Introduction
According to the World Health Organization,
approximately 43% of adults worldwide drink alcohol, and 3 million
people died from alcohol consumption-related causes in
2016(1). Alcohol is a major global
health risk factor and is considered to be the cause of many
diseases, including cardiovascular disease, diabetes, cancer, and
liver disease (2). Alcohol-related
liver disease (ALD) is a spectrum of disorders that include
alcoholic steatosis, steatohepatitis, fibrosis, and cirrhosis
(3). Since the late-ALD stage liver
damage associated with cirrhosis cannot be repaired, requiring
liver transplantation (4),
steatosis treatment should be administered in the early stage of
ALD.
Hepatic steatosis is an early symptom seen in
alcohol-related conditions, and it occurs in more than 90% of binge
drinkers (5). Alcoholic fatty liver
is characterized by the accumulation of fat in hepatocytes
(5). Ethanol (EtOH) induces lipid
accumulation by inducing the synthesis of fatty acids in the liver
or reducing fatty acid oxidation (6). Additionally, it promotes triglyceride
accumulation in liver through lipogenesis, leading to alcoholic
steatosis (7). Fatty acids are
synthesized by fat-producing enzymes, including fatty acid synthase
(FASN) and stearoyl-CoA desaturase 1 (SCD1), which are regulated by
the transcription factor sterol regulatory element-binding protein
1 (SREBP-1) (8). Ethanol induces
SREBP-1 cleavage increasing SREBP-1c production, and activated
SREBP-1c induces the expression of lipogenic enzymes that enhance
fatty acid synthesis in the liver (9). Fatty acids produced in the liver are
broken down via β-oxidation; however, EtOH interferes with this
process (9). Once activated,
peroxisome proliferator-activated receptor alpha (PPARα) enhances
the expression of proteins involved in fatty acid oxidation, but
EtOH reduces the expression of PPARα, inhibiting fatty acid
oxidation, and thus, fat accumulates in the liver (10). Inhibiting EtOH-induced increases in
lipid synthesis or decreases in fatty acid oxidation is a promising
treatment strategy for EtOH-induced hepatic steatosis.
Recent studies have shown that physiologically
active compounds derived from natural resources, such as seaweed,
can regulate metabolic pathways and alleviate liver damage
(11,12). Seaweed contains compounds with a
variety of effects, such as antioxidant (13), anti-inflammation (14), and anti-obesity (15) activities; therefore, it is widely
used in metabolic disease research. One of the most consumed
seaweed species in Korea is Capsosiphon fulvescens. Previous
studies have shown that C. fulvescens affects lipid
metabolism and obesity (12,16).
Islam et al (17)
investigated the association between three compounds separated from
C. fulvescens by EtOH extraction, along with an aldose
reductase inhibitor related to diabetes. Chalinasterol (CA;
24-methylenecholesterol), one of the three extracted compounds,
showed potential as an aldose reductase inhibitor but was less
effective than the other two. However, the pharmacological
functions of CA beyond aldose reductase inhibition remain unclear,
and its effects on hepatic lipid metabolism have not been
investigated. Thus, this study aimed to determine whether CA
attenuates EtOH-induced lipid accumulation in the liver and
investigate the mechanisms underlying its effects in cell and mouse
models.
Materials and methods
Cell cultures and treatments
Mouse hepatocyte cell line AML12 was cultured in
Dulbecco's modified Eagle's medium (DMEM)/F-12 nutrient mixture
medium. Human hepatocyte cell line Huh7 was cultured in DMEM
containing 15 mM HEPES, L-glutamine, sodium bicarbonate. Both media
were supplemented with fetal bovine serum at 10% and
penicillin/streptomycin at 1%, and all cultures were kept at 37˚C
in a humidified incubator with 5% CO2. To establish
early-stage alcoholic liver injury (steatosis) in vitro,
both AML12 and Huh7 cells were treated with 100 mM EtOH, a
well-established concentration for inducing intracellular lipid
accumulation without causing excessive cell death, for 24 h with or
without a 12 h pre-treatment with 4 µM CA. To minimize the effects
of EtOH evaporation and ensure consistent exposure, the culture
medium was replaced with fresh medium containing 100 mM EtOH every
12 h. The CA was purchased from MedChemExpress (Monmouth Junction,
USA) and dissolved in dimethyl sulfoxide (DMSO) to prepare a stock
solution, which was then diluted in the culture medium to a final
concentration of 4 µM. As a positive control for AMP-activated
protein kinase (AMPK) activation, cells were pre-treated with a 500
µM 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR)
solution for 12 h and then exposed to 100 mM EtOH for 24 h,
following the same treatment schedule used for CA.
Animals
Seven-week-old male C57BL/6 mice (Central Lab.
Animal Inc., Korea) were maintained in an air-conditioned room
(21-25˚C, 40-60% humidity) with a 12 h light/dark cycle. To
investigate the early stages of ALD, particularly hepatic
steatosis, liver injury was induced using the
chronic-plus-single-binge EtOH feeding model (NIAAA model) as
described in a previous study (18). This model was specifically chosen
because it more effectively replicates the acute-on-chronic
clinical features of human steatosis than traditional chronic
feeding models (18). The mice were
fed with the Lieber-DeCarli (LDC) diet ad libitum for the
first 5 days to allow them to adapt to a liquid diet. Subsequently,
they were randomly assigned to one of four groups: (1) control, (2) control+CA, (3) EtOH, and (4) CA+EtOH. For 10 days, the EtOH-fed
groups were given free access to an LDC diet containing 5% (v/v)
EtOH, while the control groups were fed with the standard LDC diet.
The mice in the CA-treated group were intraperitoneally (i.p.)
injected with 37.5 µg/kg CA once every 2 days. To prepare the
injection solution, CA was initially dissolved in DMSO and then
diluted to the required concentration in sterile phosphate-buffered
saline (PBS). The in vivo dose of CA was determined based on
its maximum solubility while strictly maintaining the final DMSO
concentration below 0.1% (v/v) in the PBS-based vehicle to
eliminate any potential vehicle-induced hepatotoxicity. On day 11,
the mice were orally administered 5 g/kg of EtOH for the EtOH-fed
groups or 9 g/kg of maltose dextrin for the control-fed groups.
After 9 h, the mice were sacrificed using CO2 with a
displacement rate of 30% of the chamber volume per minute. The
animal experiments were approved by the Chonnam National University
Institutional Animal Care and Use Committee (CNU
IACUC-YB-2024-123).
Serum chemistry measurement
To obtain mouse serum samples, CO2 was
administrated to anesthetize the mice, and then, their blood was
collected via cardiac puncture. The levels of alanine transaminase
(ALT) and aspartate transaminase (AST) in the serum were determined
using a Catalyst Dx chemistry analyzer (IDEXX Laboratories,
Korea).
Histological techniques
Excised livers were fixed in 10% formaldehyde in a
phosphate buffer and processed into frozen sections via routine
methods. The specimens were sectioned at 4 µm using a cryostat
microtome (Leica Biosystems, Nussloch, Germany; CM1950) from the
Department of Biological Sciences, College of Natural Sciences,
Chonnam National University. The tissues were stained with
hematoxylin and eosin (H&E) for histological assessment or 0.5%
oil red o solution for the analysis of hepatic lipid accumulation.
The stained samples were visualized using a Cytation™ 5 cell
imaging multimode reader (BioTek, USA), and quantification analyses
were performed using ImageJ software (National Institutes of
Health, USA).
MTT assay
An MTT assay was conducted using the MTT reagent
(Sigma-Aldrich, USA) to analyze cell viability. Cells were seeded
in 96-well plates at a density of 1x104 and cultured for
14-16 h. They were incubated with 0, 50, 100, 500 nM, 1, and 4 µM
CA for 24 h. Then, 10 µl of MTT was added to each well, and the
plate was incubated at 37˚C for 4 h. The medium was discarded, 100
µl of DMSO was added to each well, and the plate was shaken at
20-25˚C for 15 min. Absorbance was then measured at 550 nm by using
a microplate reader (BioTek, USA). The entire MTT assay was
performed in the dark.
Quantitative PCR
Total RNA was extracted from cell lysates and mouse
tissues using the TRIzol reagent, and cDNA was synthesized from 2
µg of RNA in a total volume of 20 µl using a random primer cocktail
and reverse transcriptase (Invitrogen, USA). The primers employed
for quantitative PCR were designed using PrimerBank (Table SI), and the specificity of each
primer was determined by a BLAST search. Quantitative PCR was
performed in 15 µl reaction volumes using SYBR Green/Rox qPCR Mix
(Enzynomics, Korea; cat. no. RT500M). The mRNA expression levels of
target genes were normalized relative to β-actin and compared among
treatment groups via the ΔΔCt method. The specificity of each
reaction was confirmed by melting curve analysis.
Western blot analysis
Proteins were extracted using RIPA buffer
(Elabscience, China) containing protease and phosphatase inhibitors
(Thermo Scientific, USA), and the extracted proteins were
quantified using the Bradford assay. One hundred µg of total
proteins were then separated via sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and transferred to
polyvinylidene fluoride membranes. The membranes were blocked with
5% (v/v) skim milk and then incubated overnight at 4˚C with the
primary antibody (diluted at 1:1,000): rabbit anti-phospho-AMPKα
(Cell Signaling Technology, USA; cat. no. #2531), rabbit anti-AMPKα
(Cell Signaling Technology; cat. no. #2532) or mouse anti-β-actin
(Santa Cruz Biotechnology, USA; cat. no. sc-47778). Afterward, they
were washed and incubated with either horseradish
peroxidase-conjugated goat anti-rabbit IgG secondary antibody
(Santa Cruz Biotechnology, USA; cat. no. sc-2357) or m-IgGκ binding
protein-horseradish peroxidase (Santa Cruz Biotechnology; cat. no.
sc-516102), both diluted at 1:10,000, at room temperature for 1 h.
They were washed again, and protein bands were detected using the
FUSION SOLO 2X imaging system (Vilber, France). Quantification
analyses were performed using ImageJ software.
Oxidative stress assays
To measure malondialdehyde (MDA), an indicator of
lipid peroxidation, cells were collected and homogenized in
ice-cold PBS. The MDA levels were then determined using a TBARS
assay kit (Cayman Chemical, USA) according to the manufacturer's
instructions. The resulting MDA values were normalized to the total
protein concentration.
To measure hydrogen peroxide
(H2O2) levels, intracellular
H2O2 was quantified using a Hydrogen Peroxide
Assay kit (Biomax, Republic of Korea) following the manufacturer's
protocol. Cells were lysed using the provided assay buffer, and the
supernatants were collected for the assay, with
H2O2 concentrations normalized to the total
protein content.
Hepatic triglyceride
quantification
Hepatic triglyceride (TG) contents were quantified
using a commercial colorimetric assay kit (Biomax) according to the
manufacturer's instructions. Liver tissues were homogenized in
lysis buffer and centrifuged to remove insoluble debris. The
supernatants were collected for the assay, and TG concentrations
were normalized to total protein content. Results were expressed as
nmol TG/mg protein.
Statistical analyses
Data were statistically analyzed using GraphPad
Prism 5 (GraphPad Software, USA). Results are expressed as means ±
the standard error of the mean (SEM). The term ‘n’ indicates
independent biological replicates (independent experiments
performed on separate occasions). Unless otherwise specified,
within each biological replicate, samples were measured in
technical duplicates, and the values were averaged to generate a
single value per biological replicate. Normality and homogeneity of
variance were assessed for each variable prior to statistical
testing. To assess statistical significance, unpaired two-tailed
Student's t-tests were used for comparisons between two groups,
while one-way analyses of variance (ANOVAs) followed by Tukey's
post hoc tests were employed for multiple-group comparisons.
Significance was accepted at P<0.05.
Results
CA reduces ethanol-induced lipid
accumulation in hepatocytes
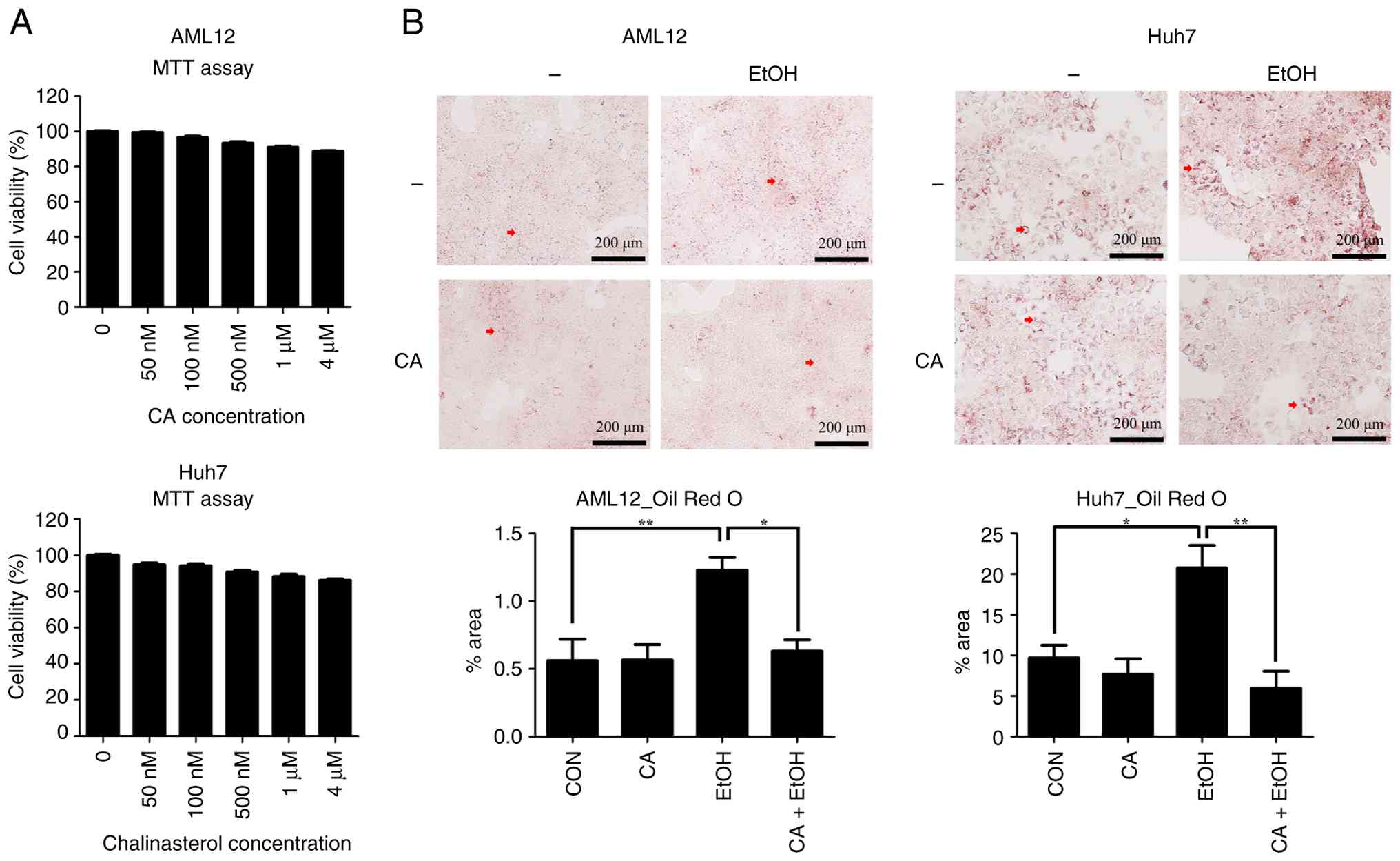
The effect of CA on the cell viability of
hepatocytes was assessed. Specifically, the viability of AML12 and
Huh7 cells exposed to 50, 100, 500 nM, 1, and 4 µM CA for 24 h was
evaluated. Up to 4 µM, CA did not affect cell viability (Fig. 1A). Therefore, 4 µM was selected as
the treatment concentration in later assays.
Previous studies have reported that fat accumulation
in hepatocytes is induced as an initial liver injury response to
EtOH (5,19). Thus, we evaluated the effect of CA
on EtOH-induced lipid accumulation in hepatocytes. We found that
EtOH treatment induced lipid droplet accumulation, but the CA
pre-treatment reduced this accumulation (Fig. 1B). Treatment with CA alone had no
effect. Therefore, CA attenuated EtOH-induced lipid
accumulation.
CA does not affect lipogenesis but
enhances β-oxidation gene expression to attenuate lipid
accumulation
EtOH-induced lipid accumulation is induced by
increased lipogenesis and/or decreased β-oxidation. To investigate
the mechanism by which CA regulates hepatic lipid accumulation, we
examined the expression levels of the genes involved in lipogenesis
and β-oxidation in human and mouse hepatocytes. The mRNA levels of
SREBP-1c, FASN, and SCD1 were not
significantly altered by CA treatment either alone or in
combination with EtOH treatment (Fig.
2A). This result indicates that CA did not suppress
EtOH-induced hepatic lipid accumulation by inhibiting lipogenesis.
Similarly, the CA treatment did not attenuate EtOH-induced reactive
oxygen species (ROS) production, as measured by
H2O2 and MDA levels (Fig. S1). Therefore, the protective effect
of CA was unlikely mediated by oxidative stress reduction.
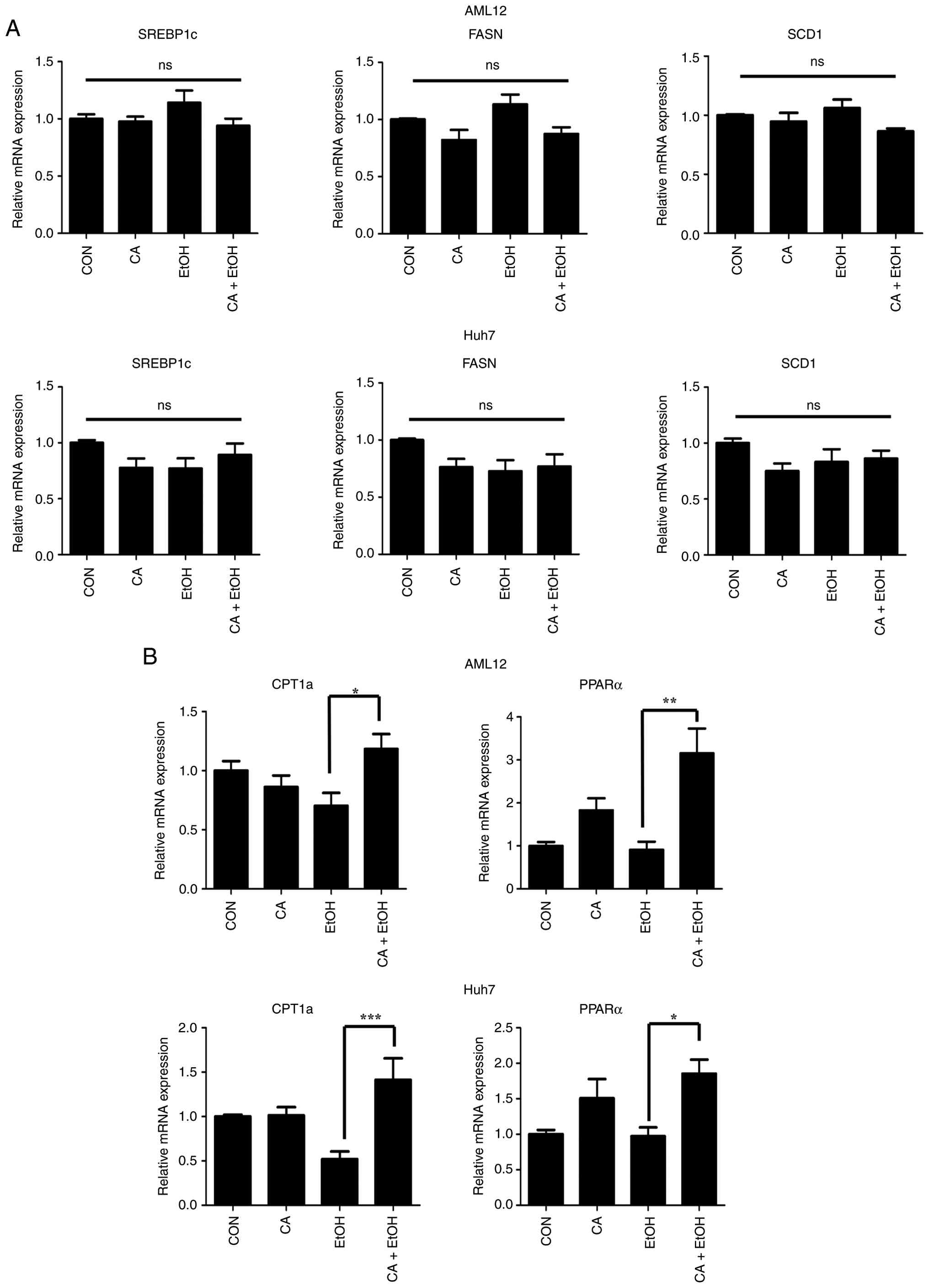 | Figure 2CA enhances β-oxidation, reducing
lipid accumulation. (A) The mRNA expression of lipogenesis-related
genes SREBP-1c, FASN and SCD1 in AML12 and
Huh7 cells treated with 100 mM EtOH for 24 h with or without a 12 h
4 µM CA pre-treatment. (B) The mRNA expression of
β-oxidation-related genes CPT1a and PPARα in AML12
and Huh7 cells treated with 100 mM of EtOH for 24 h after 12 h
pre-treatment with 4 µM CA. Data are presented as means ± the SEM.
*P<0.05, **P<0.01 and
***P<0.001. CA, Chalinasterol; EtOH, ethanol; ns, not
significant; CON, control; SREBP-1, sterol regulatory
element-binding protein 1; FASN, fatty acid synthase; SCD1,
stearoyl-CoA desaturase 1; CPT1A, carnitine palmitoyltransferase
1A; PPARα, peroxisome proliferator-activated receptor α. |
In contrast, co-treatment with CA and EtOH
significantly upregulated the expression of β-oxidation-related
genes, including carnitine palmitoyltransferase 1A (CPT1a)
and PPARα, compared with that of EtOH treatment alone
(Fig. 2B). These findings show that
CA ameliorated EtOH-induced lipid accumulation by promoting fatty
acid oxidation. In addition, AICAR, used as a positive control,
increased AMPK phosphorylation in EtOH-treated hepatocytes, and CA
similarly elevated p-AMPK levels (Fig.
S2), supporting involvement of AMPK signaling in CA-induced
upregulation of β-oxidation under EtOH exposure.
CA alleviates early EtOH-induced
hepatic injury and lipid accumulation in vivo
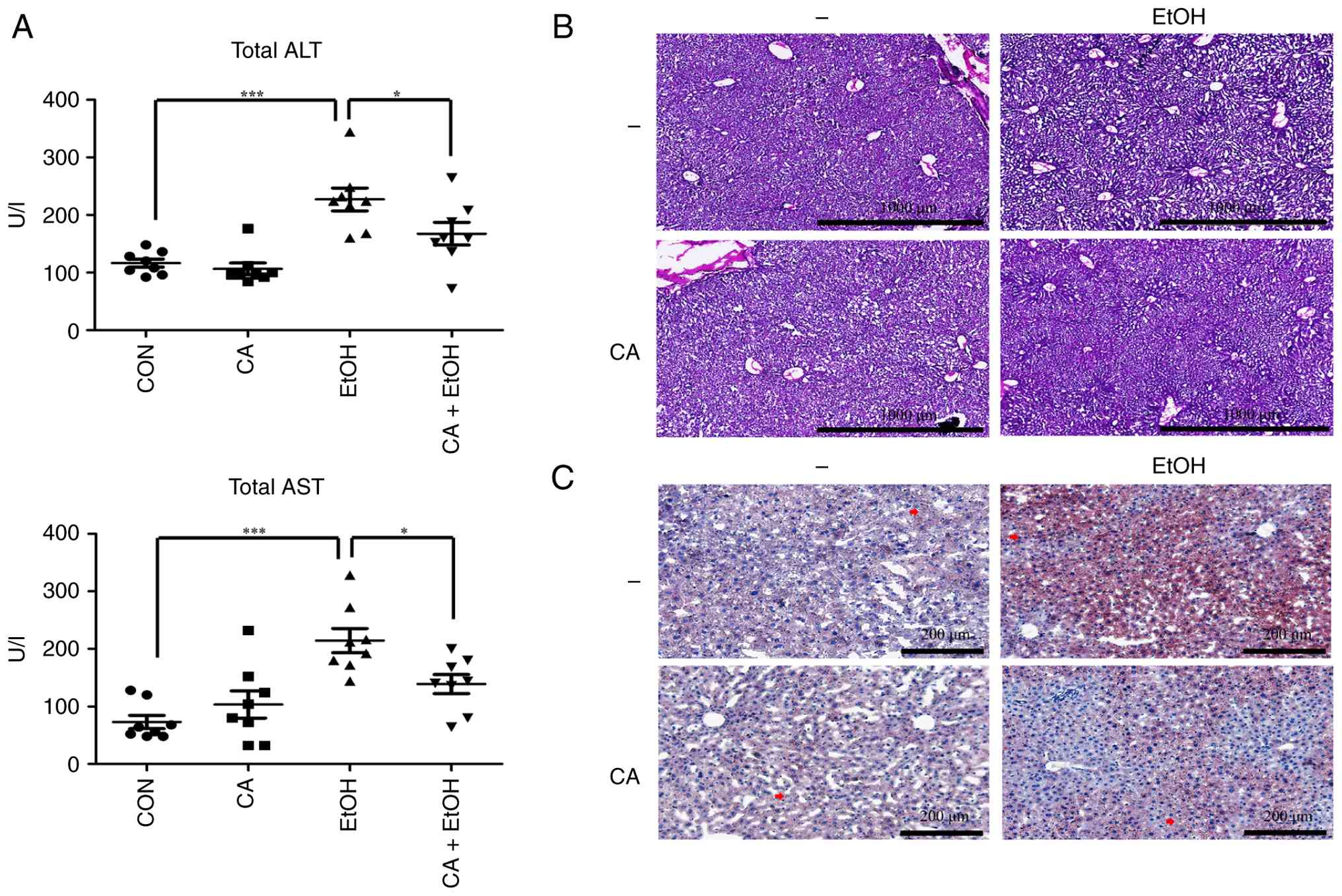
To evaluate the protective effect of CA against
EtOH-induced hepatic steatosis in vivo, we assessed the
serum levels of liver injury markers. Intraperitoneal
administration of CA alone did not significantly alter serum ALT
and AST levels compared to those in the control mice, indicating
that the selected dose of CA did not induce systemic hepatotoxicity
(Fig. 3A). In contrast, EtOH
administration significantly increased serum ALT and AST levels,
whereas the co-treatment with CA attenuated this increase (Fig. 3A). This suggests that CA mitigates
early EtOH-induced liver injury.
While histological analyses revealed no hepatic
damage in the EtOH-treated mice (Fig.
3B), EtOH administration caused lipid droplets to accumulate in
liver tissues. The CA treatment produced no detectable difference
in lipid distribution compared to the control group, but CA
administration markedly reduced the EtOH-induced lipid accumulation
(Fig. 3C). Consistent with these
histological findings, biochemical quantification showed that EtOH
increased hepatic TG levels, and the CA co-treatment significantly
reduced EtOH-induced TG accumulation (Fig. S3). Collectively, these data
indicate that CA attenuate EtOH-induced liver injury and steatosis
primarily by suppressing hepatic lipid accumulation.
CA enhances β-oxidation-related gene
expression and AMPK activation in vivo
To elucidate the molecular mechanism underlying the
protective effect of CA in vivo, we examined the expression
levels of lipid metabolism-related genes in liver tissues.
Consistent with the in vitro findings, compared to the
control group, CA administration did not significantly affect the
expression of lipogenic genes SREBP-1c, FASN, and
SCD1 (Fig. 4A). Quantitative
PCR analyses showed that CA also did not alter the mRNA levels of
these lipogenic genes in EtOH-treated mice (Fig. 4A). However, while showing no effect
on lipogenesis, the CA treatment significantly upregulated the
expression of genes related to fatty acid oxidation. While CA
administration alone slightly elevated the mRNA levels of
CPT1a compared to the control group, a significant
upregulation of both CPT1a and PPARα was seen in the
CA+EtOH group when compared to the EtOH group (Fig. 4B). Therefore, CA promoted fatty acid
β-oxidation in the liver, particularly under EtOH-induced stress
conditions.
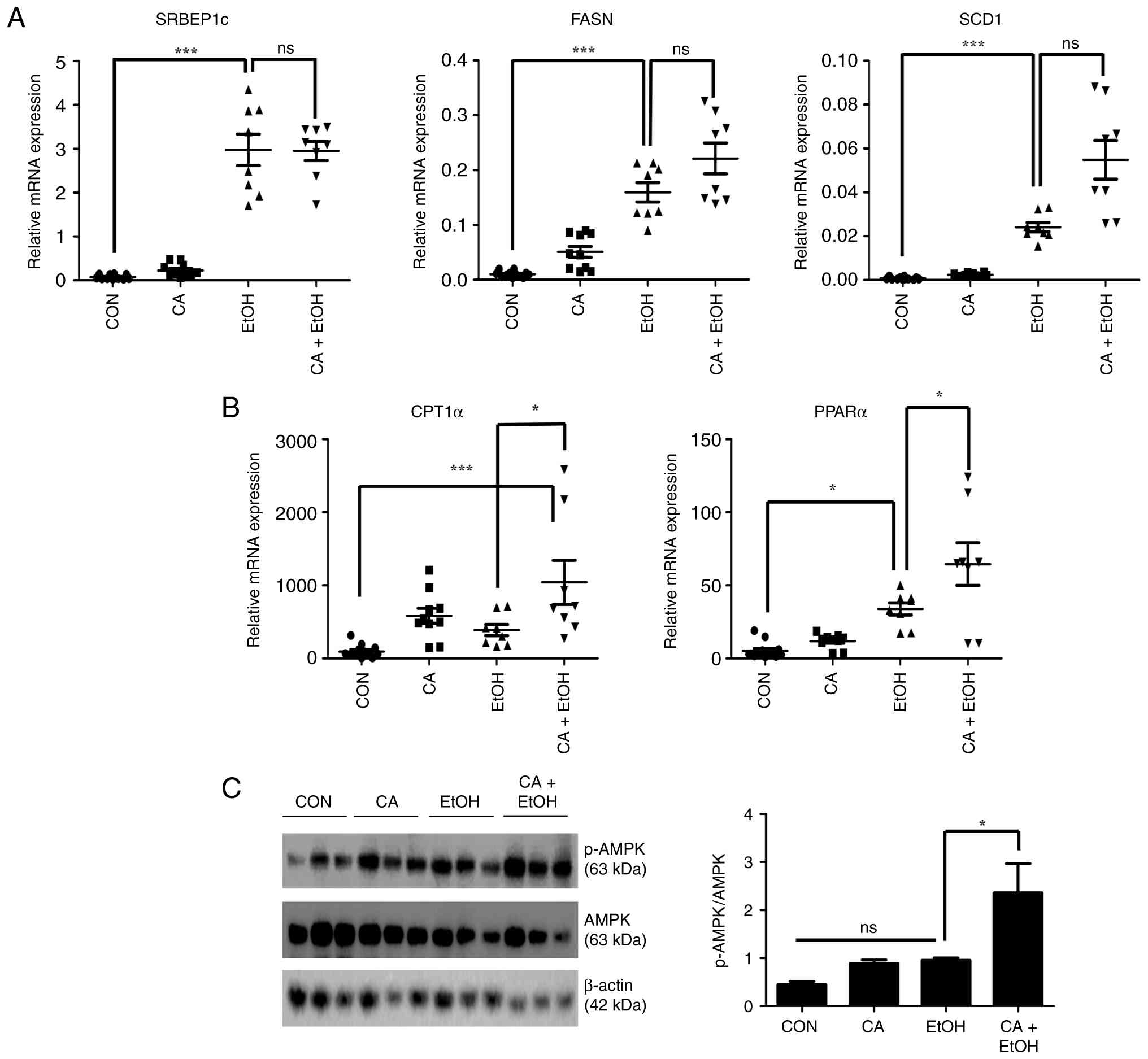 | Figure 4CA enhances β-oxidation-related gene
expression and activates AMPK signaling in vivo. (A) The
relative mRNA expression levels of lipogenesis-related genes
SREBP-1c, FASN, and SCD1 in liver tissues of
ethanol (EtOH)-treated mice with or without CA administration at
37.5 µg/kg, determined using quantitative PCR (n=8 per group). (B)
The relative mRNA expression levels of β-oxidation-related genes
CPT1a and PPARα in liver tissues of EtOH-treated mice
with or without CA administration at 37.5 µg/kg (n=8 per group).
(C) Protein levels of phosphorylated AMPK and AMPK in liver tissues
of control and EtOH-fed mice with or without CA administration at
37.5 µg/kg. Data are presented as means ± the SEM.
*P<0.05, and ***P<0.001. CA,
Chalinasterol; AMPK, AMP-activated protein kinase; CPT1A, carnitine
palmitoyltransferase 1A; EtOH, ethanol; FASN, fatty acid synthase;
PPARα, peroxisome proliferator-activated receptor α; SCD1,
stearoyl-CoA desaturase 1; SREBP-1, sterol regulatory
element-binding protein 1; ns, not significant. |
Protein analysis revealed the status of AMPK
activation. Alone, CA administration showed a tendency to increase
p-AMPK levels compared to the control group, but notably, p-AMPK
levels were clearly greater in CA+EtOH group mice than in EtOH
group mice (Fig. 4C). Thus, CA
appears to activate the AMPK pathway, which may contribute to the
upregulation of β-oxidation and the attenuation of hepatic
EtOH-induced liver injury due to fat accumulation.
Discussion
Alcohol-related liver disease remains a major global
health issue, and the earliest and most reversible stage in its
progression is alcoholic steatosis (20). Because late-stage ALD has no
effective therapy other than liver transplantation (4), treatments targeting hepatic steatosis
are critical. In the present study, CA attenuated EtOH-induced
hepatic lipid accumulation in vitro and in vivo
primarily by enhancing fatty acid β-oxidation rather than
suppressing lipogenesis. These findings suggest that CA may help
manage lipid metabolism during the early stages of EtOH
exposure.
Previous studies have established that ethanol
promotes hepatic steatosis by increasing lipogenesis via SREBP-1
signaling (9) and by impairing
lipid oxidation through the suppression of PPARα signaling
(10). Consistent with this
finding, our results showed that EtOH treatment induced lipid
droplet accumulation in hepatocytes and liver tissues. However, the
CA pre-treatment did not affect the expression of the lipogenic
genes SREBP-1c, FASN, and SCD1; thus, CA did
not act by inhibiting EtOH-induced fatty acid synthesis.
Furthermore, while oxidative stress is a known driver of ALD, CA
treatment did not significantly reduce EtOH-induced ROS production
in our cell models. This finding could be attributed to several
factors, including the specific ROS targeted or the time point used
for detection in our assays. Since EtOH-induced ROS generation
occurs in waves, our snapshot measurement might have missed
transient antioxidant effects. However, CA may exhibit a regulatory
role on lipid metabolism pathways that is distinct from direct
antioxidant scavenging. Thus, our results suggest that the
protective effect of CA is primarily mediated through metabolic
reprogramming rather than a direct reduction of oxidative stress.
It significantly upregulated the expression levels of CPT1a
and PPARα, key regulators of fatty acid β-oxidation, in
EtOH-treated hepatocytes and the liver tissues of EtOH-fed mice.
Additionally, the CA co-treatment significantly increased levels of
phosphorylated AMPK, a major regulator of energy metabolism that
facilitates lipid catabolism under stress conditions (21). Therefore, CA may activate the
AMPK-PPARα axis, promoting fatty acid oxidation and, consequently,
reducing lipid accumulation in the liver.
Interestingly, CA-induced AMPK activation is
consistent with the mechanism of metformin, a classical AMPK
activator widely used for metabolic disorders (22). However, metformin frequently causes
gastrointestinal intolerance and although rare, carries a risk of
lactic acidosis in susceptible patients (23). As a bioactive dietary compound, CA
may have potential as a nutritional adjunct or preventive strategy
for early-stage alcoholic steatosis, particularly in settings where
long-term tolerability is prioritized. Nevertheless, direct
head-to-head comparisons with metformin, to assess efficacy,
dose-response patterns, and safety/toxicology profiling, will be
required to define its translational value.
Importantly, our in vivo data showed that the
serum levels of the liver injury markers ALT and AST were increased
by EtOH exposure, and this increase was significantly attenuated by
CA administration, although histological evidence of liver damage
was not observed at the EtOH dose used. Considering that ALT and
AST serum levels are used as sensitive early indicators of liver
injury (24), even though
histological changes were not evident, these altered serum levels
suggest that CA has the potential to alleviate early-stage liver
damage, highlighting its potential role in supporting liver health
during early EtOH-induced stress.
Our findings are consistent with previous reports,
which showed that natural compounds can modulate hepatic lipid
metabolism via AMPK and PPARα pathways. Various natural chemicals,
including total saponins from Panax japonicas (25) and gallic acid (26) weaken lipid accumulation in
hepatocytes by activating AMPK signaling and promoting fatty acid
oxidation. These studies support the potential of natural compounds
as therapeutic agents in lipid metabolism disorders. Therefore, CA
promotes fatty acid oxidation and reduces EtOH-induced fat
accumulation. Consequently, lipid metabolism in the liver is
restored, showing that CA is a potential candidate for further
studies related to alcohol-associated liver injury.
Nonetheless, this study has several limitations.
Only the early-stage effects of CA were evaluated, and its
long-term effects on fibrosis or cirrhosis were not characterized.
Further studies using chronic ALD models will be required to
evaluate CA's efficacy against steatohepatitis and fibrosis.
Furthermore, while our study focused on the downstream effects of
CA on lipid metabolism, we cannot entirely rule out the possibility
that CA indirectly influences hepatic steatosis by modulating
EtOH-metabolizing enzymes, such as alcohol dehydrogenase or
aldehyde dehydrogenase. Further research should investigate whether
CA affects the rate of EtOH metabolism to fully elucidate its
protective mechanisms. Moreover, although AMPK was activated, we
did not directly confirm the link between AMPK signaling and
downstream gene regulation through inhibitor or knockout
approaches. Future studies involving AMPK or PPARα loss-of-function
models should be employed to validate the proposed mechanism.
Lastly, the pharmacokinetic properties and bioavailability of CA
were not assessed. Although effective concentrations were used
in vitro and in vivo in this study, the mechanism
through which CA is absorbed, metabolized, and distributed in the
body remains unclear. This information is crucial for devising
dosing strategies and determining clinical potential. Despite these
limitations, our study provides important insights into the early
intervention potential of CA in EtOH-induced lipid accumulation and
a basis for performing future mechanistic and translational
investigations.
In conclusion, our study provides evidence that CA
reduces EtOH-induced hepatic lipid accumulation and early liver
injury by enhancing β-oxidation through AMPK-PPARα pathway
activation. Given the global burden of ALD and the lack of
effective treatments for advanced stages, CA represents a potential
candidate for early preventive interventions aimed at mitigating
alcoholic steatosis.
Supplementary Material
H2O2/MDA assay
results for AML12 and Huh7 cells with or without 100 mM EtOH and 4
μM CA treatment. Data are presented as means ± SEM. *
*P<0.01. CA, Chalinasterol; EtOH, ethanol; MDA,
malondialdehyde; ns, not significant; CON, control.
Protein analysis results for
phosphorylated AMPK and AMPK in AML12 and Huh7 cells. Cells were
treated with 4 μM CA or 500 μM AICAR (positive
control) for 12 h and then treated with 100 mM EtOH for 24 h. Data
are presented as means ± the SEM. *P<0.05 and
***P<0.001. CA, Chalinasterol; AICAR,
5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside; EtOH, ethanol;
AMPK, AMP-activated protein kinase.
Hepatic TG contents in liver tissues
of EtOH-treated mice with or without CA at 37.5 μg/kg (n=8
per group). Data are presented as means ± the SEM.
*P<0.05, and ***P<0.001. TG,
triglyceride; EtOH, ethanol; CA, Chalinasterol; CON, control.
Sequences of oligonucleotide
primers.
Acknowledgements
Not applicable.
Funding
Funding: This work was supported by the National Research
Foundation of Korea (grant no. RS-2023-00251463) and Chonnam
National University (grant no. 2024-1145-01).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
JHK conceptualized and designed this study,
collected, assembled and analyzed data, and wrote the manuscript.
MYK collected, assembled and analyzed data. SO conceptualized and
designed this study. DHL conceptualized and designed this study,
collected, assembled, analyzed and interpreted data, wrote the
manuscript, and provided financial support. JHK and DHL confirm the
authenticity of all the raw data. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
The animal experiments were approved by the Chonnam
National University Institutional Animal Care and Use Committee
(approval no. CNU IACUC-YB-2024-123).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
World Health Organization: Global status
report on alcohol and health 2018. World Health Organization,
Geneva, 2018.
|
|
2
|
Rehm J, Baliunas D, Borges GL, Graham K,
Irving H, Kehoe T, Parry CD, Patra J, Popova S, Poznyak V, et al:
The relation between different dimensions of alcohol consumption
and burden of disease: An overview. Addiction. 105:817–843.
2010.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ohashi K, Pimienta M and Seki E: Alcoholic
liver disease: A current molecular and clinical perspective. Liver
Res. 2:161–172. 2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Schuppan D and Afdhal NH: Liver cirrhosis.
Lancet. 371:838–851. 2008.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Gao B and Bataller R: Alcoholic liver
disease: Pathogenesis and new therapeutic targets.
Gastroenterology. 141:1572–1585. 2011.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Lieber CS: Effects of ethanol upon lipid
metabolism. Lipids. 9:103–116. 1974.PubMed/NCBI
|
|
7
|
You M and Arteel GE: Effect of ethanol on
lipid metabolism. J Hepatol. 70:237–248. 2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Horton JD, Goldstein JL and Brown MS:
SREBPs: Activators of the complete program of cholesterol and fatty
acid synthesis in the liver. J Clin Invest. 109:1125–1131.
2002.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
You M and Crabb DW: Recent advances in
alcoholic liver disease II. Minireview: Molecular mechanisms of
alcoholic fatty liver. Am J Physiol Gastrointest Liver Physiol.
287:G1–G6. 2004.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Galli A, Pinaire J, Fischer M, Dorris R
and Crabb DW: The transcriptional and DNA binding activity of
peroxisome proliferator-activated receptor alpha is inhibited by
ethanol metabolism: A novel mechanism for the development of
ethanol-induced fatty liver. J Biol Chem. 276:68–75.
2001.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Menaa F, Wijesinghe U, Thiripuranathar G,
Althobaiti NA, Albalawi AE, Khan BA and Menaa B: Marine
algae-derived bioactive compounds: A new wave of nanodrugs? Mar
Drugs. 19(484)2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Yun MY, Lee JS, Kim BS and Choi HJ:
Capsosiphon fulvescens extracts improve obesity-associated
metabolic disorders and hepatic steatosis in high-fat diet-induced
obese mice. Anim Sci J. 89:589–596. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
de Almeida CL, Falcão Hde S, Lima GR,
Montenegro Cde A, Lira NS, de Athayde-Filho PF, Rodrigues LC, de
Souza Mde F, Barbosa-Filho JM and Batista LM: Bioactivities from
marine algae of the genus Gracilaria. Int J Mol Sci. 12:4550–4573.
2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Fernando IS, Nah JW and Jeon YJ: Potential
anti-inflammatory natural products from marine algae. Environ
Toxicol Pharmacol. 48:22–30. 2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Wan-Loy C and Siew-Moi P: Marine algae as
a potential source for anti-obesity agents. Mar Drugs.
14(222)2016.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Kwon MJ and Nam TJ: Effects of mesangi
(Capsosiphon fulvecens) powder on lipid metabolism in high
cholesterol fed rats. J Korean Soc Food Sci Nutr. 35:530–535.
2006.
|
|
17
|
Islam MN, Choi SH, Moon HE, Park JJ, Jung
HA, Woo MH, Woo HC and Choi JS: The inhibitory activities of the
edible green alga Capsosiphon fulvescens on rat lens aldose
reductase and advanced glycation end products formation. Eur J
Nutr. 53:233–242. 2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Bertola A, Mathews S, Ki SH, Wang H and
Gao B: Mouse model of chronic and binge ethanol feeding (the NIAAA
model). Nat Protoc. 8:627–637. 2013.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Tsukamoto H, Machida K, Dynnyk A and
Mkrtchyan H: ‘Second hit’ models of alcoholic liver disease. Semin
Liver Dis. 29:178–187. 2009.PubMed/NCBI View Article : Google Scholar
|
|
20
|
O'shea RS, Dasarathy S and McCullough AJ:
Practice Guideline Committee of the American Association for the
Study of Liver Diseases; Practice Parameters Committee of the
American College of Gastroenterology. Alcoholic liver disease.
Hepatology. 51:307–328. 2010.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Hardie DG: AMP-activated/SNF1 protein
kinases: Conserved guardians of cellular energy. Nat Rev Mol Cell
Biol. 8:774–785. 2007.PubMed/NCBI View
Article : Google Scholar
|
|
22
|
Viollet B, Guigas B, Sanz Garcia N,
Leclerc J, Foretz M and Andreelli F: Cellular and molecular
mechanisms of metformin: An overview. Clin Sci (Lond). 122:253–270.
2011.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Mazumder A, Singh A and Sujeet JHA: A
Review on metformin: Clinical significance and side effects. Int J
Pharm Res. 13(60)2021.
|
|
24
|
Louvet A and Mathurin P: Alcoholic liver
disease: Mechanisms of injury and targeted treatment. Nat Rev
Gastroenterol Hepatol. 12:231–242. 2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Qiu L, Feng R, Wu QS, Wan JB and Zhang QW:
Total saponins from Panax japonicus attenuate acute alcoholic liver
oxidative stress and hepatosteatosis by p62-related Nrf2 pathway
and AMPK-ACC/PPARα axis in vivo and in vitro. J Ethnopharmacol.
317(116785)2023.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zhang J, Zhang W, Yang L, Zhao W, Liu Z,
Wang E and Wang J: Phytochemical gallic acid alleviates
nonalcoholic fatty liver disease via AMPK-ACC-PPARa axis through
dual regulation of lipid metabolism and mitochondrial function.
Phytomedicine. 109(154589)2023.PubMed/NCBI View Article : Google Scholar
|