1. Introduction
Sphingolipids are bioactive lipids that function
both as structural components of eukaryotic membranes and as
signaling mediators that regulate cell fates and tissue homeostasis
(1). Among these metabolites,
sphingosine-1-phosphate (S1P) has emerged as a central regulator of
proliferation, survival, migration, vascular integrity and immune
cell trafficking (2). Together with
ceramide and ceramide-1-phosphate, S1P occupies a pivotal position
within the sphingolipid network, in which shifts in the metabolite
balance can profoundly influence cell behaviors (3).
In cancer, the biological effects of S1P are
especially complex (4). Rather than
generating a uniform downstream response, S1P propagates signals
through five G protein-coupled receptors, S1PR1-S1PR5, each of
which differs in expression patterns, downstream coupling and
biological output (5). As a result,
consequences of S1P signaling in tumors are determined not only by
ligand availability but also by receptor identity and the receptor
state (6). Depending on the tumor
type and cellular context, individual S1P receptors can promote or
restrain proliferation, survival, invasion, angiogenesis, immune
evasion and metastatic dissemination (7).
Much of the research has focused on elevated S1P
production as a driver of tumor progression, particularly through
increased sphingosine kinase (SphK) activity and altered
sphingolipid catabolism (8).
Although this metabolic dimension is clearly important, a
ligand-centric framework does not adequately explain the
receptor-specific and context-dependent outputs observed across
cancers (7). Instead, the
pleiotropic effects of S1P signaling are shaped by multilevel
reprogramming of the S1P-S1PR axis, in which isoform-specific
receptor expression is integrated with transcriptional and
epigenetic regulation, microRNA (miRNA)-mediated repression and
post-translational modifications (6,9). These
regulatory layers converge with altered spatiotemporal
organization, including receptor trafficking and
compartmentalization, to convert a homeostatic signaling system
into a context-dependent driver of malignancy (5,10).
Malignant progression is therefore driven not simply by activation
of the S1P pathway, but by reprogramming of the regulatory
architecture that governs it (6).
The present review examined the S1P-S1PR system as a
dynamically reprogrammed signaling network in cancer. It first
summarized the receptor-specific functions of S1PR1-S1PR5 across
tumor and microenvironmental contexts and then discussed the
mechanisms through which tumors reprogram this axis, including
transcriptional activation, epigenetic remodeling, loss of
miRNA-mediated restraint, altered receptor trafficking and
compartmentalization and coupling to ligand-rich metabolic states.
Finally, it considered how this framework may inform therapeutic
strategies that more effectively target both S1P production and
receptor-mediated signaling in a biomarker-guided manner.
2. The S1P-S1PR axis in physiology and
cancer
S1P is generated from sphingosine by the sphingosine
kinases sphingosine kinase 1 (SphK1) and SphK2 and signals through
five G protein-coupled receptors, S1PR1-S1PR5 (2,11,12). A
defining feature of this pathway is its ‘inside-out’ signaling,
whereby intracellularly generated S1P is exported by transporters
such as SPNS2 and ATP-binding cassette family members, enabling
autocrine and paracrine activation of cell-surface S1PRs (2,13).
Under physiological conditions, this system contributes to vascular
regulation, immune cell trafficking and tissue homeostasis. In
cancer, however, it is frequently dysregulated through increased
ligand production, reduced catabolism, and altered receptor
expression (Fig. 1) (7,8,11).
Importantly, signaling output is determined not only by ligand
abundance, but also by receptor identity, localization and cellular
context (5,6,11).
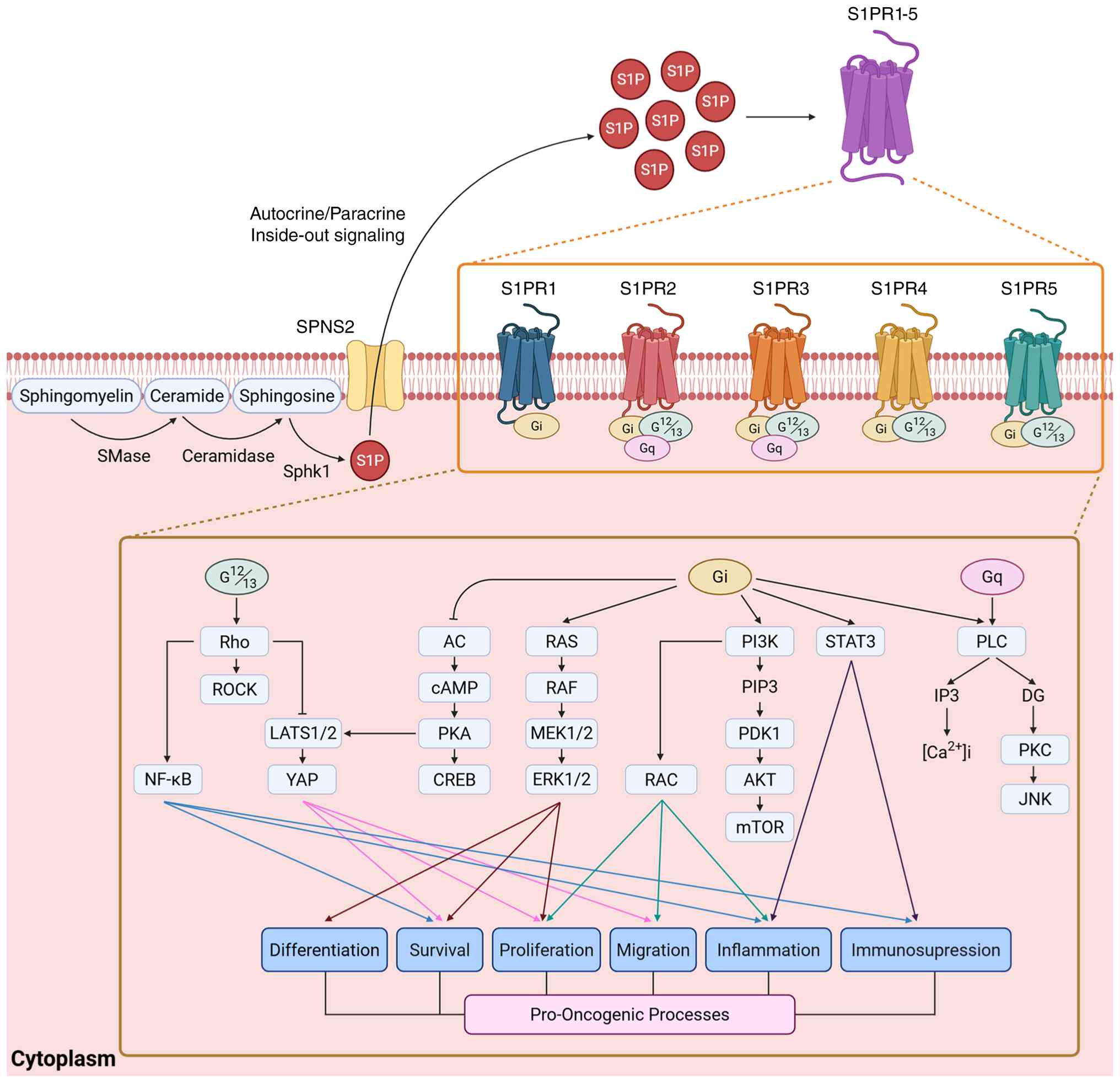 | Figure 1Overview of the S1P-S1PR signaling
axis in cancer. Sphingomyelin is hydrolyzed to ceramide by SMase
and ceramide is subsequently converted to sphingosine by
ceramidase. Sphingosine is phosphorylated by SphK1 to generate S1P,
which is exported by transporters such as SPNS2. Extracellular S1P
binds S1PR isoforms and activates downstream Gi-, Gq- and
G12/13-dependent signaling pathways, including Rho, AC, Ras, PI3K,
STAT3 and PLC, thereby promoting tumor-associated processes. S1P,
sphingosine-1-phosphate; S1PR, sphingosine-1-phosphate receptor;
SMase, sphingomyelinase; SphK1, sphingosine kinase 1; SPNS2,
spinster homolog 2; Gi, G protein alpha i; Gq, G protein alpha q;
G12/13, G protein alpha 12/13; AC, adenylyl cyclase; PI3K,
phosphatidylinositol 3-kinase; STAT3, signal transducer and
activator of transcription 3; PLC, phospholipase C. |
3. Receptor-specific outputs of S1PR
signaling in cancer
Activation of S1PR1-S1PR5 engages
phosphatidylinositol 3-kinase (PI3K)-AKT, mitogen-activated protein
kinase-extracellular signal-regulated kinase (ERK), Rho-Rho kinase
(ROCK), phospholipase C (PLC)-Ca²+, signal transducer
and activator of transcription 3 (STAT3) and nuclear factor-κB
signaling, thereby shaping malignant phenotypes that include
proliferation, survival, migration, epithelial-to-mesenchymal
transition (EMT), angiogenesis, immune modulation and therapeutic
responses (Table I) (8,11,14,15).
However, these outputs are highly context dependent and vary
according to receptor subtype, cellular lineage and
microenvironmental state (5,7,8,14,16).
 | Table ITumor-associated functions, G-protein
coupling and principal downstream signaling pathways of S1P
receptors (S1PR1-S1PR5). |
Table I
Tumor-associated functions, G-protein
coupling and principal downstream signaling pathways of S1P
receptors (S1PR1-S1PR5).
| Receptor | Primary role in
tumors | Main G-protein
coupling | Major downstream
pathways | Key tumor-relevant
functions | (Refs.) |
|---|
| S1PR1 | Mostly
pro-oncogenic | Gi | PI3K-AKT,
Ras-ERK1/2, PLC, Rac, STAT3, YAP | Promotes
proliferation, survival, migration, the EMT, metastasis, and
angiogenesis; sustains persistent STAT3 signaling and may reinforce
senescence resistance, including through YAP-associated signaling
in ovarian cancer. | (8,11,12,82) |
| S1PR2 | Mainly
tumor-suppressive, but strongly context-dependent | G12/13, Gq, weak
Gi | Rho-ROCK, Rac
inhibition; in some liver settings PI3K-AKT-mTOR | Restrains
proliferation, migration, invasion, and the EMT; promotes
cell-cycle arrest and tissue confinement; may also support
senescence and metabolic or epigenetic homeostasis, although it can
exert pro-tumorigenic effects in specific contexts such as
NAFLD-associated HCC. | (8,14,24,26,83) |
| S1PR3 | Largely
pro-oncogenic | Gi, Gq, G12/13 | PI3K-AKT, ERK,
PLC-Ca²+, Rho/ROCK, mTOR, STAT3 | Drives tumor
growth, stemness, the EMT, invasion, metastasis, inflammatory
signaling, immune suppression, and therapy resistance; also
contributes to cytoprotective autophagy and may promote T-cell
exhaustion and resistance to PD-1 blockade. | (8,11,78,84) |
| S1PR4 | Primarily immune
modulatory, with protumor effects in selected cancers | Gi, G12/13 | ERK1/2;
HER2-related crosstalk in breast cancer | Shapes the tumor
immune microenvironment by limiting CD8+ T-cell
abundance and survival in a cell-intrinsic manner; loss of S1PR4
can enhance anti-tumor immunity, whereas tumor-intrinsic S1PR4 may
promote progression in ER-negative breast cancer. | (8,36) |
| S1PR5 | Highly
context-dependent; immune-regulatory and occasionally
tumor-supportive | Gi, G12 | ERK2,
Ca²+ mobilization, Rac, AC, FAK, and PLC; involves
CXCR4-associated signaling/crosstalk. | Regulates NK-cell
egress and immune surveillance; in cancer, it has been linked to
immune responsiveness and aggressive behavior in colorectal cancer,
may exert protective effects in lung adenocarcinoma, and may
influence NK-cell desensitization and macrophage polarization in a
context-dependent manner. | (8,39,41,85) |
S1PR1 is the receptor most consistently linked to
malignant phenotypes across solid and hematologic cancers (6,7,17).
Acting predominantly through Gi, it activates ERK, PI3K-AKT, Rac
and STAT3 to promote proliferation, survival, motility and
inflammatory fitness (8,12,15).
Particularly important is the S1PR1-STAT3 feedforward circuit,
which sustains interleukin (IL)-6-dependent inflammatory signaling
(18,19). S1PR1 also contributes to endothelial
signaling, underscoring its role as a recurrent protumorigenic
signaling node (10,20-22).
By contrast, S1PR2 often opposes S1PR1 and is
generally linked to tumor-restraining functions (7,20).
Through RhoA-ROCK signaling and inhibition of Rac activity, S1PR2
can restrict cytoskeletal plasticity, migration and invasion and
its loss or downregulation is associated with more-aggressive
behavior in several malignancies (7,11,23).
However, this profile is not fixed. In selected settings, including
epidermal growth factor receptor-driven tumors and liver disease,
S1PR2 can be redirected towards protumorigenic outputs depending on
the disease stage, signaling environment and metabolic context
(16,24). In hepatocytes, S1PR2 contributes to
metabolic and epigenomic homeostasis through conjugated bile
acid-dependent activation of SphK2 and accumulation of nuclear S1P,
a pathway linked to histone acetylation and hepatic lipid and
sterol gene regulation; in nonalcoholic steatohepatitis-associated
liver injury, disruption of this axis was associated with a glycine
N-methyltransferase/S-adenosylmethionine imbalance, aberrant
methylation and enhanced susceptibility to hepatocarcinogenesis
(25-27).
S1PR2 therefore illustrates that receptor identity alone does not
determine biological outcomes.
S1PR3 is more consistently associated with
aggressive disease and phenotypic plasticity (14,28).
Through Gi- and Gq-dependent signaling, S1PR3 activates PI3K-AKT,
ERK, PLC and calcium-dependent pathways that support survival,
invasion and metastatic progression (12,15,23).
In numerous solid tumors, its upregulation is linked to
inflammatory signaling, matrix remodeling, the EMT and
stemness-associated programs (7,19,28-30).
Collectively, these observations place S1PR3 among the receptor
programs most closely linked to invasive behaviors, adaptive
plasticity and microenvironmental support in cancer (11,12).
S1PR4 and S1PR5 have more restricted physiological
expression patterns, but increasing evidence indicates that they
are important regulators of the immune context of cancer (7,31-33).
S1PR4, which is enriched in hematopoietic tissues, is linked to
immunosuppressive states and may also contribute to tumor-intrinsic
signaling in aggressive breast cancer (15,19,34-37).
S1PR5 is best known for its role in natural killer cell
trafficking, but emerging evidence suggests context-dependent
functions in solid tumors, including associations with immune
surveillance, checkpoint responsiveness and clinical outcomes
(38-41).
Although their functions remain less well defined than those of
S1PR1-S1PR3, current evidence suggests that both receptors
influence cancer progression chiefly through immune and
microenvironmental regulation (7,31,39).
Taken together, these receptor-resolved observations
indicate that the S1P-S1PR axis is neither uniformly oncogenic nor
uniformly tumor suppressive (7).
Rather, each receptor defines a distinct signaling module whose
output depends on the cellular compartment, microenvironmental
state, and regulatory architecture that governs receptor expression
and function (6,19). This diversity raises a central
question for cancer biology: How do tumors selectively reshape
receptor abundance, localization, and signaling competence to favor
malignant outputs? Addressing that question requires moving beyond
receptor cataloging toward the upstream mechanisms that reprogram
the axis in cancer.
4. Multilevel reprogramming of the S1P-S1PR
axis in cancer
Although the tumor-promoting effects of S1P receptor
signaling have been extensively described, the upstream mechanisms
that dysregulate this axis in cancer remain incompletely understood
(8). In malignant cells, the
S1P-S1PR system is reprogrammed at multiple levels, including
transcriptional and epigenetic control, posttranscriptional
regulation, receptor trafficking and posttranslational
modifications and changes in ligand availability (2,8).
Together, these mechanisms alter receptor expression levels,
receptor localization, and signaling competence, thereby sustaining
aberrant pathway output (Fig. 2)
(2,11). Understanding how these regulatory
layers converge is essential for explaining how tumors convert a
physiological homeostatic system into a context-dependent driver of
survival, progression, and therapeutic resistance (11).
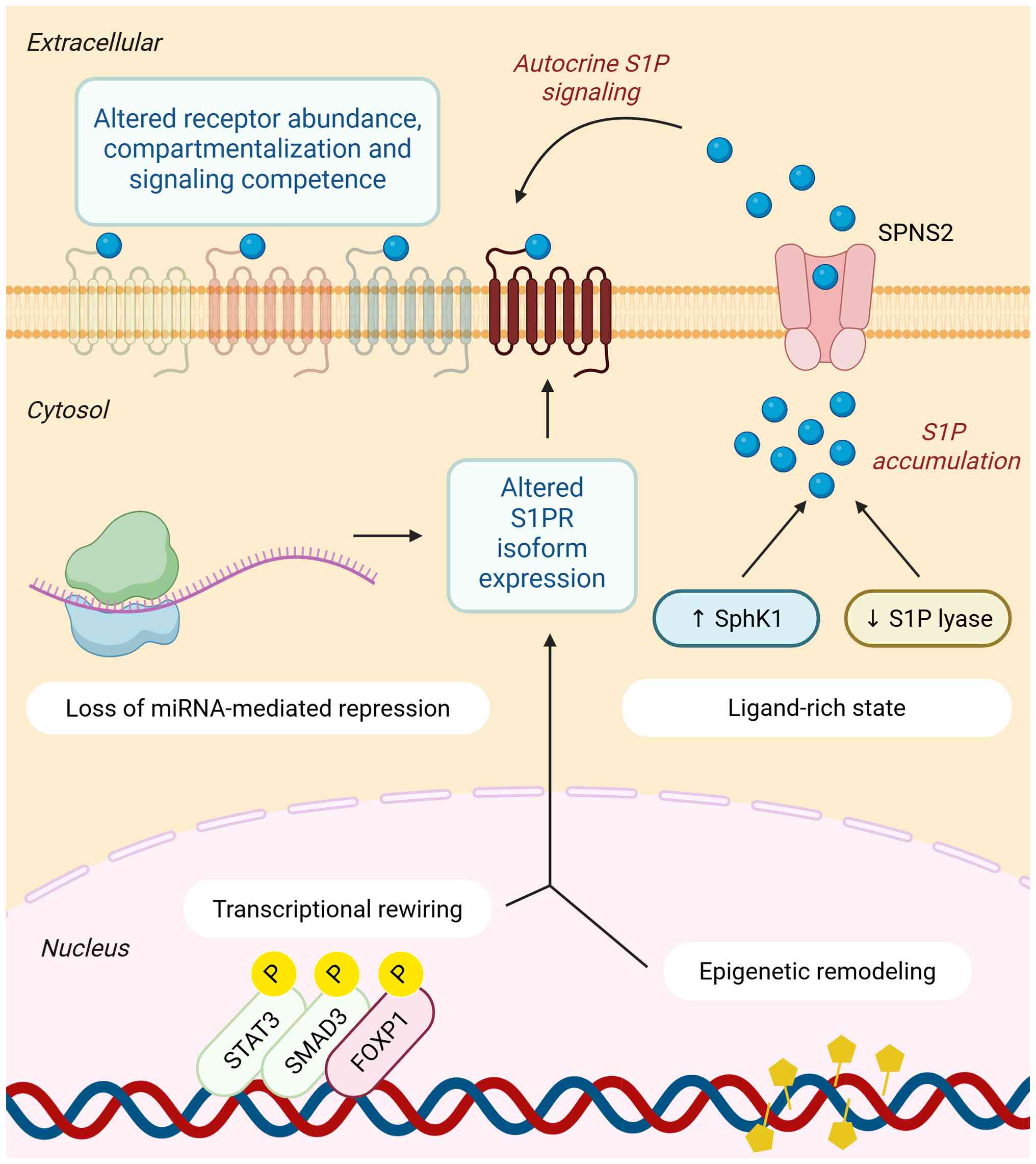 | Figure 2Multilevel reprogramming of the
S1P-S1PR axis in cancer. Within tumor cells, the S1P-S1PR axis is
reprogrammed at multiple levels. Transcriptional rewiring,
epigenetic remodeling and loss of miRNA-mediated repression reshape
S1PR isoform expression and receptor abundance. In parallel,
increased SphK1 activity and reduced S1P lyase activity create a
ligand-rich state that promotes intracellular S1P accumulation and
S1P signaling in the tumor microenvironment. Together, these
changes alter receptor abundance, compartmentalization and
signaling competence at the plasma membrane, converting a
homeostatic pathway into a context-dependent driver of malignancy.
S1P, sphingosine-1-phosphate; S1PR, sphingosine-1-phosphate
receptor; SphK1, sphingosine kinase 1; miRNA, microRNA. |
Transcriptional and epigenetic
reprogramming
Under physiological conditions, expression of S1PR
isoforms is tightly regulated in a tissue- and context-specific
manner (11). In cancer, this
architecture is frequently subverted by oncogenic and inflammatory
signaling pathways that directly reshape receptor transcription
(13,42). For example, IL-6-STAT3 signaling can
induce S1PR1 transcription through promoter binding, thereby
establishing a feedforward circuit that sustains aberrant STAT3
activation in tumor cells (18).
Similarly, transforming growth factor-β (TGFβ)-mothers against
decapentaplegic homolog 3 signaling upregulates S1PR3 in lung
adenocarcinoma, promoting the EMT, migration and metastatic
behavior (43).
By contrast, receptor subtypes with
tumor-restraining functions are often transcriptionally suppressed.
S1PR2 provides a prominent example (13). In diffuse large B cell lymphoma
(DLBCL), forkhead box protein P1 (FOXP1) represses S1PR2
transcription, thereby relieving constraints on motility and
proliferation (44). In other
settings, viral oncogenesis may also indirectly alter receptor
expression through changes in chromatin accessibility or promoter
activity, as suggested for S1PR3 in Epstein-Barr virus-associated
nasopharyngeal carcinoma (8,45).
These observations indicate that tumor-associated signaling does
not simply activate downstream pathways; it can selectively reshape
the receptor landscape through transcriptional rewiring (8).
Epigenetic regulation provides an additional layer
of control (8). DNA methylation,
histone modification and chromatin remodeling can alter
accessibility to S1PR loci and thereby reinforce malignant
signaling states (8,46). In hematologic malignancies,
disruption of the S1PR2 axis often occurs through multiple
convergent mechanisms (47,48). In DLBCL, S1PR2 may be lost through
recurrent mutations in the germinal center B cell-like subtype or
through FOXP1-mediated repression in the activated B cell-like
subtype (47). This loss can be
further reinforced by hypermethylation of SMAD1, which impairs
TGFβ-dependent induction of tumor-suppressive signaling (13,47).
More broadly, nuclear S1P itself can influence chromatin regulation
by inhibiting histone deacetylase (HDAC) 1 and HDAC2, thereby
promoting histone acetylation and transcription of genes involved
in cell-cycle control and survival (49). These findings place sphingolipid
metabolism and chromatin regulation within the same regulatory
framework and suggest that epigenetic remodeling is a central
mechanism through which tumors reprogram receptor output (2,8).
Posttranscriptional regulation
Posttranscriptional mechanisms, particularly those
mediated by miRNAs, are also important determinants of receptor
abundance (50). Several
tumor-suppressive miRNAs, including miR-148a, miR-363 and miR-133b,
directly target the 3' untranslated region of S1PR1 mRNA and
thereby limit receptor expression (12,50).
Loss of these constraints can increase S1PR1 abundance and
reinforce oncogenic phenotypes (51). In hepatocellular carcinoma,
depletion of miR-148a promotes invasion and metastasis, whereas in
nasopharyngeal carcinoma, downregulation of miR-133b was more
strongly linked to proliferative signaling (50-52).
This regulatory logic extends across the receptor
family (50). miR-9 suppresses both
S1PR1 and S1PR3 and miR-125b is also reported to negatively
regulate S1PR1 in lung carcinoma (53,54).
In cancer, these interactions may be disrupted by genomic loss of
miRNA loci, defective Dicer or Drosha processing, or repression by
oncogenic signaling pathways (54,55).
The result is not merely loss of fine-tuning, but a permissive
state in which increased receptor abundance amplifies downstream
signaling (54).
Other forms of posttranscriptional regulation may
further stabilize this rewired state (42). Long non-coding (lnc)RNAs have been
implicated in multidrug resistance and related oncogenic
phenotypes, although their receptor-specific roles within the S1P
axis remain less clearly defined (2,56).
Taken together, these findings indicate that posttranscriptional
deregulation can serve as a critical intermediate between oncogenic
signaling and receptor overactivity, enabling tumors to sustain
pathological S1P signaling even in the absence of direct genomic
alterations (11,50).
Receptor localization, trafficking,
and posttranslational control
Tumor-associated rewiring of the S1P-S1PR axis also
occurs at the level of receptor trafficking, membrane organization
and post-translational modifications (57,58).
Under physiological conditions, receptor internalization and
recycling help constrain signaling duration and preserve
responsiveness to extracellular gradients (59). In cancer, these controls may be
altered in ways that prolong receptor residence at the plasma
membrane or otherwise favor sustained signaling output (57).
S1PR1 provides one example of this principle. Its
localization within lipid raft or caveolin-enriched membrane
domains is linked to receptor clustering, internalization and
efficient downstream coupling. Disruption of this spatial
organization may impair normal desensitization and favor persistent
G protein-mediated signaling (59,60).
Similarly, adaptor proteins such as phosphatidylinositol
3,4,5-triphosphate-dependent Rac exchanger 1 protein can interfere
with agonist-induced receptor endocytosis and thereby prolong
receptor signaling at the cell surface (57). These observations support the
broader view that the strength of receptor signaling depends not
only on how much receptor is expressed, but also on whether the
receptor remains properly trafficked and spatially constrained
(57,61).
Post-translational modifications add further
complexity. N-Linked glycosylation is implicated in the efficient
surface delivery and stabilization of S1PR1 and S1PR5 (60,62,63).
Lipid modifications, particularly palmitoylation, may likewise
promote receptor partitioning into signaling-competent membrane
microdomains, thereby supporting efficient downstream coupling and
pathway output (60,64). Emerging evidence also suggests that
non-canonical forms of GPCR organization, including altered
β-arrestin dynamics, may modulate S1PR desensitization in selected
systems (57,59). Although these mechanisms remain less
established than transcriptional or epigenetic reprogramming, they
raise the possibility that tumors enhance signaling not only by
increasing receptor expression levels but also by preserving
receptors in a signaling-competent state (2,57).
De novo receptor expression vs.
functional reactivation
Cancer cells may exploit the S1P-S1PR axis through
two complementary strategies (11).
The first is ectopic expression of receptor isoforms that are
normally restricted to other tissues (34,35).
The second is functional reactivation or stabilization of receptors
that are already constitutively expressed but are normally subject
to tighter regulatory control (18,57).
Examples of de novo receptor expression
highlight how tumors can expand the signaling repertoire available
to them (11). In T-cell large
granular lymphocyte leukemia, aberrant induction of the S1PR5
lineage-restricted receptor is linked to constitutive ERK1/2
activation and enhanced pro-survival signaling, suggesting that
acquisition of this receptor program may create a selective
therapeutic vulnerability (62,65).
Similarly, S1PR4, which is typically associated with hematopoietic
tissues, can be induced in estrogen receptor-negative breast
cancer, where it amplifies human epidermal growth factor receptor
2-linked signaling and correlates with a poor prognosis (2,35). In
such settings, tumors do not simply intensify an existing signaling
axis; they acquire receptor programs that are not normally part of
the tissue context (34).
At the same time, S1PR1-S1PR3 can be functionally
reprogrammed without requiring de novo expression (2). Ligand-rich tumor microenvironments,
altered receptor trafficking and changes in membrane organization
may all favor persistent activation of constitutively expressed
receptors (11,57). In this way, tumors can strengthen
signaling output either by acquiring new receptor modules or by
stabilizing existing ones in a hyperactive state (34,57).
This distinction between de novo receptor expression and
functional reactivation provides a useful conceptual framework for
understanding how cancers expand or intensify S1P signaling in
different settings (11).
Ligand availability and receptor
signaling state cooperate to drive hyperactivation
Persistent activation of the S1P-S1PR axis in cancer
reflects the convergence of metabolic dysregulation and
receptor-level rewiring (2,11). In numerous tumors, SphK1 is
upregulated, increasing S1P production, whereas reduced activity of
sphingosine-1-phosphate lyase promotes further accumulation of
intracellular and extracellular S1P (8). These changes create ligand-rich
conditions that favor chronic pathway activation, tumor
progression, and therapeutic resistance (2).
However, ligand abundance alone does not fully
explain the signaling output (11).
Functional studies indicated that receptor abundance, localization
and signaling competence are equally important determinants of
pathway strength (57,59). In Epstein-Barr virus-positive
nasopharyngeal carcinoma, for example, both elevated S1P and
increased S1PR3 expression are required to drive migration and
silencing S1PR3 markedly attenuates S1P-dependent effects (45). Likewise, in breast cancer,
co-expression of S1P and S1PR1 establishes an autocrine signaling
loop that promotes invasion and angiogenesis (2,18).
These examples illustrate a broader principle: Tumors exploit both
metabolic and receptor-level mechanisms to amplify signaling beyond
what either mechanism could achieve alone (11).
This convergence has important conceptual and
therapeutic implications (8). It
suggests that the malignant consequences of S1P signaling emerge
not simply from excess ligands, nor solely from altered receptor
expression, but from coordinated rewiring of both (2,11).
Accordingly, effective interventions may require simultaneous
consideration of ligand production, the receptor state and cellular
context (8).
Genetic alterations as
context-specific exceptions
Although the preceding sections emphasize that most
tumor-associated rewiring of the S1P-S1PR axis is driven by
non-genetic mechanisms, focal genomic alterations provide
instructive exceptions that help clarify the oncogenic or
tumor-suppressive functions of specific receptor isoforms (13,66).
In most solid tumors, receptor dysregulation appears to
predominantly arise through transcriptional, epigenetic,
posttranscriptional, metabolic and post-translational reprogramming
rather than through recurrent coding alterations (47). Even so, selected genetic lesions
offer important insight into how individual receptors constrain or
promote malignant behaviors in defined disease contexts (48).
The clearest example is S1PR2 in DLBCL, where
recurrent loss-of-function mutations disrupt a tumor-suppressive
signaling axis involved in growth control, cytoskeletal
organization and tissue compartmentalization (48). These lesions often coexist with
additional perturbations, including guanine nucleotide-binding
protein subunit alpha-13 inactivation or SMAD1 loss, which further
weaken receptor-dependent restraint (47,48).
In this setting, genetic disruption of S1PR2 helps define a
receptor program that normally limits malignant expansion and
dissemination (48).
By contrast, S1PR3 amplification was described in
ependymomas, where recurrent gain of the 9q22.1 locus, often
together with SHC-transforming protein 3, promotes signaling
programs linked to tumor growth and survival (67,68).
Although this appears to be a more context-restricted event, it is
consistent with the broader association of S1PR3 with aggressive
and invasive phenotypes (69).
These examples are informative, but they do not
define the dominant pattern across cancers (11). Rather, they underscore that although
genomic lesions can shape receptor output in selected settings,
most tumor-associated changes in the S1P-S1PR axis arise through
non-genetic rewiring (2,47,66).
Beyond these genetic exceptions, the broader reprogramming of the
S1P-S1PR axis is likely sustained by interacting non-genetic
regulatory layers. Transcriptional activation may be stabilized by
epigenetic remodeling, whereas loss of miRNA-mediated repression
can further increase receptor abundance once permissive chromatin
states are established (42,70).
In parallel, altered trafficking and posttranslational regulation
may retain receptors in signaling-competent membrane compartments,
allowing ligand-rich microenvironments to convert increased
receptor abundances into sustained pathway activation (2,57). The
S1P-S1PR axis is therefore most plausibly reprogrammed through
convergent, mutually reinforcing layers of control rather than
through any single dominant lesion, a framework that may help
explain how modest changes across multiple levels collectively
sustain oncogenic signaling and therapeutic resistance (2,11).
5. Therapeutic implications of targeting a
reprogrammed signaling axis
If malignant progression depends on multilevel
reprogramming of the S1P-S1PR axis, then therapeutic interventions
must account not only for receptor identity but also for ligand
availability, receptor signaling competence and cellular context
(8,11). This perspective shifts the
therapeutic question away from whether the pathway is active and
toward how it has been rewired in a given tumor setting (13).
Direct receptor targeting
Pharmacological modulation of S1PRs represents one
major strategy for therapeutic interventions (11). Several S1PR-targeting agents,
initially developed or approved for autoimmune diseases, are now
being explored for antitumor applications (Table II) (71,72).
For example, the selective S1PR1/5 modulator, siponimod, is
reported to overcome chemoresistance in ovarian carcinoma by
suppressing S1PR1-dependent p-STAT3 signaling (73). Other studies link a
receptor-directed intervention to inhibition of S1PR3-dependent
AKT-ERK signaling in renal cell carcinoma or to suppression of
metabolic programs such as Yes-associated protein-c-MYC signaling
in osteosarcomas (74,75).
| Table IIS1PR-targeting agents and S1P-axis
inhibitors with relevance to cancer. |
Table II
S1PR-targeting agents and S1P-axis
inhibitors with relevance to cancer.
| Drug | Target | Cancer
relevance | (Refs.) |
|---|
| Fingolimod | S1PR1/3/4/5
modulator | Broad preclinical
antitumor activity; suppresses stemness | (11,86) |
| Siponimod | S1PR1/5
modulator | Selective
next-generation comparator; limited direct cancer data | (86) |
| Ozanimod | S1PR1/5
modulator | Clinically
validated selectivity; potential translational relevance | (86,87) |
| Ponesimod | S1PR1
modulator | Highly selective
S1PR1 reference drug | (88) |
| ACT-209905 | S1PR1
modulator | Glioblastoma
growth/migration inhibition; TMZ sensitization | (77) |
| JTE-013 | S1PR2
antagonist | Mechanistic probe;
worsens NASH-HCC in vivo | (26) |
| KRX-725-II | S1PR3
antagonist |
Anti-angiogenic/antitumor concept | (78) |
| Opaganib | SphK2
inhibitor | Upstream anti-S1P
therapeutic strategy | (12) |
| PF-543 | SphK1
inhibitor | Preclinical
anti-tumor S1P-axis inhibition | (12,89) |
Next-generation compounds with greater receptor
selectivity may offer additional advantages (76). ACT-209905, a selective S1PR1
modulator, has shown activity in glioblastoma models and can
enhance sensitivity to temozolomide (77). Likewise, S1PR3 antagonists,
including TY-52156 and related pepducins, have demonstrated
anti-angiogenic and immunomodulatory effects in preclinical systems
(28,78). These findings support the idea that
receptor-selective targeting may interrupt discrete malignant
signaling modules rather than broadly suppressing all S1P signaling
(23,76).
Targeting upstream S1P production
As elevated S1P availability is a recurring driver
of receptor activation, upstream metabolic targeting provides a
complementary therapeutic approach (8). SphK1 is of particular interest given
its frequent upregulation in cancer and its role in maintaining
ligand-rich microenvironments (2).
In principle, inhibition of SphK activity can attenuate both tumor
cell-intrinsic and microenvironmental S1P signaling (12). Similarly, S1P-neutralizing
strategies, including antibody-based approaches such as sphingomab,
have shown antitumor activity in preclinical models (79).
This upstream approach is especially attractive
within the framework advanced in the present review, because it
addresses one major component of pathway reprogramming: Metabolic
amplification of the ligand supply (2,8).
However, metabolic targeting alone may be insufficient in settings
where receptor abundance, receptor localization, or downstream
coupling have also been rewired to favor persistent signaling
(8,42).
Combined targeting strategies
The coordinated nature of S1P axis reprogramming
argues strongly for combination approaches (42). If tumors exploit both increased
ligand production and receptor-level rewiring, then simultaneous
targeting of S1P synthesis and receptor-mediated signaling may
produce more-durable pathway suppression than either approach alone
(42). This concept is supported by
experimental observations in which ligand availability and receptor
expression cooperate to drive malignant phenotypes (35).
Combination strategies may also extend beyond the
S1P pathway itself (42). Pairing
S1PR-directed agents with chemotherapy, radiotherapy, targeted
kinase inhibitors, or immune checkpoint blockade could prove
particularly effective in tumors where S1P signaling contributes to
therapeutic resistance, stromal crosstalk, or immune exclusion
(12,80). Similarly, selective S1PR3 antagonism
may complement immunotherapy by reducing macrophage polarization
toward suppressive states and by mitigating T-cell exhaustion in
selected settings (28). Although
these approaches remain largely preclinical, they align closely
with the reprogramming model developed in this review.
Biomarker and safety
considerations
Despite its promise, therapeutic targeting of the
S1P-S1PR axis is complicated by the physiological importance of
this pathway in vascular and immune homeostasis (69). Even receptor-selective agents can
produce organ-specific on-target toxicities, most notably
bradycardia and macular edema (71,76). A
distinct liability arises from interference with physiological
lymphocyte trafficking, which may lead to functional
immunosuppression rather than direct organ toxicity (11,76).
These liabilities highlight the challenge of targeting a pathway
whose physiological roles are closely intertwined with its
pathological functions (23).
For this reason, future progress will depend not
only on improved pharmacology but also on improved patient
stratification (23). Biomarkers
that capture receptor expression, ligand-rich states,
transcriptional programs such as STAT3 activation, or
immune-context features may help identify tumors most likely to
benefit from pathway-directed interventions (12,28).
More-selective agonists, antagonists, or biased modulators may
further improve the therapeutic window by preferentially targeting
disease-relevant signaling outputs while minimizing adverse effects
linked to normal physiology (62,76).
Taken together, these considerations suggest that
the most effective therapeutic strategies will likely be those that
treat the S1P-S1PR axis not as a uniform pathway, but as a
reprogrammed signaling network whose vulnerabilities vary across
tumor types and biological contexts (13).
6. Conclusions
The S1P-S1PR axis is best understood not as a
simple ligand-driven pathway, but as a dynamically reprogrammable
signaling network in cancer (11).
Its output is determined not only by S1P abundance, but also by
receptor identity and by the regulatory mechanisms that govern
receptor expression, localization and signaling competence across
tumor and microenvironmental compartments (11,13).
Tumors exploit this plasticity through coordinated transcriptional,
epigenetic, posttranscriptional, trafficking and metabolic
alterations, thereby converting a homeostatic signaling system into
a context-dependent driver of tumor progression and therapeutic
resistance (42,81). This framework supports
biomarker-guided strategies that target both ligand production and
receptor-mediated signaling according to the specific configuration
of the axis in a given tumor (12,23).
Acknowledgements
The authors would like to thank Dr. Yu-Chu Chang
(Max Planck Institute for Biology, Tübingen, Germany) for his
insightful feedback on the manuscript and continued support for
this work. Figures were created with BioRender.com.
Funding
Funding: The present study was supported by grant no.
TMU112-AE1-B29 from Taipei Medical University.
Availability of data and materials
Not applicable.
Author contributions
YTC, CFL, and IHW contributed to the
conceptualization and development of the review. YTC and CFL
drafted the manuscript and prepared the figures. CFL conducted the
literature analysis and HYL critically revised the manuscript. Data
authentication is not applicable. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Li T, He H, Zhang E, Hu F, Wang Z, Xu J,
Zeng M and Peng B: The physiological and pathological effects of
sphingolipid metabolism and signaling in the central nervous
system. Brain Pathol. 36(e70033)2026.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Alkafaas SS, Elsalahaty MI, Ismail DF,
Radwan MA, Elkafas SS, Loutfy SA, Elshazli RM, Baazaoui N, Ahmed
AE, Hafez W, et al: The emerging roles of sphingosine 1-phosphate
and SphK1 in cancer resistance: A promising therapeutic target.
Cancer Cell Int. 24(89)2024.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Hannun YA, Merrill AH and Luberto C: The
bioactive sphingolipid playbook. A primer for the uninitiated as
well as sphingolipidologists. J Lipid Res.
66(100813)2025.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Mao C: Sphingolipid metabolism
dysregulation: A cause for lung cancer development, progression,
and resistance to therapies. Chin Med J Pulm Crit Care Med.
3:88–96. 2025.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sukocheva OA, Neganova ME, Aleksandrova Y,
Burcher JT, Chugunova E, Fan R, Tse E, Sethi G, Bishayee A and Liu
J: Signaling controversy and future therapeutical perspectives of
targeting sphingolipid network in cancer immune editing and
resistance to tumor necrosis factor-α immunotherapy. Cell Commun
Signal. 22(251)2024.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Xiong X, Zeng L, Zeng F, Huang Y and Jia
L: Bioinformatics exploration of the S1PR1 receptor in various
human cancers and its clinical relevance. Discov Oncol.
16(449)2025.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wang Z, Zhang HM, Guo YR, Li LL and Zhang
GZ: Role of sphingosine-1-phosphate receptors in the tumor
microenvironment: Prospects for cancer immunotherapy. Eur Rev Med
Pharmacol Sci. 27:713–727. 2023.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Rufail ML, Bassi R and Giussani P:
Sphingosine-1-Phosphate metabolic pathway in cancer: Implications
for therapeutic targets. Int J Mol Sci. 26(1056)2025.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Chai Y, Xiang H, Ma Y, Feng W, Jiang Z,
Zhu Q, Chen Y, Liu Q, Zhang J, Ouyang J, et al: S1PR1 suppresses
lung adenocarcinoma progression through p-STAT1/miR-30c-5 p/FOXA1
pathway. J Exp Clin Cancer Res. 43(304)2024.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Jia W, Yuan J, Zhang J, Li S, Lin W and
Cheng B: Bioactive sphingolipids as emerging targets for signal
transduction in cancer development. Biochim Biophys Acta Rev
Cancer. 1879(189176)2024.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Powell JA and Pitson SM: Sphingosine
1-phosphate signalling in cancer stem cells. Oncogenesis.
14(42)2025.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Xu X, Li H, Li R, Xu Y, Xu Y, Huang H, Lv
X, Liao C, Ye J and Bo L: Mechanisms and potential therapeutic
targets of SphK1 and SphK2 in hepatocellular carcinoma. Front Med.
12(1617401)2025.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Pyne NJ and Pyne S: Recent advances in the
role of sphingosine 1-phosphate in cancer. FEBS Lett.
594:3583–3601. 2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Limbu KR, Chhetri RB, Kim S, Shrestha J,
Oh YS, Baek DJ and Park EY: Targeting sphingosine 1-phosphate and
sphingosine kinases in pancreatic cancer: Mechanisms and
therapeutic potential. Cancer Cell Int. 24(353)2024.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Hii LW, Chung FFL, Mai CW, Ng PY and Leong
CO: Sphingosine Kinase 1 signaling in breast cancer: A potential
target to tackle breast cancer stem cells. Front Mol Biosci.
8(748470)2021.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Wongviriya A, Shelton RM, Cooper PR,
Milward MR and Landini G: The relationship between
sphingosine-1-phosphate receptor 2 and epidermal growth factor in
migration and invasion of oral squamous cell carcinoma. Cancer Cell
Int. 23(65)2023.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Rostami N, Nikkhoo A, Ajjoolabady A, Azizi
G, Hojjat-Farsangi M, Ghalamfarsa G, Yousefi B, Yousefi M and
Jadidi-Niaragh F: S1PR1 as a novel promising therapeutic target in
cancer therapy. Mol Diagn Ther. 23:467–487. 2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Lee H, Deng J, Kujawski M, Yang C, Liu Y,
Herrmann A, Kortylewski M, Horne D, Somlo G, Forman S, et al:
STAT3-induced S1PR1 expression is crucial for persistent STAT3
activation in tumors. Nat Med. 16:1421–1428. 2010.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Nagahashi M and Miyoshi Y: Targeting
Sphingosine-1-Phosphate signaling in breast cancer. Int J Mol Sci.
25(3354)2024.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Cartier A, Leigh T, Liu CH and Hla T:
Endothelial sphingosine 1-phosphate receptors promote vascular
normalization and antitumor therapy. Proc Natl Acad Sci.
117:3157–3166. 2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Laroche FJF, Li S, Shen N, Hwang SK,
Nguyen G, Yu W, Wong CK, Quinton RJ, Berman JN, Liu CT, et al: S1P1
Threonine 236 phosphorylation mediates the invasiveness of
triple-Negative breast cancer and sensitivity to FTY720. Cells.
12(980)2023.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Tao YP, Zhu HY, Shi QY, Wang CX, Hua YX,
Hu HY, Zhou QY, Zhou ZL, Sun Y, Wang XM, et al: S1PR1 regulates
ovarian cancer cell senescence through the PDK1-LATS1/2-YAP
pathway. Oncogene. 42:3491–3502. 2023.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Riboni L, Abdel Hadi L, Navone SE,
Guarnaccia L, Campanella R and Marfia G: Sphingosine-1-Phosphate in
the tumor microenvironment: A signaling hub regulating cancer
hallmarks. Cells. 9(337)2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wang G, Zhang X, Zhou Z, Song C, Jin W,
Zhang H, Wu W, Yi Y, Cui H, Zhang P, et al: Sphingosine 1-phosphate
receptor 2 promotes the onset and progression of non-alcoholic
fatty liver disease-related hepatocellular carcinoma through the
PI3K/AKT/mTOR pathway. Discov Oncol. 14(4)2023.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Nagahashi M, Takabe K, Liu R, Peng K, Wang
X, Wang Y, Hait NC, Wang X, Allegood JC, Yamada A, et al:
Conjugated bile acid activated S1P Receptor 2 is a key regulator of
sphingosine kinase 2 and hepatic gene expression. Hepatology.
61:1216–1226. 2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Yoshida T, Tsuchiya A, Kumagai M, Takeuchi
S, Nojiri S, Watanabe T, Ogawa M, Itoh M, Takamura M, Suganami T,
et al: Blocking sphingosine 1-phosphate receptor 2 accelerates
hepatocellular carcinoma progression in a mouse model of NASH.
Biochem Biophys Res Commun. 530:665–672. 2020.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Quinn C, Rico MC, Merali C and Merali S:
Dysregulation of S-adenosylmethionine metabolism in nonalcoholic
steatohepatitis leads to polyamine flux and oxidative stress. Int J
Mol Sci. 23(1986)2022.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Gao G, Liao W, Shu P, Ma Q, He X, Zhang B,
Qin D and Wang Y: Targeting sphingosine 1-phosphate receptor 3
inhibits T-cell exhaustion and regulates recruitment of
proinflammatory macrophages to improve antitumor efficacy of CAR-T
cells against solid tumor. J Immunother Cancer.
11(e006343)2023.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Hirata N, Yamada S, Shoda T, Kurihara M,
Sekino Y and Kanda Y: Sphingosine-1-phosphate promotes expansion of
cancer stem cells via S1PR3 by a ligand-independent Notch
activation. Nat Commun. 5(4806)2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Kim MH, Huh B, Park JW and Park WJ:
Targeting sphingolipids in breast cancer: From tumor biology to
therapeutic strategies. Oncol Res. 34(6)2026.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Yuan Y, Jia G, Wu C, Wang W, Cheng L, Li
Q, Li Z, Luo K, Yang S, Yan W, et al: Structures of signaling
complexes of lipid receptors S1PR1 and S1PR5 reveal mechanisms of
activation and drug recognition. Cell Res. 31:1263–1274.
2021.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Wang Z, Pan F and Zhang G: Expression and
prognostic role of sphingosine 1-phosphate receptor 4 (S1PR4) as a
biomarker of skin cutaneous melanoma. Heliyon.
10(e27505)2024.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Huang C, Zhu F, Zhang H, Wang N and Huang
Q: Identification of S1PR4 as an immune modulator for favorable
prognosis in HNSCC through machine learning. iScience.
26(107693)2023.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Long JS, Edwards J, Watson C, Tovey S,
Mair KM, Schiff R, Natarajan V, Pyne NJ and Pyne S: Sphingosine
Kinase 1 induces tolerance to human epidermal growth factor
receptor 2 and prevents formation of a migratory phenotype in
response to Sphingosine 1-Phosphate in estrogen receptor-positive
breast cancer cells. Mol Cell Biol. 30:3827–3841. 2010.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Ohotski J, Long JS, Orange C, Elsberger B,
Mallon E, Doughty J, Pyne S, Pyne NJ and Edwards J: Expression of
sphingosine 1-phosphate receptor 4 and sphingosine kinase 1 is
associated with outcome in oestrogen receptor-negative breast
cancer. Br J Cancer. 106:1453–1459. 2012.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Olesch C, Sirait-Fischer E, Berkefeld M,
Fink AF, Susen RM, Ritter B, Michels BE, Steinhilber D, Greten FR,
Savai R, et al: S1PR4 ablation reduces tumor growth and improves
chemotherapy via CD8+ T cell expansion. J Clin Invest.
130:5461–5476. 2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Wang B, Wu X, Cheng J, Ye J, Zhu H and Liu
X: Regulatory role of S1P and its receptors in sepsis-induced liver
injury. Front Immunol. 16(1489015)2025.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Jenne CN, Enders A, Rivera R, Watson SR,
Bankovich AJ, Pereira JP, Xu Y, Roots CM, Beilke JN, Banerjee A, et
al: T-bet-dependent S1P5 expression in NK cells promotes egress
from lymph nodes and bone marrow. J Exp Med. 206:2469–2481.
2009.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Pei A, Lu L and Wu X: S1PR5 as a
prognostic biomarker in colon cancer: Insights into
efferocytosis-related mechanisms and immune modulation. J Mol
Histol. 56(315)2025.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Puig-Gámez M, Van Attekum M, Theis T, Dick
A and Park JE: Transcriptional signature of rapidly responding NK
cells reveals S1P5 and CXCR4 as anti-tumor response disruptors. Sci
Rep. 15(10769)2025.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Xing Z, Liu Y, Yang X, Yao Y, Chang T,
Zhou L, Luo R, Jiang L and Xue J: Multi-omics and Mendelian
randomization identify S1PR5 as a causal protective gene and NK
cell-mediated prognostic biomarker in lung adenocarcinoma. Transl
Lung Cancer Res. 14:3553–3576. 2025.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Hu Y, Dong Z and Liu K: Unraveling the
complexity of STAT3 in cancer: Molecular understanding and drug
discovery. J Exp Clin Cancer Res. 43(23)2024.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Zhao J, Liu J, Lee JF, Zhang W, Kandouz M,
VanHecke GC, Chen S, Ahn YH, Lonardo F and Lee MJ: TGF-β/SMAD3
pathway stimulates Sphingosine-1 Phosphate Receptor 3 Expression
IMPLICATION OF SPHINGOSINE-1 PHOSPHATE RECEPTOR 3 IN LUNG
ADENOCARCINOMA PROGRESSION. J Biol Chem. 291:27343–27353.
2016.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Flori M, Schmid CA, Sumrall ET, Tzankov A,
Law CW, Robinson MD and Müller A: The hematopoietic oncoprotein
FOXP1 promotes tumor cell survival in diffuse large B-cell lymphoma
by repressing S1PR2 signaling. Blood. 127:1438–1448.
2016.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Lee HM, Lo KW, Wei W, Tsao SW, Chung GTY,
Ibrahim MH, Dawson CW, Murray PG, Paterson IC and Yap LF: Oncogenic
S1P signalling in EBV-associated nasopharyngeal carcinoma activates
AKT and promotes cell migration through S1P receptor 3. J Pathol.
242:62–72. 2017.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Fong CY, Morison J and Dawson MA:
Epigenetics in the hematologic malignancies. Haematologica.
99:1772–1783. 2014.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Stelling A, Hashwah H, Bertram K, Manz MG,
Tzankov A and Müller A: The tumor suppressive TGF-β/SMAD1/S1PR2
signaling axis is recurrently inactivated in diffuse large B-cell
lymphoma. Blood. 131:2235–2246. 2018.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Muppidi JR, Schmitz R, Green JA, Xiao W,
Larsen AB, Braun SE, An J, Xu Y, Rosenwald A, Ott G, et al: Loss of
signaling via Gα13 in germinal center B cell-derived lymphoma.
Nature. 516:254–258. 2014.
|
|
49
|
Hait NC, Allegood J, Maceyka M, Strub GM,
Harikumar KB, Singh SK, Luo C, Marmorstein R, Kordula T, Milstien S
and Spiegel S: Regulation of histone acetylation in the nucleus by
Sphingosine-1-Phosphate. Science. 325:1254–1257. 2009.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Xu G, Yang Z, Sun Y, Dong H and Ma J:
Interaction of microRNAs with sphingosine kinases, sphingosine-1
phosphate, and sphingosine-1 phosphate receptors in cancer. Discov
Oncol. 12(33)2021.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Cheng N and Wang GH: miR-133b, a microRNA
targeting S1PR1, suppresses nasopharyngeal carcinoma cell
proliferation. Exp Ther Med. 11:1469–1474. 2016.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Zhang SL and Liu L: microRNA-148a inhibits
hepatocellular carcinoma cell invasion by targeting
sphingosine-1-phosphate receptor 1. Exp Ther Med. 9:579–584.
2015.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Yao X, Xie L and Zeng Y: MiR-9 Promotes
angiogenesis via targeting on Sphingosine-1-Phosphate receptor 1.
Front Cell Dev Biol. 8(755)2020.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Zhang X, Liu Y, Huang WC and Zheng LC:
MiR-125b-1-3p exerts antitumor functions in lung carcinoma cells by
targeting S1PR1. Chin Med J (Engl). 131:1909–1916. 2018.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Chen Y, Lin T, Tang L, He L and He Y:
MiRNA signatures in nasopharyngeal carcinoma: Molecular mechanisms
and therapeutic perspectives. Am J Cancer Res. 13:5805–5824.
2023.PubMed/NCBI
|
|
56
|
Majumdar S, Chakraborty A, Das S, Gorain
M, Chatterjee S, Dey I, Bhowmik S, Ghosh S, Banerjee S, Ahammed SM,
et al: Sponging of five tumour suppressor miRNAs by lncRNA-KCNQ1OT1
activates BMPR1A/BMPR1B-ACVR2A/ACVR2B signalling and promotes
chemoresistance in hepatocellular carcinoma. Cell Death Discov.
10(274)2024.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Baker MJ, Hampson E, Islam P, Moral RP,
Maunders EA, Hornigold K, Tsonou E, Malliri A, Hornigold DC,
Hubbard RE, et al: P-Rex1 limits the agonist-induced
internalization of GPCRs independently of its Rac-GEF activity.
Cell Rep. 44(116403)2025.PubMed/NCBI View Article : Google Scholar
|
|
58
|
D'Aprile C, Prioni S, Mauri L, Prinetti A
and Grassi S: Lipid rafts as platforms for sphingosine 1-phosphate
metabolism and signalling. Cell Signal. 80(109929)2021.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Anwar M and Mehta D: Post-translational
modifications of S1PR1 and endothelial barrier regulation. Biochim
Biophys Acta Mol Cell Biol Lipids. 1865(158760)2020.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Patwardhan A, Cheng N and Trejo J:
Post-Translational modifications of G Protein-Coupled receptors
control cellular signaling dynamics in space and time. Pharmacol
Rev. 73:120–151. 2021.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Ripoll L, von Zastrow M and Blythe EE:
Intersection of GPCR trafficking and cAMP signaling at
endomembranes. J Cell Biol. 224(e202409027)2025.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Lyapina E, Marin E, Gusach A, Orekhov P,
Gerasimov A, Luginina A, Vakhrameev D, Ergasheva M, Kovaleva M,
Khusainov G, et al: Structural basis for receptor selectivity and
inverse agonism in S1P5 receptors. Nat Commun.
13(4736)2022.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Kohno T, Wada A and Igarashi Y: N-Glycans
of sphingosine 1-phosphate receptor Edg-1 regulate ligand-induced
receptor internalization. FASEB J. 16:983–992. 2002.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Badawy SMM, Okada T, Kajimoto T, Ijuin T
and Nakamura S: DHHC5-mediated palmitoylation of S1P receptor
subtype 1 determines G-protein coupling. Sci Rep.
7(16552)2017.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Park SH, Lee YJ, Kim Y, Kim HK, Lim JH and
Jo JC: T-large granular lymphocytic leukemia. Blood Res. 58
(Suppl):S52–S57. 2023.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Bozzini N, Avnet S, Baldini N and Cortini
M: Epigenetic regulation mediated by sphingolipids in cancer. Int J
Mol Sci. 24(5294)2023.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Magrassi L, Marziliano N and Inzani F,
Magrassi L, Marziliano N and Inzani F: EDG3 and SHC3 on chromosome
9q22 are co-amplified in human ependymomas. Cancer Lett. 290:36–42.
2010.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Seo SH, Paul SK, Shikder M, Khanam M,
Ghosh P, Hasib TA, Ahmed KA, Sikdar S, Uddin MJ and Kwon Y: An
insight into pathophysiological features and therapeutic advances
on ependymoma. Cancers. 13(3221)2021.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Chen H, Wang J, Zhang C, Ding P, Tian S,
Chen J, Ji G and Wu T: Sphingosine 1-phosphate receptor, a new
therapeutic direction in different diseases. Biomed Pharmacother.
153(113341)2022.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Pajares MJ, Alemany-Cosme E, Goñi S,
Bandres E, Palanca-Ballester C and Sandoval J: Epigenetic
regulation of microRNAs in cancer: Shortening the distance from
bench to bedside. Int J Mol Sci. 22(7350)2021.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Pérez-Jeldres T, Alvarez-Lobos M and
Rivera-Nieves J: Targeting Sphingosine-1-phosphate signaling in
Immune-mediated diseases: Beyond multiple sclerosis. Drugs.
81:985–1002. 2021.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Perry TA, Masand N, Vrzalikova K, Pugh M,
Wei W, Hollows R, Bouchalova K, Nohtani M, Fennell E, Bouchal J, et
al: The oncogenic lipid Sphingosine-1-Phosphate impedes the
phagocytosis of tumor cells by M1 macrophages in diffuse large B
cell lymphoma. Cancers. 16(574)2024.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Gong K, Dong Y, Wang L and Duan Y, Yu J,
Sun Y, Bai M and Duan Y: Nanoparticle BAF312@CaP-NP overcomes
Sphingosine-1-Phosphate Receptor-1-mediated chemoresistance through
inhibiting S1PR1/P-STAT3 axis in ovarian carcinoma. Int J
Nanomedicine. 15:5561–5571. 2020.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Yan Y, Bao G, Pei J, Cao Y, Zhang C, Zhao
P, Zhang Y and Damirin A: NF-κB and EGFR participate in
S1PR3-mediated human renal cell carcinomas progression. Biochim
Biophys Acta Mol Basis Dis. 1868(166401)2022.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Shen Y, Zhao S, Wang S, Pan X, Zhang Y, Xu
J, Jiang Y, Li H, Zhang Q, Gao J, et al: S1P/S1PR3 axis promotes
aerobic glycolysis by YAP/c-MYC/PGAM1 axis in osteosarcoma.
EBioMedicine. 40:210–223. 2019.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Ye T, Yu J, Fang Y, Xu Z, Guo S, Zhao Z,
Li H, He H and Zhu L: Discovery and SAR study of highly selective
and potent 1,2,4-oxadiazole-based S1PR1 agonists. Eur J Med Chem.
300(118097)2025.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Bien-Möller S, Chen F, Xiao Y, Köppe H,
Jedlitschky G, Meyer U, Tolksdorf C, Grube M, Marx S, Tzvetkov MV,
et al: The putative S1PR1 modulator ACT-209905 impairs growth and
migration of glioblastoma cells in vitro. Cancers.
15(4273)2023.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Avnet S, Mizushima E, Severino B, Lipreri
MV, Scognamiglio A, Corvino A, Baldini N and Cortini M:
Antagonizing the S1P-S1P3 axis as a promising Anti-angiogenic
strategy. Metabolites. 15(178)2025.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Visentin B, Vekich JA, Sibbald BJ, Cavalli
AL, Moreno KM, Matteo RG, Garland WA, Lu Y, Yu S, Hall HS, et al:
Validation of an anti-sphingosine-1-phosphate antibody as a
potential therapeutic in reducing growth, invasion, and
angiogenesis in multiple tumor lineages. Cancer Cell. 9:225–238.
2006.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Shen H, Deng Q, Chen Z, Zhang Q, Zhou X,
Chen Q and Fan J: In situ-formed immunotherapeutic hydrogel
containing sphingosine-1-phosphate for enhanced lung cancer
immunotherapy. Sci Adv. 11(eadw5001)2025.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Mishra A, Hourigan D and Lindsay AJ:
Inhibition of the endosomal recycling pathway downregulates HER2
activation and overcomes resistance to tyrosine kinase inhibitors
in HER2-positive breast cancer. Cancer Lett. 529:153–167.
2022.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Anu B, Namitha NN and Harikumar KB:
Chapter Nine-S1PR1 signaling in cancer: A current perspective. In:
Apoptosis in Health and Disease-Part A. vol. 125 Donev R (ed.)
Academic Press, pp259-274, 2021.
|
|
83
|
Zhang Y, Wang H, Lu J, Lv Q, Yun B, Ge Z
and Yan L: Down-regulation of S1PR2 is correlated with poor
prognosis and immune infiltrates in cervical squamous cell
carcinoma and endocervical adenocarcinoma. Int J Immunopathol
Pharmacol. 37(03946320231178131)2023.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Zhou X, Liu J, Chen X, Zhou X, Xu B, Gan G
and Chen F: S1PR3 inhibition impairs cell cycle checkpoint via the
AKT/WEE1 pathway in oral squamous cell carcinoma. J Transl Med.
23(573)2025.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Evrard M, Wynne-Jones E, Peng C, Kato Y,
Christo SN, Fonseca R, Park SL, Burn TN, Osman M, Devi S, et al:
Sphingosine 1-phosphate receptor 5 (S1PR5) regulates the peripheral
retention of tissue-resident lymphocytes. J Exp Med.
219(e20210116)2021.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Coyle PK, Freedman MS, Cohen BA, Cree BAC
and Markowitz CE: Sphingosine 1-phosphate receptor modulators in
multiple sclerosis treatment: A practical review. Ann Clin Transl
Neurol. 11:842–855. 2024.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Shields N, Colwill M, Raspa V, Twum-Danso
Y, Poullis A, Patel K and Honap S: Sphingosine-1-Phosphate (S1P)
receptor modulators for the treatment of inflammatory bowel disease
(IBD): Mechanisms, clinical evidence, and practical insights.
Biomedicines. 13(2655)2025.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Chen S, Wu L, Lang B, Zhao G and Zhang W:
Sphingosine 1-phosphate receptor 1 modulators exert neuroprotective
effects in central nervous system disorders. Front Pharmacol.
16(1516991)2025.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Kruschel RD, Malone K, Walsh AN, Waeber C
and McCarthy FO: Discovery of sphingosine kinase inhibition by
modified Quinoline-5,8-Diones. Pharmaceuticals (Basel).
18(268)2025.PubMed/NCBI View Article : Google Scholar
|














