai:
Evidence based medicine: The PICO framework. https://libguides.mssm.edu/ebm/ebp_pico. Accessed
July 2, 2024.
|
|
34
|
MIT Libraries: Database search tips:
Boolean operators. https://libguides.mit.edu/c.php?g=175963&p=1158594.
Accessed July 2, 2024.
|
|
35
|
Cochrane Methods Bias: RoB 2: A revised
Cochrane risk-of-bias tool for randomized trials. https://methods.cochrane.org/bias/resources/rob-2-revised-cochrane-risk-bias-tool-randomized-trials.
Aaccessed July 12, 2024.
|
|
36
|
Marušić MF, Fidahić M, Cepeha CM, Farcaș
LG, Tseke A and Puljak L: Methodological tools and sensitivity
analysis for assessing quality or risk of bias used in systematic
reviews published in the high-impact anesthesiology journals. BMC
Med Res Methodol. 20(121)2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Kabir R, Syed HZ, Hayhoe R, Parsa AD,
Sivasubramanian M, Mohammadnezhad M, Sathian B, Kizhessery R, Jain
M, Gandhi PA, et al: Meta-analysis using SPSS: A simple guide for
clinicians, public health, and allied health specialists. Evidence.
2:1–24. 2024.
|
|
38
|
Kwak S: Are only P-values less than 0.05
significant? A P-value greater than 0.05 is also significant! J
Lipid Atheroscler. 12:89–95. 2023.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Page MJ, Moher D, Bossuyt PM, Boutron I,
Hoffmann TC, Mulrow CD, Shamseer L, Tetzlaff JM, Akl EA, Brennan
SE, et al: PRISMA 2020 explanation and elaboration: updated
guidance and exemplars for reporting systematic reviews. BMJ.
372(n160)2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Innocenti T, Feller D, Giagio S, Salvioli
S, Minnucci S, Brindisino F, Cosentino C, Piano L, Chiarotto A and
Ostelo R: Adherence to the PRISMA statement and its association
with risk of bias in systematic reviews published in rehabilitation
journals: A meta-research study. Braz J Phys Ther.
26(100450)2022.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Patel SP, Othus M, Chae YK, Giles FJ,
Hansel DE, Singh PP, Fontaine A, Shah MH, Kasi A, Baghdadi TA, et
al: A phase II basket trial of dual anti-CTLA-4 and anti-PD-1
blockade in rare tumors (DART SWOG 1609) in patients with
nonpancreatic neuroendocrine tumors. Clin Cancer Res. 26:2290–2296.
2020.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Marabelle A, Le DT, Ascierto PA, Di
Giacomo AM, De Jesus-Acosta A, Delord JP, Geva R, Gottfried M,
Penel N, Hansen AR, et al: Efficacy of pembrolizumab in patients
with noncolorectal high microsatellite instability/mismatch
repair-deficient cancer: Results from the phase II KEYNOTE-158
study. J Clin Oncol. 38:1–10. 2020.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Tawbi HA, Schadendorf D, Lipson EJ,
Ascierto PA, Matamala L, Castillo Gutiérrez E, Rutkowski P, Gogas
HJ, Lao CD, De Menezes JJ, et al: Relatlimab and nivolumab versus
nivolumab in untreated advanced melanoma. N Engl J Med. 386:24–34.
2022.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Puca L, Gavyert K, Sailer V, Conteduca V,
Dardenne E, Sigouros M, Isse K, Kearney M, Vosoughi A, Fernandez L,
et al: Delta-like protein 3 expression and therapeutic targeting in
neuroendocrine prostate cancer. Sci Transl Med.
11(eaav0891)2019.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Larkin J, Chiarion-Sileni V, Gonzalez R,
Grob JJ, Rutkowski P, Lao CD, Cowey CL, Schadendorf D, Wagstaff J,
Dummer R, et al: Five-year survival with combined nivolumab and
ipilimumab in advanced melanoma. N Engl J Med. 381:1535–1546.
2019.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Jonasch E, Donskov F, Iliopoulos O,
Rathmell WK, Narayan VK, Maughan BL, Oudard S, Else T, Maranchie
JK, Welsh SJ, et al: Belzutifan for renal cell carcinoma in von
hippel-lindau disease. N Engl J Med. 385:2036–2046. 2021.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Morizane C, Machida N, Honma Y, Okusaka T,
Boku N, Kato K, Nomura S, Hiraoka N, Sekine S, Taniguchi H, et al:
Effectiveness of etoposide and cisplatin vs irinotecan and
cisplatin therapy for patients with advanced neuroendocrine
carcinoma of the digestive system: The TOPIC-NEC phase 3 randomized
clinical trial. JAMA Oncol. 8:1447–1455. 2022.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Amaria RN, Reddy SM, Tawbi HA, Davies MA,
Ross MI, Glitza IC, Cormier JN, Lewis C, Hwu WJ, Hanna E, et al:
Neoadjuvant immune checkpoint blockade in high-risk resectable
melanoma. Nat Med. 24:1649–1654. 2018.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Motzer RJ, McDermott DF, Escudier B,
Burotto M, Choueiri TK, Hammers HJ, Barthélémy P, Plimack ER, Porta
C, George S, et al: Conditional survival and long-term efficacy
with nivolumab plus ipilimumab versus sunitinib in patients with
advanced renal cell carcinoma. Cancer. 128:2085–2097.
2022.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Ott PA, Hu-Lieskovan S, Chmielowski B,
Govindan R, Naing A, Bhardwaj N, Margolin K, Awad MM, Hellmann MD,
Lin JJ, et al: A phase Ib trial of personalized neoantigen therapy
plus anti-PD-1 in patients with advanced melanoma, non-small cell
lung cancer, or bladder cancer. Cell. 183:347–362.e24.
2020.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Dummer R, Hauschild A, Santinami M,
Atkinson V, Mandalà M, Kirkwood JM, Chiarion Sileni V, Larkin J,
Nyakas M, Dutriaux C, et al: Five-year analysis of adjuvant
dabrafenib plus trametinib in stage III melanoma. N Engl J Med.
383:1139–1148. 2020.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Eggermont AMM, Blank CU, Mandala M, Long
GV, Atkinson V, Dalle S, Haydon A, Lichinitser M, Khattak A,
Carlino MS, et al: Adjuvant pembrolizumab versus placebo in
resected stage III melanoma. N Engl J Med. 378:1789–1801.
2018.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Taylor MH, Lee CH, Makker V, Rasco D,
Dutcus CE, Wu J, Stepan DE, Shumaker RC and Motzer RJ: Phase IB/II
trial of lenvatinib plus pembrolizumab in patients with advanced
renal cell carcinoma, endometrial cancer, and other selected
advanced solid tumors. J Clin Oncol. 38:1154–1163. 2020.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Olson DJ, Eroglu Z, Brockstein B,
Poklepovic AS, Bajaj M, Babu S, Hallmeyer S, Velasco M, Lutzky J,
Higgs E, et al: Pembrolizumab plus ipilimumab following
anti-PD-1/L1 failure in melanoma. J Clin Oncol. 39:2647–2655.
2021.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Chauhan A, Farooqui Z, Murray LA, Weiss
HL, War Myint Z, Raajasekar AKA, Evers BM, Arnold S and Anthony L:
Capecitabine and temozolomide in neuroendocrine tumor of unknown
primary. J Oncol. 2018(3519247)2018.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Topalian SL, Bhatia S, Amin A, Kudchadkar
RR, Sharfman WH, Lebbé C, Delord JP, Dunn LA, Shinohara MM,
Kulikauskas R, et al: Neoadjuvant nivolumab for patients with
resectable merkel cell carcinoma in the CheckMate 358 trial. J Clin
Oncol. 38:2476–2487. 2020.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Pavel M, Lahner H, Hörsch D, Rinke A,
Denecke T, Koch A, Regnault B, Helbig D, Hoffmanns P and Raderer M:
Combined lanreotide autogel and temozolomide treatment of
progressive pancreatic and intestinal neuroendocrine tumors: The
phase II SONNET study. Oncologist. 29:e643–e654. 2024.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Leidner R, Conlon K, McNeel DG,
Wang-Gillam A, Gupta S, Wesolowski R, Chaudhari M, Hassounah N, Lee
JB, Ho Lee L, et al: First-in-human phase I/Ib study of NIZ985, a
recombinant heterodimer of IL-15 and IL-15Rα, as a single agent and
in combination with spartalizumab in patients with advanced and
metastatic solid tumors. J Immunother Cancer.
11(e007725)2023.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Reidy-Lagunes D, Pandit-Taskar N,
O'Donoghue JA, Krebs S, Staton KD, Lyashchenko SK, Lewis JS, Raj N,
Gönen M, Lohrmann C, et al: Phase I trial of well-differentiated
neuroendocrine tumors (NETs) with radiolabeled somatostatin
antagonist 177Lu-satoreotide tetraxetan. Clin Cancer Res.
25:6939–6947. 2019.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Iyer RV, Konda B, Fountzilas C, Mukherjee
S, Owen D, Attwood K, Wang C, Maguire O, Minderman H, Suffren SA,
et al: Multicenter phase 2 trial of nintedanib in advanced
nonpancreatic neuroendocrine tumors. Cancer. 126:3689–3697.
2020.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Rinke A, Maintz C, Müller L, Weber MM,
Lahner H, Pavel M, Saeger W, Houchard A, Ungewiss H and Petersenn
S: Multicenter, observational study of lanreotide autogel for the
treatment of patients with neuroendocrine tumors in routine
clinical practice in Germany and Austria. Exp Clin Endocrinol
Diabetes. 129:500–509. 2021.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Singh S, Halperin D, Myrehaug S, Herrmann
K, Pavel M, Kunz PL, Chasen B, Tafuto S, Lastoria S, Capdevila J,
et al: [177Lu]Lu-DOTA-TATE plus long-acting octreotide
versus high-dose long-acting octreotide for the treatment of newly
diagnosed, advanced grade 2-3, well-differentiated,
gastroenteropancreatic neuroendocrine tumours (NETTER-2): An
open-label, randomised, phase 3 study. Lancet. 403:2807–2817.
2024.
|
|
63
|
Rich JT, Neely JG, Paniello RC, Voelker
CC, Nussenbaum B and Wang EW: A practical guide to understanding
Kaplan-Meier curves. Otolaryngol Head Neck Surg. 143:331–336.
2010.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Dudley WN, Wickham R and Coombs N: An
introduction to survival statistics: Kaplan-Meier analysis. J Adv
Pract Oncol. 7:91–100. 2016.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Goel MK, Khanna P and Kishore J:
Understanding survival analysis: Kaplan-Meier estimate. Int J
Ayurveda Res. 1:274–278. 2010.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Riffenburgh RH (ed): Chapter 25 - Survival
and time-series analysis. In: Statistics in Medicine. 2nd edition.
Academic Press, pp487-519, 2006.
|
|
67
|
Turkson AJ, Ayiah-Mensah F, Nimoh V and
Tang N: Handling censoring and censored data in survival analysis:
A standalone systematic literature review. Int J Math Math Sci.
2021:1–16. 2021.
|
|
68
|
Ro C, Chai W, Yu VE and Yu R: Pancreatic
neuroendocrine tumors: Biology, diagnosis, and treatment. Chin J
Cancer. 32:312–324. 2013.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Kurakawa KI, Okada A, Manaka K, Konishi T,
Jo T, Ono S, Uda K, Michihata N, Matsui H, Fushimi K, et al:
Clinical characteristics and incidences of benign and malignant
insulinoma using a national inpatient database in Japan. J Clin
Endocrinol Metab. 106:3477–3486. 2021.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Pánczél G, Horváth P, Temaj E, Czirbesz K,
Kispál MT, Fröhlich G and Balatoni T: Real-world outcomes of
ipilimumab-nivolumab vs Anti-PD-1 monotherapy in metastatic uveal
melanoma: A single-center retrospective study. Cancers (Basel).
17(3521)2025.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Hektoen HH, Tsuruda KM, Fjellbirkeland L,
Nilssen Y, Brustugun OT and Andreassen BK: Real-world evidence for
pembrolizumab in non-small cell lung cancer: A nationwide cohort
study. Br J Cancer. 132:93–102. 2025.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Choucair K, Odabashian R, Reddy SN, Azmi
AS and Saif MW: An update on novel pharmacotherapies for the
treatment of neuroendocrine tumors. Int J Mol Sci.
26(11095)2025.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Wild D, Grønbæk H, Navalkissoor S, Haug A,
Nicolas GP, Pais B, Ansquer C, Beauregard JM, McEwan A, Lassmann M,
et al: A phase I/II study of the safety and efficacy of
[177Lu]Lu-satoreotide tetraxetan in advanced
somatostatin receptor-positive neuroendocrine tumours. Eur J Nucl
Med Mol Imaging. 51:183–195. 2023.PubMed/NCBI View Article : Google Scholar
|