1. Introduction
As a major global health concern, non-alcoholic
fatty liver disease (NAFLD), now termed metabolic
dysfunction-associated steatotic liver disease (MASLD), is strongly
associated with sedentary lifestyles and suboptimal dietary habits
(1). High-fat, high-fructose,
Western diets may promote lipid accumulation in the liver, while
early-life nutritional insufficiency contributes to increased
susceptibility to metabolic disease in later life (2,3).
Despite the substantial differences in the nutritional profiles of
high-fat/sucrose diets and nutritional insufficiency), both
conditions result in hepatic mitochondrial dysfunction and
defective mitochondrial quality control (4). The metabolic function of the liver
depends on dynamic mitochondrial regulation that is affected by
fluctuating nutrient availability (5). Lipid-induced oxidative stress in
obesity or energy deprivation in malnutrition may contribute to
hepatocellular injury and lead to non-alcoholic steatohepatitis
(NASH) and fibrosis (6). Thus,
preserving mitochondrial health through nutritional adjustments is
key to maintaining normal liver function.
Mitochondrial health is maintained by mitophagy,
which selectively degrades damaged or dysfunctional mitochondria in
the liver (7). Efficient mitophagy
in liver cells may prevent the accumulation of excess reactive
oxygen species (ROS) and pro-apoptotic signaling (8). Two major mechanisms of mitophagy are
the ubiquitin (Ub)-dependent pathways [PTEN-induced kinase 1
(PINK1) and Parkin] and receptor-dependent pathways [Bcl-2
interacting protein 3 (BNIP3), NIP3-like protein X (NIX; also known
as BNIP3L) and FUN14 domain-containing protein 1 (FUNDC1)]
(8,9). These pathways result in mitochondrial
sequestration within autophagosomes, followed by lysosomal fusion
that eventually leads to degradation and recycling of mitochondrial
constituents (10). Mitophagic flux
is commonly inferred from autophagic markers, including LC3
lipidation on autophagosome membranes and concomitant degradation
of sequestome-1 (also known as p62) (11).
Mitochondrial renewal and quality control are
strongly modulated by exercise (12). By promoting mitochondrial biogenesis
through peroxisome proliferator-activated receptor (PPAR)γ
coactivator 1-α (PGC-1α) and maintaining fusion-fission balance,
exercise can modulate mitochondrial dynamics (13). A recent literature review reported
that mitophagic flux is stimulated under certain conditions
(14). In addition to
exercise-induced effects, diet composition and overall energy
balance influence mitophagy. Autophagy and mitophagy are activated
by caloric restriction (CR) and fasting, and 24-h fasting strongly
enhances BNIP3-associated mitophagy in the murine liver (15). Conversely, excessive high-calorie,
high-fat intake impairs autophagy, which is evidenced by p62
accumulation and defective autophagic flux in the steatotic livers
of obese animals and humans (16,17).
Biological sex and maternal or paternal programming
are also recognized as modifiers of mitophagy in the liver.
Sex-specific differences in mitochondrial function, hormone
signaling and metabolic flexibility may influence basal mitophagy
activity and its responsiveness to metabolic stress or exercise
(18). Maternal and paternal
nutritional status or physical activity before and during early
development can also induce long-lasting metabolic programming
effects that influence hepatic mitochondrial quality control later
in life (19,20). The interplay of these variables
likely accounts for much of the variability observed across
experimental models, underscoring the need to account for sex and
developmental background when drawing conclusions from mitophagy
data.
Given the growing global burden of metabolic
disease, understanding how exercise and nutritional factors jointly
regulate mitophagy has become increasingly urgent. The present
review examines the effect of different exercise modalities
combined with nutritional interventions on hepatic mitophagy and
liver recovery. Particular focus is placed on the PINK1/Parkin and
BNIP3/NIX pathways, as well as tissue-specific, sex-related and
intergenerational programming factors that help explain the
variability observed across studies. Given that impaired
mitochondrial quality control is a common pathogenic thread across
steatotic, pharmacologically induced and other metabolic forms of
liver injury, studies conducted outside the strict NAFLD/MASLD
context are incorporated where they provide mechanistic insights
relevant to the broader aims of the current review.
2. Literature review methodology
The literature search was conducted using PubMed
(https://pubmed.ncbi.nlm.nih.gov), Scopus
(https://www.scopus.com), Cochrane (https://www.cochranelibrary.com) and Google
Scholar (https://scholar.google.com) databases
for articles published between March 2016 and May 2026. The search
strategy combined key words related to mitochondrial quality
control (‘mitophagy’, ‘mitochondrial quality control’), liver
disease (‘non-alcoholic fatty liver disease’, ‘liver’,
‘metabolic-associated liver disease’), and interventions
(‘exercise’, ‘diet’, ‘fasting’, ‘caloric restriction’). The Boolean
operators ‘AND’ and ‘OR’ were used to create the key word
combinations. The study included only original articles that
investigated mitophagy or mitochondrial quality control in the
liver in relation to exercise with or without dietary or
pharmacological interventions. Studies on non-liver tissues and
non-English articles were excluded. Abstracts and titles were
screened, followed by full-text assessment. Given the heterogeneity
of models and outcomes, a narrative synthesis was adopted to
integrate findings while highlighting mechanistic insights and
knowledge gaps. A total of 19 articles were finally included in the
present review (19,21-38).
3. Mechanistic pathways of mitophagy and
their regulation by exercise and diet
Mitophagy begins with the sensing of dysfunctional
mitochondria, progresses to autophagosome formation around the
organelle and culminates in lysosomal degradation through a
coordinated multistep process (8).
Rather than functioning independently, Ub-dependent and
receptor-mediated mitophagy pathways often cooperate to ensure
efficient mitochondrial turnover (10). Different stress cues, such as
hypoxia and mitochondrial depolarization, can activate BNIP3/NIX-
and Parkin-mediated mitophagy, respectively, and converge on the
same mitochondrial target (39).
Both exercise and dietary interventions influence the balance and
coordination of these mitophagy pathways (6,40).
The PINK1/Parkin pathway is activated by
mitochondrial damage that stabilizes PINK1 on the outer membrane
and drives Parkin-dependent ubiquitination of mitochondrial
proteins, including voltage-dependent anion channel and mitofusin
(MFN)2(41). Adaptor proteins such
as p62, neighbor of BRCA1 gene 1 protein (NBR1) and optineurin
recognize Ub signals and link damaged mitochondria to LC3-positive
autophagosomes, which promote their degradation (11). p62 is degraded together with
mitochondrial cargo, and its protein levels are commonly used as an
indicator of mitophagic flux (11).
Exercise is associated with increased LC3 lipidation and reduced
p62 levels, which reflects active autophagic flux. However,
high-fat feeding alone often results in hepatic p62 accumulation,
which is consistent with disrupted mitochondrial turnover (16).
An alternative mitophagy pathway involves
receptor-mediated processes that do not require Parkin activation
(9). BNIP3 and NIX are
stress-inducible proteins that respond to hypoxia, energetic stress
and developmental cues by localizing to the mitochondrial outer
membrane (39). These receptors
link mitochondria directly to autophagosomes in a Ub-independent
manner via LC3-interacting regions, while FUNDC1 adds further
regulation through hypoxia-dependent phosphorylation (9). Exercise can swiftly engage
receptor-mediated mitophagy, as BNIP3 levels rise in hepatic
mitochondria immediately post-exercise (0 h), potentially preceding
robust Parkin involvement (31).
Consistent with this, several studies have reported that exercise
preferentially activates BNIP3-dependent mitophagy rather than the
Parkin pathway (42-44).
In a weight-loss context, obese mice that exercised had marked
upregulation of BNIP3 in the liver, which coincided with
restoration of mitophagic flux, whereas diet-induced weight loss
without exercise did not upregulate BNIP3 and yielded less
mitophagy improvement (28). Thus,
exercise performed under fasting conditions may engage
receptor-mediated pathways more strongly than exercise during the
fed state. These distinctions may partly explain inconsistencies
among intervention studies.
These two pathway arms are linked by several
bidirectional regulatory interactions rather than operating in
parallel. BNIP3 interacts with PINK1 to inhibit its proteolytic
cleavage, stabilizes PINK1 and reinforces Ub-dependent mitophagy.
Conversely, Parkin can ubiquitinate NIX, and ubiquitinated NIX
subsequently recruits NBR1 to further promote mitophagy (45). AMP-activated protein kinase (AMPK)
adds further complexity: Beyond promoting unc-51-like autophagy
activating kinase 1 (ULK1)-dependent autophagosome formation, AMPK
can phosphorylate and stabilize BNIP3, directly linking energy
sensing to receptor-mediated mitophagy independently of PINK1
(46,47). Nicotinamide adenine dinucleotide
(NAD)+-driven activation of sirtuin (SIRT)1, operating
downstream of AMPK, links cellular energy status to mitophagy gene
expression: SIRT1 deacetylates forkhead box O3 (FOXO3) to
transcriptionally upregulate BNIP3 and LC3, while also targeting
LC3 directly to facilitate autophagosome maturation (48,49).
When engaged by exercise or fasting, these molecules [AMPK, SIRT1,
FOXO3 and mammalian target of rapamycin (mTOR)] function as an
integrated regulatory network governing both mitophagy arms in
concert but not as independent switches. High-fat diet
(HFD)-induced insulin resistance or lysosomal dysfunction can break
down network coordination, explaining the paradoxical accumulation
of upstream mitophagy markers alongside defective clearance
commonly observed in NAFLD/MASLD (50).
Exercise intersects with the mitophagy machinery at
more than one regulatory level. High-intensity exercise or
prolonged endurance exercise can acutely stress mitochondria (such
as through increased calcium cycling, ROS and fluctuations in
membrane potential). In the liver, a single treadmill exercise bout
does not immediately elevate Parkin on mitochondria, but evidence
of Parkin-independent ubiquitination has emerged within ~2 h
post-exercise (31). This
Parkin-independent ubiquitination likely involves alternative E3
ligases, such as mitochondrial Ub ligase 1, which ubiquitinates
mitochondrial substrates including MFN2 and ULK1 to facilitate
mitophagic clearance independently of the canonical PINK1/Parkin
axis (9,51). This suggests that mitochondrial
disturbances during exercise may first activate receptor pathways
(such as BNIP3) and engage PINK1/Parkin only later, if needed. ROS
generated during exercise should not be viewed solely as harmful
by-products; they serve as both triggers and effector molecules in
the mitochondrial quality control pathways (45,52).
Moderate increases in mitochondrial ROS serve as signaling
molecules that stabilize PINK1 and activate BNIP3/NIX, whereas
excessive ROS production due to accumulated defective mitochondria
induces lipid peroxidation, inflammation and cell death (45,53).
Exercise training can induce the expression of
autophagy and mitophagy-related genes (14,40).
Endurance training elevates PGC-1α, which co-activates
transcription factor EB and other transcription factors for
lysosomal and autophagy genes (13). A previous study showed that chronic
exercise increases hepatic BNIP3 protein concentration (24). Additionally, exercise can affect
FOXO3 activity: FOXO3 is activated by endurance exercise and is
known to transcriptionally induce BNIP3 and LC3 in skeletal muscle,
linking exercise to increased autophagy gene expression (54). Diets also serve a role: Fasting
increases BNIP3 expression in the liver and muscle via FOXO3 and
hypoxia-inducible factor 1α (HIF-1α) signaling, whereas a HFD may
downregulate BNIP3 (observed as ~30% lower BNIP3 protein in the
livers of Western diet-fed mice) (22,55).
LC3-II levels, as a proxy for autophagosome
formation, and downstream lysosomal clearance are responsive to
exercise status and dietary composition (14). A recurring dissociation in metabolic
liver disease is intact PINK1/Parkin signaling alongside blocked
lysosomal clearance; doxorubicin-treated livers exemplify this,
where exercise preconditioning restores mitochondrial function via
SIRT3-mediated deacetylation rather than through enhanced
mitophagic flux as such (26).
These data collectively underscore that upstream pathway
activation, while required, does not guarantee functional
mitochondrial removal unless the full degradative cascade proceeds
to completion. ‘Stalled’ mitophagy has been documented in the
livers of HFD-fed mice, where abundant phosphorylated (p)-Ub serine
65 (Ub Ser65) and p62 coexist with minimal Parkin activation,
indicating that mitophagy is initiated but cannot proceed to full
execution. When paired with weight loss, exercise resolves this
block, with concurrent reductions in p62 and phosphorylated (p)-Ub
serving as evidence of restored flux (28).
The direction of exercise-induced changes in hepatic
BNIP3, Parkin and LC3-II varies considerably across studies,
ranging from marked upregulation to normalization or suppression
following training (21,24,26,29).
Baseline disease severity, exercise modality, duration, sex,
nutritional status and tissue sampling timing all likely contribute
to this variability. Notably, acute exercise transiently amplifies
mitophagy signaling, whereas repeated training appears to dampen
pathological overactivation by improving mitochondrial efficiency
(24,31). What appears contradictory may thus
reflect distinct adaptive phases rather than genuine biological
discordance.
Gonçalves et al (21) and Deng et al (37) reported that HFD suppressed hepatic
PINK1 and Parkin while exercise restored them, indicating recovery
clearance capacity (21,37) Hinkley et al (26) reported that exercise preconditioning
improved mitochondrial function in doxorubicin-treated livers
through SIRT3-mediated deacetylation, even as PINK1 and p62
remained persistently elevated. This introduces further
interpretative complexity, as the concentrations of markers and
functional recovery were dissociated. Zou et al (33) observed that HFD-fed zebrafish
developed a suppressed mitophagy profile of reduced PINK1 and
Parkin, and elevated p62. Notably, chronic swimming exercise did
not restore PINK1 but increased Parkin beyond control levels;
however, p62 declined, reflecting reactivation of flux rather than
simple marker normalization (33)
This pattern suggests that exercise can restore effective
mitophagic flux even when upstream markers appear dysregulated at
baseline, reinforcing the concept that marker concentrations alone
are insufficient indicators of functional mitophagy without
concurrent assessment of downstream clearance.
McCoin et al (31) and Dethlefsen et al (24) highlighted how the temporal scale of
exercise shapes its mitophagic signature. Acute exercise activated
flux within 2 h post-exercise, whereas chronic training produced
LC3-II/LC3-I normalization that, without proper flux assessment,
could be misattributed to reduced mitophagy (24,31).
Finally, Li et al (34)
demonstrated that moderate-intensity continuous training (MICT) and
high-intensity interval training (HIIT) had different primary
effectors. MICT preferentially restored PINK1-dependent signaling
and reduced endoplasmic reticulum (ER) stress, whereas HIIT more
strongly promoted optic atrophy protein 1 (OPA1)-mediated
mitochondrial fusion. This highlights that exercise can yield
divergent marker profiles that are not contradictory but rather
reflect different mechanistic entry points into the mitophagy
network (34).
In conclusion, exercise and diet can modulate
mitophagy at the level of initiating signals (such as PINK1
stabilization and BNIP3 expression), upstream signaling (for
example, AMPK, mTOR and SIRTs) and downstream execution (including
autophagosome formation and lysosomal degradation). Fig. 1 summarizes how exercise- and
nutrition-related signals regulate mitophagy through Ub-dependent
and receptor-mediated pathways. Exercise, fasting and CR activate
AMPK, which suppresses mTOR, phosphorylates ULK1 and engages
SIRT1-FOXO3 signaling to transcriptionally induce BNIP3 and LC3.
They also affect mitochondrial stress signals and promote
PINK1/Parkin-mediated ubiquitination and LC3-positive autophagosome
formation, while stress cues such as hypoxia and estrogen (via
FOXO3 and HIF-1α) also engage receptor-mediated mitophagy via
BNIP3, NIX and FUNDC1. While coordinated pathway activity supports
mitochondrial quality and respiration, chronic consumption of
high-fat or Western diets compromises lysosomal function, causing
impaired mitophagy and buildup of dysfunctional mitochondria
commonly observed in NAFLD.
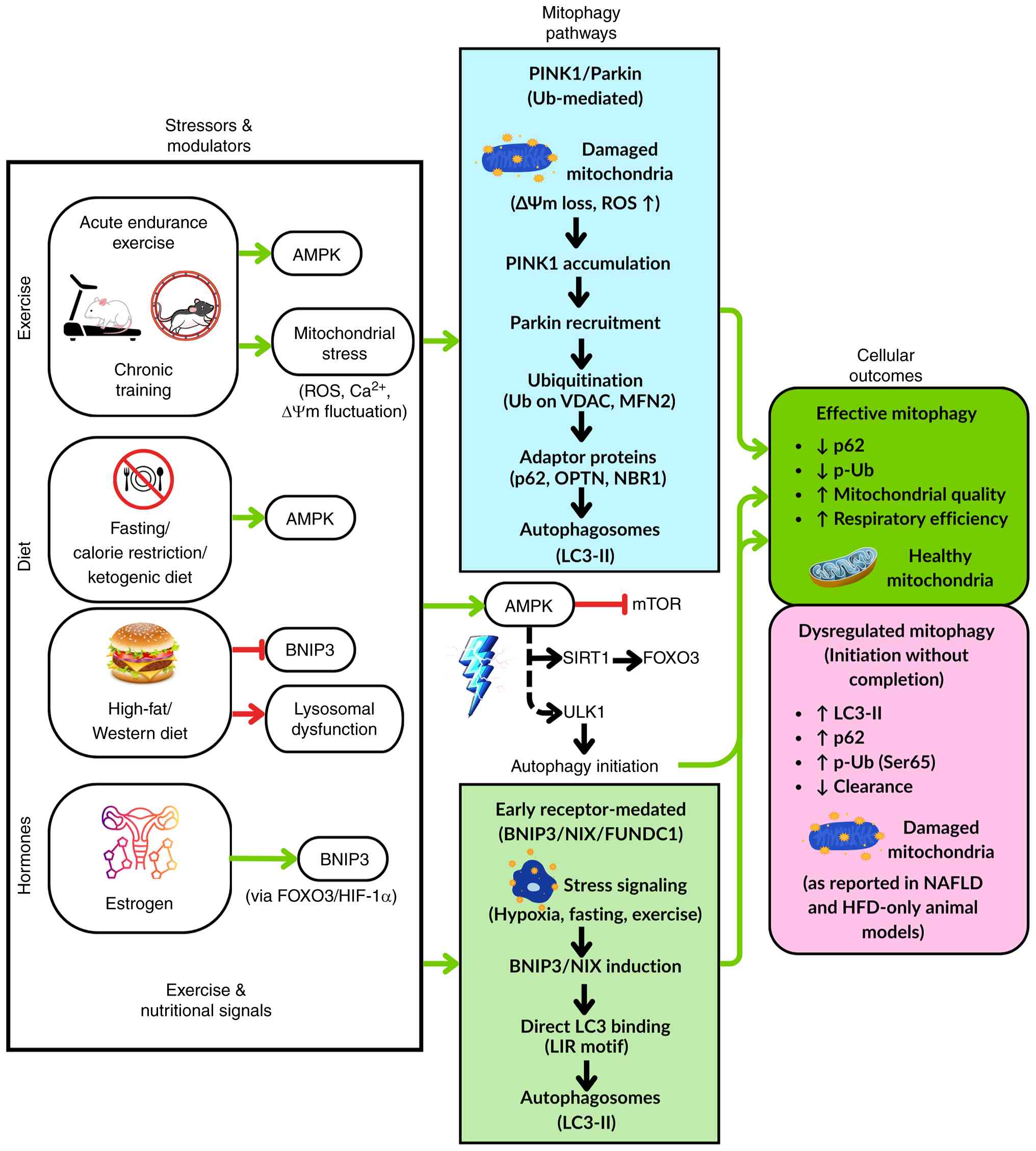 | Figure 1Integrated regulation of hepatic
mitophagy by exercise and nutritional signals. AMPK, AMP-activated
protein kinase; BNIP3, Bcl-2 interacting protein 3; FOXO3, forkhead
box O3; FUNDC1, FUN14 domain-containing protein 1; HFD, high-fat
diet; HIF-1α, hypoxia-inducible factor 1α; LIR, LC3-interacting
region; MFN2, mitofusin 2; mTOR, mechanistic target of rapamycin;
NAFLD, non-alcoholic fatty liver disease; NBR1, neighbor of BRCA1
gene 1 protein; NIX, NIP3-like protein X; OPTN, optineurin; p-Ub,
phosphorylated-ubiquitin; PINK1, PTEN-induced kinase 1; ROS,
reactive oxygen species; SIRT1, sirtuin 1; ULK1, unc-51 like
autophagy activating kinase 1; VDAC, voltage-dependent anion
channel; ΔΨm, mitochondrial membrane potential. |
4. Sex- and tissue-specific differences in
exercise-induced mitophagy
Sex differences influence basal hepatic mitophagy
and adaptive response to exercise. Higher intrinsic mitochondrial
quality and lower mitophagic flux have been found in female
individuals, whereas male individuals have higher baseline
mitophagic flux that coincides with reduced coupling efficiency and
elevated oxidative stress (23,25).
Even under sedentary conditions, female mice have higher electron
transport chain content, greater respiratory capacity and reduced
ROS production, despite comparable or elevated BNIP3 and Parkin
levels, this suggests a reduced reliance on active mitochondrial
turnover (23,25). After a period of voluntary exercise,
male mice have been shown to exhibit notable improvements:
Mitochondrial coupling improves and their previously elevated
mitophagy markers normalize to ‘female-like’ levels (25). In female mice, exercise does less in
terms of mitophagy marker changes (since they are already
considered optimal); however, the capacity to further increase
mitochondrial respiration with training is blunted in female mice
with BNIP3 knockout. This indicates that BNIP3-mediated mitophagy
is important for females to gain maximal benefit from exercise,
even if their baseline mitophagic flux is low. In addition, partial
PGC-1α deficiency has been reported to not have as large an effect
on blunting exercise-induced mitochondrial respiratory adaptations
compared with BNIP3 knockout in female mice, indicating that
quality control (mitophagy) rather than just biogenesis may be the
limiting factor for female adaptation (25).
However, the characterization of female individuals
as having uniformly ‘lower’ mitophagic flux requires qualification.
Moore et al (27) reported
that female Wistar rats had higher baseline LC3-II/I,
autophagy-related (ATG)12-ATG5 conjugate and p-AMPK/AMPK ratios
than male rats, thus suggesting greater basal autophagy and
energy-sensing activity, a finding that appears to contradict the
lower flux reported by McCoin et al (25). This apparent contradiction is best
explained by the methodological gap between the studies. Von
Schulze et al (23) and
McCoin et al (31) isolated
hepatic mitochondria and blocked lysosomal degradation to measure
mitophagy-specific flux, whereas Moore et al (27) relied on whole-liver homogenates for
general autophagy markers, a distinction that precludes direct
comparison (23,27,31).
In whole-tissue lysates, elevated LC3-II or ATG12-ATG5 more
probably captures global autophagic turnover than
mitochondria-targeted clearance, an interpretation reinforced by
the observation that general autophagy markers were elevated as a
sex effect rather than tracking mitophagy-specific clearance in the
study (27). These observations
collectively suggest that female individuals operate with higher
general autophagic activity, but lower and more tightly regulated
mitophagy-specific flux, a functional division that investigators
should account for when comparing mitophagy between the sexes.
When female mice undergo ovariectomy (OVX), the
liver mitophagy response to acute exercise is notably impaired,
especially if they are also being fed a HFD. In sham-operated
female mice (with intact ovaries), an acute treadmill run has been
reported to induce robust mitophagic flux in the liver, evidenced
by increased mitochondrial p62 accumulation under lysosomal
blockade and, by proteomics, greater recruitment of LC3-II and
adaptor proteins such as optineurin, NBR1 and nuclear dot protein
52 than in OVX mice. Female mice undergoing OVX and being fed a
low-fat diet have been reported to still show a partial increase in
mitophagy with exercise, whereas those fed a HFD exhibit almost no
increase: Exercise fails to induce the normal signs of mitophagy in
the absence of estrogen under lipid overload. Concomitantly,
HFD-fed mice undergoing OVX had blunted exercise-induced
improvements in mitochondrial respiration and redox signaling. This
suggests estrogen is a permissive factor for effective mitophagy
during exercise. In support of this, loss of ovarian function via
OVX blunted exercise-induced recruitment of DRP1 to hepatic
mitochondria and reduced mitochondrial H2O2
signaling, thereby impairing the activation of exercise-induced
mitophagic flux, particularly under HFD feeding (36). Franczak et al (36) revealed that impaired mitophagic flux
in OVX animals fed a HFD extends beyond autophagosome formation to
the fission machinery that precedes it. Diminished DRP1 and
mitochondrial fission factor (MFF) recruitment to mitochondria
suggests that estrogen loss may interfere with the segregation of
damaged organelles before they can be targeted for autophagic
clearance (36). Post-menopausal
women and those with ovarian dysfunction may therefore derive less
mitochondrial benefit from exercise, a gap that hormonal or dietary
support could help to close.
Sex-specific hepatic mitophagy is further shaped by
metabolic context. A single exercise bout has been reported to fail
to alter whole-liver autophagy markers in chow-fed female mice;
however, lysosomal inhibition in isolated mitochondrial fractions
reveals a clear flux increase during the early post-exercise
recovery period (~2 h post-exercise) (31). The discrepancy highlights that
transient mitophagic events in the liver require assays performed
in isolated mitochondrial fractions combined with lysosomal
inhibition, rather than whole-liver homogenates, and some reported
sex differences may reflect methodology rather than true biology.
Male individuals appear to rely more heavily on exercise-induced
mitophagy for quality control, whereas female individuals may
benefit more from interventions that preserve baseline
mitochondrial integrity.
Fig. 2 summarizes
how sex, ovarian function and diet interact to influence
exercise-induced hepatic mitophagy. In female mice with intact
ovaries, high baseline mitochondrial quality shifts the exercise
response toward biogenesis rather than mitophagy, although
BNIP3-dependent clearance remains necessary for full adaptation.
Male mice rely more on mitophagic turnover to achieve comparable
mitochondrial quality, which normalizes their elevated markers to
female-like levels with training. When OVX is combined with HFD,
impaired DRP1 and MFF recruitment disrupts fission-dependent
organelle segregation and abolishes exercise responsiveness, a
deficit partially rescued by low-fat feeding. Sex, hormonal status
and diet therefore act together to determine hepatic mitophagic
capacity.
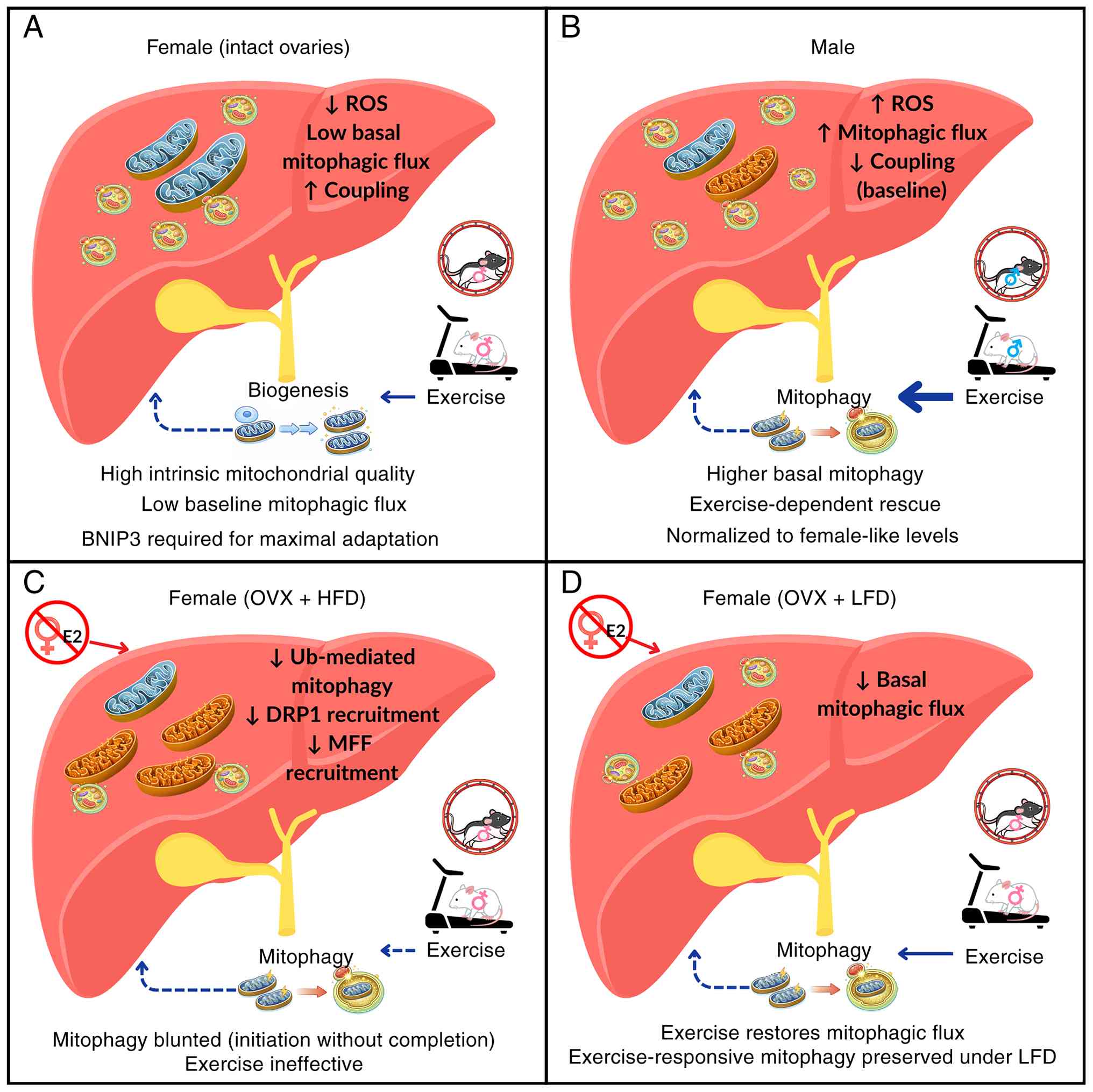 | Figure 2Sex-, hormone- and diet-dependent
regulation of hepatic mitophagy and mitochondrial quality control.
(A) Female mice with intact ovaries have high mitochondrial quality
and low basal mitophagic flux, which favors exercise-induced
biogenesis. (B) Male mice have higher basal mitochondrial stress
and mitophagy, with exercise enhancing mitochondrial turnover. (C)
OVX combined with HFD blunts Ub-mediated mitophagy and renders
exercise ineffective, (D) whereas ovariectomized females on an LFD
retain exercise-responsive mitophagy. BNIP3, Bcl-2 interacting
protein 3; DRP1, dynamin-related protein 1; E2, estradiol; HFD,
high-fat diet; LFD, low-fat diet; OVX, ovariectomy; Ub, ubiquitin;
MFF, mitochondrial fission factor; ROS, reactive oxygen
species. |
5. Effects of exercise modality, intensity
and duration on mitophagy
Not all exercise engages mitophagy to the same
degree. The mitophagic response depends heavily on the nature of
the exercise stimulus. Mild daily activity preserves basal
autophagic function but falls short of reversing the substantial
mitophagy suppression observed in NASH (21). Voluntary wheel running has been
shown to improve general autophagy in Western diet-fed mice but
fails to restore BNIP3, pointing to a stimulus threshold that
separates general autophagic flux from mitophagy-specific induction
(22). LC3 lipidation and p62
clearance respond to modest AMPK-ULK1 activation, whereas BNIP3
upregulation requires the sustained HIF-1α and FOXO3 signaling that
only higher-intensity or longer-duration exercise reliably
generates. Dethlefsen et al (24) confirmed that this threshold can be
crossed: Treadmill training elevated total hepatic BNIP3
concentration in high-fat high-fructose diet (HFF)-fed mice despite
the adverse dietary background.
Acute and chronic exercise engage mitophagy through
different temporal patterns. McCoin et al (31) reported that bulk autophagy markers
were unchanged immediately after a 1-h treadmill run, whereas
hepatic mitophagic flux was clearly detectable within 2 h in
appropriately measured fractions. The absence of Parkin or DRP1
changes alongside simultaneous BNIP3 and optineurin recruitment
indicated concurrent rather than sequential engagement of both
mitophagy arms through Parkin-independent ubiquitination. Chronic
repetition of the same stimulus, however, blunted this acute flux
response (31). Dethlefsen et
al (24) observed that an acute
bout of exercise activated AMPK and increased PGC-1α mRNA in
sedentary mice fed a HFF, but the same acute stimulus yielded a
much smaller spike in those signals for mice that had undergone 5
weeks of training. Training also normalized the elevated
LC3-II/LC3-I ratio observed in fatty liver and increased total
BNIP3 levels without further increasing its active dimeric form,
indicating an expansion of mitophagy capacity rather than sustained
flux at rest (24). In this
context, acute exercise serves as a transient stimulus for
mitophagy, whereas chronic training resets the baseline capacity
for mitochondrial turnover.
Mitophagy-induced adaptation to exercise is also
influenced by its intensity. In male Wistar rats with
dexamethasone-induced NAFLD, both moderate- and high-intensity
treadmill training attenuated markers of excessive mitophagy
signaling, significantly reducing the elevated hepatic Bcl-2 and
LC3 gene expression seen in untreated animals with NAFLD, while
concurrently increasing p62 protein levels (35). In a zebrafish model of NAFLD, Zou
et al (33) reported that a
HFD reduced PINK1 and Parkin while increasing p62, a pattern
consistent with suppressed mitophagy. Swimming exercise pushed
Parkin concentrations higher while reducing the levels of p62, an
outcome that reflects reactivation of a dysfunctional Parkin pool
rather than marker normalization (33). This dose-dependent benefit fits the
mitohormesis framework, in which a controlled metabolic challenge
promotes mitochondrial adaptation. The limits of this framework are
demonstrated by Mokhtari-Andani et al (35), in which dexamethasone-treated rats
exhibited paradoxically elevated p62 concentrations after exercise
despite reduced Bcl-2 and LC3gene expression.
The time of day at which exercise is performed also
adds a further layer of regulation to hepatic mitophagy outcomes.
Zhang et al (30)
demonstrated that identical exercise performed during the active
phase (evening for mice) was more effective than rest-phase
exercise in restoring mitochondrial dynamics and reducing aberrant
mitophagy signaling in diabetic mice. Specifically, diabetic mice
had elevated hepatic Parkin and LC3 concentrations (indicating
excessive or maladaptive mitophagy and apoptosis); both morning and
evening exercise significantly reduced Parkin and LC3
concentrations, with no significant difference between the two
timings for these markers. Furthermore, both morning and evening
exercise reduced Parkin and LC3 concentrations but through
different primary mechanisms. Morning exercise primarily restored
glucose handling and insulin sensitivity via glucose transporter
type 4, whereas night exercise primarily realigned the circadian
locomotor output cycles kaput (CLOCK)-mitophagy axis, which reduced
CLOCK upregulation and improved mitochondrial ultrastructure
(30). This represents a
within-study contradiction where the same intervention at different
times yields mechanistically distinct, partially non-overlapping
effects, neither of which is comprehensively superior. For humans,
this raises the question of whether morning or evening exercise may
differently impact mitophagy in the liver or muscle (especially in
metabolic disease or diabetes); however, to the best of our
knowledge, human data are not yet available.
The comparison of HIIT and MICT has attracted
growing attention, as each modality may engage distinct
mitophagy-related effectors. Li et al (34) reported that MICT more effectively
rescued PINK1 in HFD-fed male C57BL/6J mice, normalized
LC3-II/LC3-I and BNIP3, and reduced ER stress, whereas HIIT was
superior in restoring OPA1-mediated fusion and insulin signaling
via PI3K/AKT/GSK3β; taken together, this positions MICT as the
stronger driver of canonical PINK1/Parkin-dependent mitophagic
flux. HIIT that restored Parkin without restoring PINK1 suggests
Parkin can be recruited through PINK1-independent mechanisms,
possibly via alternative E3 ligases or non-PINK1 p-Ub signals under
HIIT-specific energetic conditions. Deng et al (37) further complicated this by reporting
that HIIT was superior to MICT for p-AMPK activation and
mitochondrial improvement, implying a stronger HIIT-driven upstream
stimulus for PINK1/Parkin signaling. The divergence between studies
more plausibly reflects differences in animal model, HFD duration
and outcome measures than genuine biological contradiction. Li
et al (34) centered their
analysis on mitochondria-associated membrane (MAM) integrity and ER
stress, whereas Deng et al (37) prioritized AMPK-driven metabolic
remodeling and the gut-liver axis. This suggests that MICT and HIIT
access the mitophagy network through partially distinct entry
points rather than producing opposing effects (34,37).
Wang et al (56) extended the mechanistic picture by
demonstrating that HIIT can promote M1-to-M2 macrophage
polarization in the liver of a HFD- and streptozotocin-induced type
2 diabetes mellitus (T2DM) mouse model through RAR-related orphan
receptor α/Krüppel-like factor 4 signaling. As M1 macrophages
impair mitophagic flux and lysosomal function through ROS and
cytokine release, this shift in inflammatory tone may represent an
indirect but functionally important route by which HIIT supports
the restoration of mitophagy (56).
At the transcriptional level, Durak et al
(38) reported that 24 weeks of
exercise for HFD-fed female mice restored ATG3, PINK1, MFN1 and
Parkin expression levels toward normal, supporting exercise-driven
recovery of mitophagy and fusion gene programs. The upward
direction of these changes superficially conflicts with Dethlefsen
et al (24), where chronic
training reduced LC3-II/LC3-I in HFF-fed males. In Durak et
al (38), HFD feeding
suppressed hepatic Parkin and MFN1 expression, and exercise
restored these toward control levels, whereas PINK1 and ATG3 were
further upregulated by exercise beyond both control and HFD levels.
In Dethlefsen et al (24),
mice had pathologically elevated LC3-II concentrations, and its
reduction reflected normalization. Both outcomes represent the
restoration of appropriate mitophagy regulation by exercise, and
the comparison reinforces that marker direction alone is
uninformative without knowledge of the starting point.
In conclusion, exercise must reach a certain
threshold of intensity/duration to robustly engage mitophagy. Light
physical activity appears to maintain overall health and support
baseline autophagy, yet substantial mitochondrial impairment may
only respond to more robust training stimuli. While vigorous
exercise acutely activates mitophagy, ongoing training appears to
adjust the baseline state of mitochondrial quality control.
Integrating occasional high-intensity sessions with regular
moderate activity may best balance short-term mitophagy activation
and long-term mitochondrial remodeling. These findings indicate
that exercise does not universally increase mitophagy; rather,
exercise restores mitochondrial quality control according to the
metabolic state and the degree of pre-existing mitochondrial
dysfunction.
Table I compiles
animal studies examining the interplay between exercise modalities
and dietary or metabolic factors in shaping hepatic mitophagy.
Structured endurance training, HIIT, MICT and voluntary exercise
are contrasted across metabolic stress models, with the table
mapping how the intensity and duration of each modality translate
to distinct hepatic mitochondrial outcomes.
 | Table IAnimal studies examining the effects
of exercise and dietary interventions on hepatic mitophagy and
liver recovery. |
Table I
Animal studies examining the effects
of exercise and dietary interventions on hepatic mitophagy and
liver recovery.
| First author,
year | Type of
exercise | Type of
diet/nutraceutical intervention | Method | Results | Key findings | (Refs.) |
|---|
| Gonçalves et
al, 2016 | Treadmill running
or VWR | HFD | HFD-induced NASH
rats (9 weeks) underwent VPA or ET (8 weeks); hepatic mitochondrial
permeability, biogenesis, dynamics, mitophagy and apoptosis markers
were assessed. | HFD/NASH: ↑ mPTP
susceptibility, ↑ Bax/Bcl-2 ratio, ↓ TFAM/MFN1/PINK1/Parkin VPA: ↑
PGC-1α/Beclin-1, ↓ apoptotic signaling ET: ↓ mPTP susceptibility, ↑
PGC-1α/TFAM/MFN1/MFN2/PI NK1/Parkin, ↓ Bax/Bcl-2 ratio/caspase
activity | ET more effectively
restored mitochondrial quality control than VPA by enhancing
biogenesis, fusion, mitophagy-related signaling and apoptosis
regulation in NASH. | (21) |
| Rosa-Caldwell et
al, 2017 | VWR | WD | C57BL/6J mice were
fed normal or WD (4 weeks) and assigned to SED or VWR groups (4
weeks); hepatic histology and mitochondrial biogenesis, dynamics
and autophagy/mitophagy markers were assessed. | WD: ↓ Mitochondrial
content (COX IV, Cyt c)/PGC-1α/fusion/fission markers VWR (NC and
WD): ↑ LC3-II/LC3-I, ↓ p62 WD: ↓ BNIP3 WD + VWR: partial protection
from steatosis (trend, NS vs. WD-SED) | Moderate physical
activity enhanced basal hepatic autophagy and attenuated early
steatosis, while WD suppressed mitochondrial quality control and
BNIP3-linked mitophagy capacity. | (22) |
| Von Schulze et
al, 2018 | VWR | LFD | Male and female WT,
liver-specific PGC-1α+/-, and BNIP3-/- mice;
SED vs. VWR (4 weeks); hepatic mitochondrial respiration, coupling,
ROS (H2O2) and mitophagy (LC3-II, p62) were
detected. | Female mice: ↑ ETS
content/respiratory capacity/coupling efficiency; ↓
H2O2 emission/mitophagic flux (LC3-II↓),
independent of genotype Male mice: Baseline ↓ coupling and ↑
mitophagy; exercise resulted in ↑ coupling, ↓ LC3-II
PGC-1α+/- or BNIP3-/-: Minimal effect on
sex-specific adaptations | Sex primarily
determined hepatic mitochondrial adaptation: Female mice showed
intrinsically efficient mitochondria, whereas male mice relied more
on exercise-induced mitophagy for mitochondrial quality
control. | (23) |
| Dethlefsen et
al, 2018 | Treadmill
running | HFF | Male PGC-1α LKO and
littermate control mice were fed a control diet/HFF (13 weeks),
underwent treadmill training (5 weeks), with or without an
additional acute exercise bout; hepatic autophagy and mitophagy
markers, AMPK-mTOR, and PGC-1α mRNA were assessed. | HFF diet: ↑
Parkin/BNIP3 dimer/LC3-II/LC3-I resulting in dysregulated
autophagy/mitophagy Exercise training: ↓ LC3-II/LC3-I, ↑ PGC-1α
mRNA/total BNIP3 Acute exercise: ↑ p-AMPK/PGC-1α mRNA in untrained
HFF-fed mice (minimal effect) PGC-1α LKO: Minimal impact on
mitophagy | HFF disrupted
hepatic autophagy regulation while increasing mitophagy signaling
capacity. Exercise training partially normalized autophagy and
enhanced mitophagic capacity largely independent of hepatic PGC-1α
. | (24) |
| Cunningham et
al, 2018 | VWR | WD | Female Wistar rats
assigned to normal chow diet or WD with or without VWR during
gestation. Offspring (male and female) assessed in young adulthood
for hepatic steatosis, mitochondrial biogenesis markers and
autophagy/mitophagy markers. | Maternal WD: ↑
Hepatic steatosis (male offspring) Maternal exercise: ↑ TFAM/PPARγ;
↑ NRF2 (all offspring); ↑ BNIP3/ATG12-ATG5 (WD male offspring); Sex
effect (female vs. male): ↑ p62, BNIP3, ATG12:5, LC3-II/LC3-I and
Keap1, ↓ Parkin Diet effect: ↑ LC3-II/LC3-I in male SED offspring
(WD vs. ND) | Maternal physical
activity enhanced hepatic mitochondrial biogenesis and
mitophagy-related signaling in offspring, with pronounced
sex-dependent effects, despite limited early protection against
steatosis. | (19) |
| McCoin et
al, 2019 | VWR | HFD | Male and female WT,
liver-specific PGC-1α heterozygous and BNIP3-knockout mice were fed
a HFD (16 weeks), with or without VWR (final 8 weeks). Hepatic
mitochondrial respiration, coupling efficiency, ROS emission and
mitophagy markers were assessed. | Female mice: ↑
Coupling efficiency, ↓ H2O2
emission/steatosis/mitophagy Exercise: ↑ Respiratory capacity
mainly in WT and liver-specific PGC-1α heterozygous female mice,
blunted in BNIP3-knockout female mice Male mice required exercise
to normalize coupling and mitophagy to female-like levels | Hepatic
mitochondrial responses to HFD and physical activity are primarily
sex dependent. Female mice exhibited intrinsic mitochondrial
efficiency, whereas male mice relied more on exercise-induced
mitophagy for quality control. | (25) |
| Hinkley et
al, 2019 | Treadmill
running | Pharmacological
hepatotoxicity model (DOX)-, no dietary manipulation | Female SD rats
underwent treadmill training (10 days) or were SED, followed by
acute DOX administration. Hepatic mitochondrial respiration,
coupling efficiency, oxidative capacity, mitophagy, mitochondrial
biogenesis, protein acetylation and SIRT3 were assessed. | DOX (SED): ↑ State
4 respiration (proton leak)/PINK1/p62/protein acetylation, ↓
RCR/citrate synthase/NRF1/SIRT3 Exercise + DOX: ↓ State 4
respiration, ↑ RCR/citrate synthase/mitochondrial efficiency,
attenuation of protein hyperacetylation, preservation of SIRT3,
PINK1 and p62 remained ↑, and Parkin remained ~ | Exercise
preconditioning prevented DOX-induced hepatic mitochondrial
dysfunction primarily by preserving oxidative capacity and protein
deacetylation status rather than by suppressing mitophagy
signaling. | (26) |
| Moore et al,
2020 | VWR | KD, WD | Male and female
Wistar rats with access to VWR were fed SC, WD or KD (7 weeks).
Hepatic lipid content, oxidative stress, mitochondrial biogenesis
and autophagy/mitophagy markers were assessed. | WD: ↓ LC3-II/LC3-I,
↑ p62 (impaired autophagy/mitophagy markers) KD + exercise:
Suppressed hepatic de novo lipogenesis, ↑ PGC-1α, TFAM,
citrate synthase Female rats: Higher baseline markers of
mitochondrial biogenesis, autophagy/mitophagy and energy sensing (↑
LC3-II/LC3-I, ATG12:5, p-AMPK/AMPK) than males | KD and exercise
enhanced hepatic mitochondrial remodeling and redox balance. Female
rats showed greater intrinsic mitochondrial biogenesis and general
autophagy markers, although mitophagy-specific flux was not
assessed. | (27) |
| Rosa-Caldwell et
al, 2022 | VWR | HFD with
weight-loss intervention | Male C57BL/6J mice
were fed LFD, HFD, weight loss by diet alone or D/PA. Hepatic
mitochondrial content, biogenesis, mitophagy and macroautophagy
markers were assessed by immunoblotting. | HFD: ↓ COX IV and
disrupted mitochondrial quality control (↑ p- UbSer65, ↑ p62) Diet
alone: ↓ p62 and p-UbSer65 (minimal improvement) D/PA: ↑
PGC-1α/mitochondrial content/BNIP3, normalized p62 and ↓ aberrant
p-UbSer65 accumulation, indicating improved mitophagy | Weight loss
combined with physical activity was superior to diet alone in
restoring hepatic mitochondrial quality, enhancing BNIP3-mediated
mitophagy and improving autophagy resolution in NAFLD. | (28) |
| Stevanović-Silva
et al, 2022 | Moderate-intensity
treadmill + VWR | Maternal HFHS | Female rats were
fed a control or HFHS diet, were SED or performed GE during
pregnancy. Female offspring were fed a control diet and kept SED.
Hepatic lipid accumulation, mitochondrial biogenesis, mitochondrial
dynamics and mitophagy/autophagy markers were assessed. | Maternal HFHS
(offspring): ↑ hepatic lipid stress markers/Parkin/OPA1,
↓PGC-1α/TFAM (trend)/mitochondrial dynamics mRNA GE (offspring): ↓
Hepatic triglycerides and NAFLD activity score, ↑
PGC-1α/TFAM/MFN1/2/DRP1, ↓ Parkin/OPA1 Mitophagy/autophagy: LC3,
PINK1 ~ | GE counteracted
maternal HFHS-induced mitochondrial dysregulation in female
offspring primarily by enhancing mitochondrial biogenesis and
dynamics, while preventing pathological accumulation of mitophagy
signaling. | (29) |
| Zhang et al,
2022 | Treadmill
running | No dietary
manipulation | Male db/db mice
(genetic T2DM model) underwent treadmill aerobic exercise (8
weeks), morning (rest phase) or night (active phase). Hepatic
glucose/lipid metabolism, circadian clock proteins, mitochondrial
morphology and dynamics, mitophagy and apoptosis were
assessed. | T2DM: ↑ Blood
glucose/serum cholesterol/CLOCK/Parkin/LC3/apoptosis, ↓ OPA1/Fis1
Morning exercise: ↓ Glucose/Parkin/LC3/apoptosis, ↑ insulin
sensitivity/GLUT4 Night exercise: ↓ Glucose/cholesterol/Parkin/LC3,
↓ CLOCK/apoptosis (greater than morning), ↑ mitochondrial networks
and morphology | Morning exercise
favored glucose handling and insulin sensitivity, whereas night
exercise more effectively normalized CLOCK-linked mitophagy,
mitochondrial structure and apoptosis. | (30) |
| McCoin et
al, 2022 | Treadmill
running | No dietary
manipulation | Adult female
C57BL/6J mice performed a single acute bout of treadmill exercise;
livers were collected immediately (0 h) or 2 h post-exercise.
Mitophagic flux was assessed using leupeptin (lysosomal inhibitor),
isolated hepatic mitochondria, mitophagy reporters
(Cox8-GFP-mCherry), WB and mitochondrial proteomics. | All comparisons:
Exercise vs. SED 0 h post-exercise: ~ LC3-II, ~ p62 (no flux); ↑
BNIP3/MFN2 (transient) 2 h post-exercise (+ leupeptin): ↑
LC3-II/p62/mitochondrial Ub resulting in ↑ mitophagic flux
Whole-liver homogenate (saline- and leupeptin-treated groups):
~LC3-II, ~ p62 Isolated mitochondria (WB, saline-treated groups
only): ~ Parkin, ~ DRP1 Proteomics: ↑ BNIP3/OPTN/LC3-II (saline-
and leupeptin-treated groups); ↑ Beclin-1 (leupeptin-treated
groups) | A single exercise
bout rapidly and transiently activated hepatic mitophagic flux,
detectable during early recovery and mediated by Ub- and
receptor-dependent pathways rather than global autophagy
induction. | (31) |
| McCoin et
al, 2022 | Treadmill
running | No dietary
manipulation | Female mice
performed 1 h treadmill exercise vs. SED and were sacrificed
immediately (0 h) or 2 h post-exercise. Isolated hepatic
mitochondria were analyzed by untargeted proteomics (3,241
proteins) plus targeted validation (such as p62 and Ndufa4). | Acute exercise: ↑
Mitochondrial processes for lipid localization/IL-5/protein
phosphorylation; proteome showed time-dependent clustering Exercise
+ leupeptin: Altered exercise-modulated protein profiles under
lysosomal inhibition, suggesting autophagolysosomal turnover | A single exercise
bout rapidly remodeled the hepatic mitochondrial proteome, and
lysosomal inhibition revealed autophagolysosomal turnover linked to
mitochondrial quality-control processes. | (32) |
| Zou et al,
2023 | Swimming | HFD | Zebrafish were fed
a HFD to induce NAFLD and subjected to chronic swimming exercise.
Liver pathology, mitochondrial morphology and dynamics,
mitochondrial biogenesis and mitophagy markers were assessed. | HFD: Impaired
mitochondrial morphology, ↓ PGC-1α/PINK1/Parkin and ↑ p62 Swimming
exercise: ↓ Liver injury/fibrosis/p62, ↑
AMPK/SIRT1/PGC-1α/oxidative metabolism/Parkin/mitochondrial
dynamics | Exercise improved
mitochondrial quality in NAFLD by simultaneously activating
mitochondrial biogenesis and restoring mitophagy. | (33) |
| Li et al,
2024 | Treadmill
running | HFD | Male C57BL/6J mice
divided into four groups: Normal diet + SED, HFD + SED, HFD + HIIT
and HFD + MICT. Hepatic MAM formation assessed by
immunofluorescence colocalization of IP3R and VDAC1. Mitophagy
markers, mitochondrial dynamics, ER stress markers and hepatic
insulin signaling were assessed. | HFD: ↓ IP3R-VDAC1
colocalization (MAMs)/PINK1/Parkin/p-PI3K/p-AKT/p-GSK3β, ↑
LC3-II/LC3-I/BNIP3/Fis1/p-PERK/p-eIF2α HFD + HIIT and HFD + MICT: ↑
IP3R-VDAC1 colocalization/Parkin, ↓ Fis1/p-PERK HFD + MICT only: ↑
PINK1/Parkin, ↓ LC3-II/LC3-I/BNIP3/p-eIF2α HFD + HIIT only: ↑
OPA1/Parkin | HFD-induced
reduction in hepatic MAMs formation was associated with suppressed
mitophagy and elevated ER stress, contributing to hepatic insulin
resistance. HIIT and MICT restored MAMs formation and alleviated
hepatic IR, with MICT demonstrating superior efficacy in restoring
mitophagy and reducing ER stress. | (34) |
| Mokhtari-Andani
et al, 2025 | Treadmill
running | DEX-induced NAFLD +
silymarin | Male Wistar rats
with DEX-induced NAFLD underwent moderate-intensity/high-intensity
exercise (8 weeks), with or without silymarin supplementation.
Hepatic mitophagy signaling was assessed. | DEX: ↑
PINK1/Bcl-2/Beclin-1/LC3, ↓ mTOR Moderate-intensity/high-intensity
exercise: ↓ Bcl-2/LC3 mRNA, ↑ p62 protein levels compared with DEX
Silymarin: ↓ Parkin/Bcl-2/LC3/PINK1
Moderate-intensity/high-intensity exercise + silymarin: ↓ Bcl-2/LC3
mRNA | Exercise attenuated
pathological mitophagy signaling and restored mitochondrial quality
control in pharmacologically induced NAFLD, with separate
(non-additive) benefits from nutraceutical support. | (35) |
| Franczak et
al, 2025 | Treadmill
running | HFD | Female C57BL/6J
mice undergoing sham operation or OVX and fed a 4-week LFD or HFD
performed acute exercise. Hepatic mitophagic flux,
isolated-mitochondria proteomics (HFD groups), mitochondrial
respiration and H2O2 emissions were
measured. | Sham + exercise: ↑
mitochondrial p62 (WB, leupeptin); by proteomics ↑ E3-Ub
ligases/UBB and adaptors (OPTN, NBR1, NDP52), ↑ LC3-II OVX + LFD: ↓
Basal mitophagic flux OVX + HFD: ↓↓ Exercise-induced mitophagy; ↓
mitochondrial H2O2 signaling and fatty
acid-supported respiration; ↓ DRP1/MFF recruitment | Ovarian function
was required for full activation of exercise-induced hepatic
mitophagic flux, particularly during HFD feeding. Loss of estrogen
blunted redox signaling and mitochondrial quality control responses
to exercise. | (36) |
| Deng et al,
2025 | Treadmill
running | HFD | Male SD rats
divided into four groups: Normal-fat diet, HFD, MICT and HIIT. HFD
induction (8 weeks) followed by treadmill training and HFD (8
weeks). Hepatic mitophagy markers, mitochondrial dynamics,
mitochondrial biogenesis, lipid metabolism, inflammatory cytokines,
gut microbiota and gut-liver axis parameters were assessed. | HFD: ↓
PINK1/Parkin/PGC-1α/CS/COX IV/MFN1/MFN2, ↑
Fis1/steatosis/TG/TC/ALT/AST/leptin/LPS/IL-1β/IL-6/TNF-α HIIT +
MICT: ↑ PINK1/Parkin/PGC-1α/CS/COX IV/MFN1/MFN2/p-AMPK,
↓Fis1/steatosis/TG/TC/ALT/AST/leptin/LPS/IL-1β/IL-6/TNF-α HIIT >
MICT in improving mitochondrial function and regulation of
AMPK/SREBP-1c/PPARα/CPT-1 | HIIT demonstrated
superior efficacy than MICT in improving hepatic mitochondrial
function. Improvements in MASLD were mediated through the
AMPK-PINK1/Parkin axis and gut-liver axis remodeling. | (37) |
| Durak et al,
2026 | Treadmill
running | HFD | Female C57BL/6J
mice divided into control, HFD and exercise + HFD groups. Exercise
started at week 6 until 24 weeks. Gene expression levels were
evaluated in the liver. | HFD: ↓ Parkin and
MFN1 compared with the control group Exercise + HFD: ↑
ATG3/PINK1/MFN1/Parkin compared with in the HFD group | Exercise altered
the expression of mitophagy-related genes in the liver. | (38) |
6. Dietary influences on mitophagy and
interactions with exercise
Diet markedly affects mitochondrial turnover
processes. The present review discusses how overnutrition (high-fat
or Western diets), specific macronutrient compositions [for
example, a ketogenic diet (KD)] and undernutrition (CR and fasting)
modulate mitophagy and how the effects of exercise on mitophagy
intersect with these states. The roles of weight-loss strategies
and nutraceutical approaches are also considered.
High-fat and Western-style diets are known to
rapidly induce hepatic steatosis and insulin resistance in rodent
models. These are accompanied by early mitochondrial abnormalities,
such as reduced respiratory capacity, disrupted mitochondrial
dynamics and increased oxidative stress. Disruption of
mitochondrial quality control occurs early, as even short-term
exposure to a Western diet markedly reduces key mitochondrial
proteins and BNIP3 concentrations, indicating suppressed mitophagy
capacity before overt inflammation (22). The suppression of BNIP3 by a Western
diet likely reflects hyperactivation of mTOR complex 1 by nutrient
excess (57), which inhibits
HIF-1α- and FOXO3-dependent BNIP3 transcription (58). Simultaneously, lipotoxicity-induced
oxidative stress may enhance PINK1 stabilization on depolarized
mitochondria (59), creating a
paradoxical state in which the Ub-dependent arm is primed while the
receptor-mediated arm is suppressed. While increased Parkin, BNIP3
activation and LC3-II accumulation have been documented in certain
models, such patterns are more consistent with dysfunctional
mitophagy signaling than with effective mitochondrial turnover
(24). This apparent contradiction,
Western diet suppressing BNIP3(22)
vs. HFF increasing the concentration of active BNIP3 homodimer
(24), likely reflects the severity
and composition of the dietary challenge. Short-term Western diet
may suppress BNIP3 transcription through mTOR-mediated FOXO3
inhibition, whereas prolonged HFF feeding may drive compensatory
BNIP3 dimerization as a failed attempt to clear overwhelmed
mitochondria. Lysosomal dysfunction prevents effective flux despite
elevated receptor signaling (22,24).
Exercise and dietary modification act
synergistically to restore hepatic mitophagy in NAFLD. Both
structured training and voluntary activity reduce steatosis and
enhance general autophagic markers (higher LC3-II/I ratio, lower
p62); however, BNIP3-mediated mitophagy is not consistently
restored by physical activity alone under continued high-fat
feeding (22), and robust mitophagy
improvement in weight-loss models depends critically on combining
exercise with dietary change (28).
Rosa-Caldwell et al (28)
indicated that dietary restriction alone increased the expression
of PINK1 and Parkin, and normalized p62 and p-Ub Ser65, yet failed
to restore the levels of BNIP3, outcomes achieved only when
exercise was added; thus, the two pathway arms [the
PINK1/Parkin-dependent (Ub-dependent) arm and the BNIP3-dependent
receptor-mediated arm] may be under partially distinct control.
PINK1 responds to any reduction in mitochondrial depolarization and
can be partially rescued by CR, whereas BNIP3 requires FOXO3 and
AMPK activation, which exercise alone reliably provides in the
liver. Persistent p-Ub Ser65 upregulation in response to HFD, by
contrast, indicates that PINK1 was active but Parkin-mediated
clearance remained incomplete, a downstream bottleneck that
exercise uniquely resolves.
Unlike Western-style feeding, a KD shifts hepatic
metabolism toward fatty acid oxidation and ketogenesis, generating
mitochondrial adaptations of a qualitatively different character.
In rats with running wheel access, a previous study reported that a
KD combined with exercise could suppress sterol regulatory
element-binding protein 1/FAS-driven lipogenesis, improve
glutathione redox balance, and elevate PGC-1α, TFAM and citrate
synthase, indicating robust mitochondrial biogenesis (27). Higher hepatic LC3-II/I and
ATG12-ATG5 were observed in female vs. male rats irrespective of
diet, consistent with their greater baseline AMPK sensitivity;
notably, KD did not raise LC3-II/I relative to standard chow,
whereas Western diet lowered it (27). Some hepatic triglycerides accumulate
under KD but without the inflammatory or dysfunctional features of
Western diet-induced steatosis. Mechanistically, KD and exercise
may converge on AMPK-ULK1 signaling: Ketosis suppresses mTOR
through reduced glucose and amino acid availability while raising
the AMP/ATP ratio, and exercise superimposes a further energetic
demand, which drives AMPK-ULK1 signaling beyond what either
intervention achieves alone (27).
The result is a quasi-fasted metabolic state in which autophagic
quality control is constitutively primed. As all animals in that
study had running-wheel access (27), the contribution of exercise cannot
be isolated; therefore, the possibility that, in the absence of
exercise, a KD promotes steatosis rather than mitochondrial benefit
remains to be confirmed in sedentary models.
Among longevity interventions, CR is one of the best
characterized, with autophagy and mitophagy induction recognized as
central mechanisms (60). Fasting
activates these pathways rapidly in the liver: BNIP3 concentrations
increase within 6 h and continue to increase through 24 h of
nutrient deprivation in mice, reflecting early receptor-mediated
mitophagy (61). Human skeletal
muscle requires a longer fasting period to mount a comparable
autophagic response (62). When
combined with exercise, fasting or CR produces additive mitophagy
activation through convergent AMPK and SIRT1 signaling, including
in human muscle (48,63,64),
although prolonged restriction risks impairing exercise capacity
and should be applied judiciously.
The context dependence of exercise-induced mitophagy
is illustrated by a chlorpyrifos exposure model, in which fat
mobilization during exercise released hepatically accumulated
toxicants that inhibited AMPK signaling and caused p62
accumulation, ultimately abrogating mitophagy despite continued
physical activity (65).
Toxicant-mediated AMPK inhibition appears to sever the upstream
energetic signal that normally coordinates PINK1/Parkin and BNIP3
activation, leaving mitochondria without an adequate clearance
signal even as oxidative stress continues. This scenario is
functionally analogous to the lysosomal dysfunction seen in HFD
models where upstream signaling is intact but downstream execution
fails.
Specific dietary compounds can also modulate
mitophagy. Mokhtari-Andani et al (35) revealed that exercise paradoxically
elevated the concentrations of hepatic p62 while suppressing those
of LC3 and Bcl-2 mRNA in dexamethasone-treated rats, a two-level
dysfunction not observed in dietary NAFLD models. In addition,
silymarin supplementation combined with exercise attenuated the
serum lipid profile in the same model (35). Across the interventions reviewed,
Western-style diets have been shown to consistently compromise
mitophagy, while fasting, ketogenic feeding, CR and exercise
promote mitochondrial turnover and interacted synergistically.
Effective mitochondrial quality control, therefore, depends on how
nutritional context and exercise are aligned.
7. Maternal and paternal programming of
mitophagy across generations
While maternal exercise is associated with
beneficial metabolic outcomes in offspring, the majority of studies
have prioritized traditional endpoints such as adiposity and
insulin sensitivity rather than mitochondrial quality regulation.
Cunningham et al (19)
provided novel insight into offspring liver mitochondrial markers.
In this previous study, female rats were fed either a Western diet
or normal diet during pregnancy; half of each group had access to
running wheels. The offspring were studied in young adulthood under
normal diet conditions. Maternal exercise robustly upregulated
markers of mitochondrial biogenesis (TFAM, PPARγ) and nuclear
factor erythroid 2-related factor 2 in the offspring livers of both
sexes, whereas mitophagy/autophagy markers BNIP3 and ATG12-ATG5
concentrations were increased specifically in Western diet-exposed
male offspring. Female offspring had inherently higher levels of
these markers than male offspring regardless of the maternal group,
which corresponds with protection from steatosis even under
maternal Western diet exposure (19).
Stevanović-Silva et al (29) explored whether maternal exercise
could counteract the negative effects of a high-fat high-sucrose
(HFHS) diet during pregnancy. In offspring of HFHS-fed mothers,
reduced PGC-1α and TFAM disturbed fusion-fission dynamics and
elevated Parkin expression pointed to subclinical mitochondrial
stress preceding overt steatosis. Gestational exercise corrected
these biogenic and dynamic markers and reduced Parkin expression,
with LC3 and PINK1 unaltered in any group (29). That mitophagic flux markers were
unchanged despite functional recovery supports the view that active
mitophagy induction is unnecessary when mitochondrial architecture
is adequately preserved. Two observations complicate the
mechanistic picture. Parkin expression increased without a
corresponding increase in that of PINK1, suggesting recruitment
through non-canonical routes such as p-Ub signals from alternative
kinases or calcium-mediated translocation. The concurrent increase
in the expression of OPA1, a fusion-promoting protein, under HFHS
stress most plausibly reflects compensatory mitochondrial
elongation, a protective response that resists fission-dependent
segregation of damaged organelles and paradoxically impairs quality
control. Gestational exercise reversing this OPA1 elevation would
indicate restored fission-fusion balance rather than diminished
fusion capacity.
Stevanović-Silva et al (29) and Cunningham et al (19) reported divergent marker-level
outcomes of maternal exercise in offspring liver. Cunningham et
al (19) documented
upregulation of BNIP3 and ATG12-ATG5 expression, suggesting
enhanced mitophagy capacity, whereas Stevanović-Silva et al
(29) detected reductions in Parkin
and OPA1 expression without changes in those of LC3 and PINK1, a
pattern interpreted as mitophagy restraint rather than induction.
Differences in maternal diet type, offspring sex and assessment
timing are the most plausible sources of this divergence. Notably,
they may not be true contradictions: The findings of Cunningham
et al (19) suggested that
exercise programs induce greater mitophagy capacity in offspring
(more machinery available), whereas those of Stevanović-Silva et
al (29) suggested that
exercise may prevent maladaptive mitophagy signaling in offspring
(less pathological activation). These represent two different but
compatible aspects of mitochondrial quality control programming
across generations.
A previous study supported the idea that paternal
lifestyle can induce epigenetic regulation that modulates offspring
metabolism (20). Batista et
al (20) exhibited improved
metabolic outcomes, including HFD-induced obesity and hepatic
steatosis, in offspring from fathers that underwent swim training
before conception. The increased expression levels of genes
associated with energy sensing regulation (protein kinase
AMP-activated catalytic subunit α2/AMPKα2) and fatty acid oxidation
(PPAR-1α and carnitine palmitoyltransferase 1) were also detected
in offspring. This was accompanied by increased AMPK activity,
which was consistent with improved mitochondrial stress management.
Detection of parallel metabolic adaptations in paternal
reproductive tissues suggested epigenetic mechanisms mediating
intergenerational effects (20).
Fig. 3 compiles all
19 studies in Table I into a
schematic overview illustrating how different dietary interventions
(high-fat, western diet, weight loss and KD), acute or chronic
exercise, exercise modality (HIIT vs. MICT), parental programming,
sex and hormonal modulation, as well as pathological conditions and
exercise preconditioning interact to regulate hepatic mitophagy.
Moore et al (27) measured
general autophagy markers (LC3-II/I, ATG12-ATG5) without
mitophagy-specific readouts; the findings reflected autophagic
rather than strictly mitophagic activity.
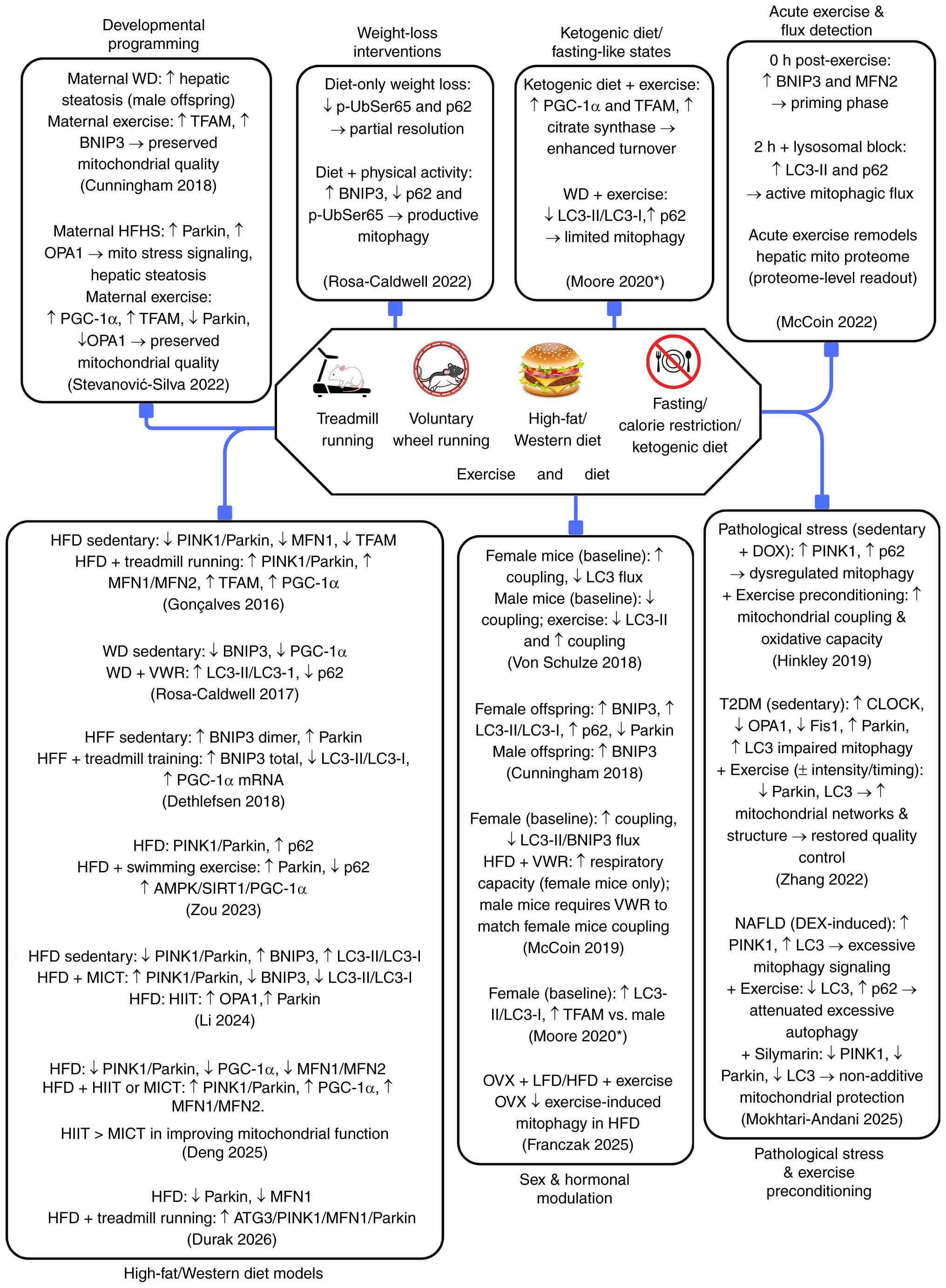 | Figure 3Integrated effects of exercise
modality, dietary context, sex, hormonal status, developmental
programming and pathological stress on hepatic mitophagy and
mitochondrial quality control. All findings are from animal models
and no human liver mitophagy data are included.
*Evidence from Moore et al reflects general
autophagy markers rather than mitophagy-specific readouts. AMPK,
AMP-activated protein kinase; ATG3, autophagy-related 3; BNIP3,
Bcl-2 interacting protein 3; CLOCK, circadian locomotor output
cycles kaput; DEX, dexamethasone; DOX, doxorubicin; Fis1,
mitochondrial fission protein 1; HFD, high-fat diet; HFF, high-fat
high-fructose diet; HFHS, high-fat high-sucrose diet; HIIT,
high-intensity interval training; LFD, low-fat diet; MFN,
mitofusin; MICT, moderate-intensity continuous training; NAFLD,
non-alcoholic fatty liver disease; OPA1, optic atrophy protein 1;
OVX, ovariectomy; PGC-1α, peroxisome proliferator-activated
receptor γ coactivator 1-α; PINK1, PTEN-induced kinase 1; p-Ub,
phosphorylated-ubiquitin; SIRT1, sirtuin 1; T2DM, type 2 diabetes
mellitus; TFAM, mitochondrial transcription factor A; VWR,
voluntary wheel running; WD, Western diet. |
The present review has several limitations. No
paired human liver study has measured hepatic PINK1, Parkin, BNIP3,
LC3-II, p62 or mitophagic flux before and after a lifestyle
intervention. All the evidence summarized herein derives from
animal models, which differ from humans in metabolism, lifespan and
susceptibility to MASLD. What exists is a looser chain of proximal
evidence. Clinical trials have demonstrated that exercise and
dietary interventions improve hepatic steatosis and, in some cases,
biopsy-based metabolic dysfunction-associated steatohepatitis
outcomes (66-70).
A 2025 randomized trial reported that 16:8 time-restricted eating
increased serum ATG5 but not Beclin-1 concentration without
liver-tissue confirmation (71).
Human observational studies have demonstrated that NAFLD/NASH is
associated with altered hepatic mitochondrial respiration,
oxidative stress and disease-state changes in mitophagy-related
pathways (5,72). Finally, an exercise study measured
the concentrations of peripheral myokines such as irisin as
surrogate signals of mitochondrial stress (73). These findings support biological
plausibility but cannot establish that exercise or diet improves
hepatic mitophagy in humans, because circulating markers reflect
systemic rather than hepatocyte-specific mitophagic clearance.
Sex and phenotype heterogeneity remain unresolved,
as human trials rarely report sex-stratified mitophagy outcomes
despite known sex differences in MASLD biology and exercise
response. A fundamental limitation is that static LC3 or BNIP3
concentrations are uninformative about flux direction: Marker
accumulation may reflect more initiation or less degradation, and
the reviewed literature contains multiple examples where the same
profile corresponded to diametrically opposite biological states.
Study heterogeneity arising from differences in diet, exercise
type, duration, sex, age and genetic background compounds this
interpretive challenge. Mitophagy itself adds further complexity,
as both inadequate and excessive clearance can be detrimental.
Progress will require delineating the adaptive-maladaptive
threshold and validating non-invasive biomarkers such as plasma
acylcarnitines, breath-test indices or cell-free mitochondrial DNA.
Crosstalk between mitophagy and related quality-control processes,
including biogenesis, fission-fusion dynamics and mitochondrial
unfolded protein response, also remains insufficiently
explored.
8. Pharmacological implications
Pharmacological and nutraceutical mitophagy inducers
represent a promising but clinically underdeveloped therapeutic
option for metabolic liver disease. These agents can be grouped by
the mitophagy pathway they engage. Metformin stands out as the
candidate with the strongest clinical evidence for mitophagy
induction. In a randomized placebo-controlled trial, patients with
T2DM who received metformin exhibited upregulation of PINK1,
Parkin, MFN2, NIX, LC3-II and lysosome-associated membrane protein
2, together with AMPK activation, suggesting mitophagy induction as
a glucose-lowering-independent effect (74). Mechanistically, metformin-driven
AMPK activation concurrently phosphorylates ULK1, stabilizes BNIP3
and suppresses mTOR, engaging the mitophagy network at multiple
levels simultaneously.
Urolithin A acts on mitophagy through PINK1
stabilization and p-Ub accumulation and has shown acceptable
safety, bioavailability, and mitochondrial health benefits in Phase
I human trials, although these findings derive from skeletal muscle
studies and liver-specific data are lacking (75,76).
NAD+ precursors such as nicotinamide mononucleotide and
nicotinamide riboside represent another mechanistically based
approach: By replenishing NAD+, they activate SIRT1,
which promotes BNIP3/NIX-dependent mitophagy via FOXO3
deacetylation and facilitates autophagosome maturation through LC3
deacetylation (77,78). These effects lead to restored
mitophagy in disease models including ATM deficiency and
neurodegeneration; however, liver-specific mitophagy endpoints have
not been assessed in human trials. More selective
mitophagy-targeting agents are also emerging; for example, TJ0113
is a first-in-class small-molecule mitophagy inducer with favorable
GLP-compliant preclinical safety data, but its current development
is still outside liver disease (79). Ub-specific protease 30 (USP30)
inhibitors represent a promising emerging class. USP30 is a
deubiquitinase localized on the outer mitochondrial membrane that
antagonizes Parkin-mediated ubiquitination, and its inhibition
promotes mitophagy without requiring mitochondrial depolarization.
MTX325, an orally bioavailable USP30 inhibitor, has entered Phase I
clinical trials, although it has not yet been tested for metabolic
liver disease (45,80,81).
In the liver-specific context, recent NASH data have identified the
tumor necrosis factor α-Myc-interacting zinc-finger protein
1/peroxiredoxin/Parkin axis as a potential therapeutic target,
because disruption of this inflammatory feedback loop may restore
Parkin-mediated hepatocyte mitophagy and reduce NASH progression
(82). Harmol, a β-carboline
alkaloid, also warrants mention here, as it mimics exercise-induced
mitophagy through transient mitochondrial depolarization and AMPK
engagement, with demonstrated efficacy in improving metabolic
health in model organisms (83).
In addition to direct mitophagy inducers,
mitochondrial dynamics regulators such as DRP1 may represent future
therapeutic targets in NAFLD. While DRP1-mediated fission is
necessary to segregate damaged organelles for mitophagic clearance,
its overactivation promotes fragmentation, inflammation and
fibrosis (8). Human NAFLD data
identifying DRP1 as a marker of hepatic inflammation and fibrosis
suggests that the therapeutic goal should be dynamic balance rather
than fission enhancement as such (84). Future management of MASLD/metabolic
dysfunction-associated steatohepatitis may require integrating
lifestyle interventions with pharmacological mitophagy modulation,
whether through AMPK activators, urolithin A or Parkin-targeted
agents, although all such strategies await rigorous validation in
liver-specific human trials.
9. Conclusion
The regulation of hepatic mitophagy is
multidetermined and influenced by exercise type, nutritional
context, biological sex and parental programming. Within the NAFLD
setting, voluntary or low-intensity activity falls short of
restoring mitophagic function, and higher-intensity protocols
combined with dietary modification or weight loss appear necessary
to drive meaningful mitochondrial improvement. Two major pathways
of mitophagy (Ub- and receptor-mediated) are modulated by exercise,
with energetic stress and hormonal milieu determining their
relative contribution. Increased mitophagy signaling is not always
associated with beneficial outcomes; therefore, efficient
completion of lysosomal clearance serves an important role in
mitochondria recovery. Adaptations to exercise and dietary
interventions are further modified by sex hormones and maternal or
paternal programming, which highlights the chronic and
intergenerational dimensions of mitochondrial regulation. In
summary, effective optimization of hepatic mitochondrial health
should integrate exercise with nutritional state, biological sex
and parental programming, recognizing mitophagy as a conditional
therapeutic target. Thus, mitophagy should be considered a
context-dependent therapeutic target rather than a pathway that
should always be stimulated.
Acknowledgements
Not applicable.
Funding
Funding: The article processing charge for the present study was
funded by Universitas Kristen Maranatha (grant no.
023/SK/ADD/UKM/V/2024).
Availability of data and materials
Not applicable.
Authors' contributions
All authors (JWG, DKJ, FK, SS and RL)
conceptualized the study. JWG supervised the work and drafted the
manuscript. DKJ and RL contributed to critical evaluation and
interpretation of the literature. FK and SS assisted in literature
organization and synthesis. Data authentication is not applicable.
All authors revised the manuscript, and read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Artificial intelligence statement
During the preparation of this work, AI tools were
used to improve the readability and language of the manuscript, and
subsequently, the authors revised and edited the content produced
by the AI tools as necessary, taking full responsibility for the
ultimate content of the present manuscript.
References
|
1
|
Roeb E: Non-alcoholic fatty liver
diseases: Current challenges and future directions. Ann Transl Med.
9(726)2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Janssen JAMJL: The pivotal role of the
western diet, hyperinsulinemia, ectopic fat, and
diacylglycerol-mediated insulin resistance in type 2 diabetes. Int
J Mol Sci. 26(9191)2025.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wang N, Wang X, Li Q, Han B, Chen Y, Zhu
C, Chen Y, Lin D, Wang B, Jensen MD and Lu Y: The famine exposure
in early life and metabolic syndrome in adulthood. Clin Nutr.
36:253–259. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Do LH, Da Costa RT and Solesio ME: Effects
of nutrients and diet on mitochondrial dysfunction: An opportunity
for therapeutic approaches in human disease. Biomed Pharmacother.
191(118493)2025.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Koliaki C, Szendroedi J, Kaul K, Jelenik
T, Nowotny P, Jankowiak F, Herder C, Carstensen M, Krausch M,
Knoefel WT, et al: Adaptation of hepatic mitochondrial function in
humans with non-alcoholic fatty liver is lost in steatohepatitis.
Cell Metab. 21:739–746. 2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Kyriazis ID, Vassi E, Alvanou M, Angelakis
C, Skaperda Z, Tekos F, Garikipati VNS, Spandidos DA and Kouretas
D: The impact of diet upon mitochondrial physiology (Review). Int J
Mol Med. 50(135)2022.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Gustafsson ÅB and Dorn GW II: Evolving and
expanding the roles of mitophagy as a homeostatic and pathogenic
process. Physiol Rev. 99:853–892. 2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Wang S, Long H, Hou L, Feng B, Ma Z, Wu Y,
Zeng Y, Cai J, Zhang DW and Zhao G: The mitophagy pathway and its
implications in human diseases. Signal Transduct Target Ther.
8(304)2023.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Yang J, Yang F, Chen G, Liu M, Yuan S and
Zhang TE: Receptor-mediated mitophagy: A new target of
neurodegenerative diseases. Front Neurol.
16(1665315)2025.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Goodall EA, Kraus F and Harper JW:
Mechanisms underlying ubiquitin-driven selective mitochondrial and
bacterial autophagy. Mol Cell. 82:1501–1513. 2022.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Mizushima N: Autophagic flux measurement:
Cargo degradation versus generation of degradation products. Curr
Opin Cell Biol. 93(102463)2025.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Sorriento D, Di Vaia E and Iaccarino G:
Physical exercise: A novel tool to protect mitochondrial health.
Front Physiol. 12(660068)2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Oliveira AN, Richards BJ, Slavin M and
Hood DA: Exercise is muscle mitochondrial medicine. Exerc Sport Sci
Rev. 49:67–76. 2021.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Zhou XH, Luo YX and Yao XQ:
Exercise-driven cellular autophagy: A bridge to systematic
wellness. J Adv Res. 76:271–291. 2025.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Springer MZ, Poole LP, Drake LE,
Bock-Hughes A, Boland ML, Smith AG, Hart J, Chourasia AH, Liu I,
Bozek G and Macleod KF: BNIP3-dependent mitophagy promotes
cytosolic localization of LC3B and metabolic homeostasis in the
liver. Autophagy. 17:3530–3546. 2021.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Korovila I, Höhn A, Jung T, Grune T and
Ott C: Reduced liver autophagy in high-fat diet induced liver
steatosis in New Zealand obese mice. Antioxidants (Basel).
10(501)2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Fukushima H, Yamashina S, Arakawa A,
Taniguchi G, Aoyama T, Uchiyama A, Kon K, Ikejima K and Watanabe S:
Formation of p62-positive inclusion body is associated with
macrophage polarization in non-alcoholic fatty liver disease.
Hepatol Res. 48:757–767. 2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Kugler BA, Thyfault JP and McCoin CS:
Sexually dimorphic hepatic mitochondrial adaptations to exercise: A
mini-review. J Appl Physiol (1985). 134:685–691. 2023.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Cunningham RP, Moore MP, Meers GM,
Ruegsegger GN, Booth FW and Rector RS: Maternal physical activity
and sex impact markers of hepatic mitochondrial health. Med Sci
Sports Exerc. 50:2040–2048. 2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Batista RO, Budu A, Alves-Silva T, Arakaki
AM, Gregnani MFS, Rodrigues Húngaro TG, Burgos-Silva M, Wasinski F,
Lanzoni VP, Camara NOS, et al: Paternal exercise protects against
liver steatosis in the male offspring of mice submitted to high fat
diet. Life Sci. 263(118583)2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Gonçalves IO, Passos E, Diogo CV,
Rocha-Rodrigues S, Santos-Alves E, Oliveira PJ, Ascensão A and
Magalhães J: Exercise mitigates mitochondrial permeability
transition pore and quality control mechanisms alterations in
nonalcoholic steatohepatitis. Appl Physiol Nutr Metab. 41:298–306.
2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Rosa-Caldwell ME, Lee DE, Brown JL, Brown
LA, Perry RA Jr, Greene ES, Carvallo Chaigneau FR, Washington TA
and Greene NP: Moderate physical activity promotes basal hepatic
autophagy in diet-induced obese mice. Appl Physiol Nutr Metab.
42:148–156. 2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Von Schulze A, McCoin CS, Onyekere C,
Allen J, Geiger P, Dorn GW II, Morris EM and Thyfault JP: Hepatic
mitochondrial adaptations to physical activity: Impact of sexual
dimorphism, PGC1α and BNIP3-mediated mitophagy. J Physiol.
596:6157–6171. 2018.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Dethlefsen MM, Kristensen CM, Tøndering
AS, Lassen SB, Ringholm S and Pilegaard H: Impact of liver PGC-1α
on exercise and exercise training-induced regulation of hepatic
autophagy and mitophagy in mice on HFF. Physiol Rep.
6(e13731)2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
McCoin CS, Von Schulze A, Allen J, Fuller
KNZ, Xia Q, Koestler DC, Houchen CJ, Maurer A, Dorn GW II, Shankar
K, et al: Sex modulates hepatic mitochondrial adaptations to
high-fat diet and physical activity. Am J Physiol Endocrinol Metab.
317:E298–E311. 2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Hinkley JM, Morton AB, Ichinoseki-Sekine
N, Huertas AM and Smuder AJ: Exercise training prevents
doxorubicin-induced mitochondrial dysfunction of the liver. Med Sci
Sports Exerc. 51:1106–1115. 2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Moore MP, Cunningham RP, Kelty TJ,
Boccardi LR, Nguyen NY, Booth FW and Rector RS: Ketogenic diet in
combination with voluntary exercise impacts markers of hepatic
metabolism and oxidative stress in male and female Wistar rats.
Appl Physiol Nutr Metab. 45:35–44. 2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Rosa-Caldwell ME, Poole KE, Seija A,
Harris MP, Greene NP and Wooten JS: Exercise during weight loss
improves hepatic mitophagy. Sports Med Health Sci. 4:183–189.
2022.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Stevanović-Silva J, Beleza J, Coxito P,
Rocha H, Gaspar TB, Gärtner F, Correia R, Fernandes R, Oliveira PJ,
Ascensão A and Magalhães J: Exercise performed during pregnancy
positively modulates liver metabolism and promotes mitochondrial
biogenesis of female offspring in a rat model of diet-induced
gestational diabetes. Biochim Biophys Acta Mol Basis Dis.
1868(166526)2022.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Zhang Z, Li X, Zhang J, Du J, Zhang Q, Ge
Z and Ding S: Chrono-Aerobic exercise optimizes metabolic state in
DB/DB mice through CLOCK-mitophagy-apoptosis. Int J Mol Sci.
23(9308)2022.PubMed/NCBI View Article : Google Scholar
|
|
31
|
McCoin CS, Franczak E, Deng F, Pei D, Ding
WX and Thyfault JP: Acute exercise rapidly activates hepatic
mitophagic flux. J Appl Physiol (1985). 132:862–873.
2022.PubMed/NCBI View Article : Google Scholar
|
|
32
|
McCoin CS, Franczak E, Washburn MP, Sardiu
ME and Thyfault JP: Acute exercise dynamically modulates the
hepatic mitochondrial proteome. Mol Omics. 18:840–852.
2022.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zou YY, Tang XB, Chen ZL, Liu B, Zheng L,
Song MY, Xiao Q, Zhou ZQ, Peng XY and Tang CF: Exercise
intervention improves mitochondrial quality in non-alcoholic fatty
liver disease zebrafish. Front Endocrinol (Lausanne).
14(1162485)2023.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Li X, Yang JY, Hu WZ, Ruan Y, Chen HY,
Zhang Q, Zhang Z and Ding ZS: Mitochondria-associated membranes
contribution to exercise-mediated alleviation of hepatic insulin
resistance: Contrasting high-intensity interval training with
moderate-intensity continuous training in a high-fat diet mouse
model. J Diabetes. 16(e13540)2024.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Mokhtari-Andani F, Talebi-Garakani E,
Nasiri K and Akbari A: Exercise training and Silymarin consumption
can ameliorate mitophagy signaling flux in hepatocytes of rats with
dexamethasone-induced non-alcoholic fatty liver disease. Sci Rep.
15(36637)2025.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Franczak E, Kugler BA, Salathe SF, Allen
JA, Sardiu ME, McCoin CS, Hevener AL, Morris EM and Thyfault JP:
Loss of ovarian function prevents exercise-induced activation of
hepatic mitophagic flux. Am J Physiol Endocrinol Metab.
328:E869–E884. 2025.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Deng D, Xu L, Liu Y, Li C, Jiang Q, Shi J,
Feng S and Lin Y: HIIT versus MICT in MASLD: Mechanisms mediated by
gut-liver axis crosstalk, mitochondrial dynamics remodeling, and
adipokine signaling attenuation. Lipids Health Dis.
24(144)2025.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Durak S, Sezgin SBA, Celik F, Yilmazer Y,
Cevik A, Kucukhuseyin O, Yaylim I and Zeybek U: Exercise Stimulates
PINK-1, PARKIN, MFN-1, and ATG-3 genes expression despite high-fat
diet: Tissue-specific responses. In Vivo. 40:1473–1484.
2026.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Ney PA: Mitochondrial autophagy: Origins,
significance, and role of BNIP3 and NIX. Biochim Biophys Acta. 1853
(10 Pt B):2775–2783. 2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Guan Y, Drake JC and Yan Z:
Exercise-induced mitophagy in skeletal muscle and heart. Exerc
Sport Sci Rev. 47:151–156. 2019.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Bayne AN and Trempe JF: Mechanisms of
PINK1, ubiquitin and Parkin interactions in mitochondrial quality
control and beyond. Cell Mol Life Sci. 76:4589–4611.
2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Ju JS, Jeon SI, Park JY, Lee JY, Lee SC,
Cho KJ and Jeong JM: Autophagy plays a role in skeletal muscle
mitochondrial biogenesis in an endurance exercise-trained
condition. J Physiol Sci. 66:417–430. 2016.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Brandt N, Gunnarsson TP, Bangsbo J and
Pilegaard H: Exercise and exercise training-induced increase in
autophagy markers in human skeletal muscle. Physiol Rep.
6(e13651)2018.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Drake JC, Laker RC, Wilson RJ, Zhang M and
Yan Z: Exercise-induced mitophagy in skeletal muscle occurs in the
absence of stabilization of Pink1 on mitochondria. Cell Cycle.
18:1–6. 2019.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Wang Q, Sun Y, Li TY and Auwerx J:
Mitophagy in the pathogenesis and management of disease. Cell Res.
36:11–37. 2026.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Mandic M, Misirkic Marjanovic M,
Janjetovic K, Bosnjak M, Harhaji-Trajkovic L, Trajkovic V and
Vucicevic L: Multifaceted role of AMPK in autophagy: More than a
simple trigger? Am J Physiol Cell Physiol. 329:C1380–C1397.
2025.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Mehrabani S, Bagherniya M, Askari G, Read
MI and Sahebkar A: The effect of fasting or calorie restriction on
mitophagy induction: A literature review. J Cachexia Sarcopenia
Muscle. 11:1447–1458. 2020.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Cantó C, Jiang LQ, Deshmukh AS, Mataki C,
Coste A, Lagouge M, Zierath JR and Auwerx J: Interdependence of
AMPK and SIRT1 for metabolic adaptation to fasting and exercise in
skeletal muscle. Cell Metab. 11:213–219. 2010.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Tang BL: Sirt1 and the mitochondria. Mol
Cells. 39:87–95. 2016.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Huang Y, Xia X, Xu J, Wang Z, You Y and Du
Q: Mitophagy as a pivotal axis in non-alcoholic fatty liver
disease: From pathogenic mechanisms to therapeutic strategies
(Review). Mol Med Rep. 32(299)2025.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Laker RC, Drake JC, Wilson RJ, Lira VA,
Lewellen BM, Ryall KA, Fisher CC, Zhang M, Saucerman JJ, Goodyear
LJ, et al: Ampk phosphorylation of Ulk1 is required for targeting
of mitochondria to lysosomes in exercise-induced mitophagy. Nat
Commun. 8(548)2017.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Sies H and Jones DP: Reactive oxygen
species (ROS) as pleiotropic physiological signalling agents. Nat
Rev Mol Cell Biol. 21:363–383. 2020.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Yamashita SI, Sugiura Y, Matsuoka Y, Maeda
R, Inoue K, Furukawa K, Fukuda T, Chan DC and Kanki T: Mitophagy
mediated by BNIP3 and NIX protects against ferroptosis by
downregulating mitochondrial reactive oxygen species. Cell Death
Differ. 31:651–661. 2024.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Liu W, Meng P, Li Z, Shen Y, Meng X, Jin S
and Hu M: The multifaceted impact of physical exercise on FoxO
signaling pathways. Front Cell Dev Biol. 13(1614732)2025.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Zimmermann A, Madeo F, Diwan A, Sadoshima
J, Sedej S, Kroemer G and Abdellatif M: Metabolic control of
mitophagy. Eur J Clin Invest. 54(e14138)2024.
|
|
56
|
Wang Y, Guo Y, Xu Y, Wang W, Zhuang S,
Wang R and Xiao W: HIIT Ameliorates inflammation and lipid
metabolism by regulating macrophage polarization and mitochondrial
dynamics in the liver of type 2 diabetes mellitus mice.
Metabolites. 13(14)2022.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Ashraf S, Ashraf N, Yilmaz G and Harmancey
R: Crosstalk between beta-adrenergic and insulin signaling mediates
mechanistic target of rapamycin hyperactivation in liver of
high-fat diet-fed male mice. Physiol Rep. 9(e14958)2021.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Lin A, Yao J, Zhuang L, Wang D, Han J, Lam
EW and Gan B: The FoxO-BNIP3 axis exerts a unique regulation of
mTORC1 and cell survival under energy stress. Oncogene.
33:3183–3194. 2014.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Okatsu K and Fukai S: Ubiquitin signaling
in PINK1/Parkin-dependent mitophagy. J Biochem. 179:145–154.
2026.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Madeo F, Pietrocola F, Eisenberg T and
Kroemer G: Caloric restriction mimetics: Towards a molecular
definition. Nat Rev Drug Discov. 13:727–740. 2014.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Glick D, Zhang W, Beaton M, Marsboom G,
Gruber M, Simon MC, Hart J, Dorn GW II, Brady MJ and Macleod KF:
BNip3 regulates mitochondrial function and lipid metabolism in the
liver. Mol Cell Biol. 32:2570–2584. 2012.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Islam H, Amato A, Bonafiglia JT, Rahman
FA, Preobrazenski N, Ma A, Simpson CA, Quadrilatero J and Gurd BJ:
Increasing whole-body energetic stress does not augment
fasting-induced changes in human skeletal muscle. Pflugers Arch.
473:241–252. 2021.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Seabright AP, Fine NHF, Barlow JP, Lord
SO, Musa I, Gray A, Bryant JA, Banzhaf M, Lavery GG, Hardie DG, et
al: AMPK activation induces mitophagy and promotes mitochondrial
fission while activating TBK1 in a PINK1-Parkin independent manner.
FASEB J. 34:6284–6301. 2020.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Schwalm C, Jamart C, Benoit N, Naslain D,
Prémont C, Prévet J, Van Thienen R, Deldicque L and Francaux M:
Activation of autophagy in human skeletal muscle is dependent on
exercise intensity and AMPK activation. FASEB J. 29:3515–3526.
2015.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Xiang Y, Tang G, Hu X, Yu X, Feng Y, Huang
Z, Luo Y, Jiang Z, Lv Y, Sun X, et al: Mobilization of the
environmental toxicant chlorpyrifos during weight loss and its
impact on liver and adipose tissue metabolism in mice. Environ
Health Perspect: Jun 13, 2025 (Epub ahead of print).
|
|
66
|
Zhang HJ, He J, Pan LL, Ma ZM, Han CK,
Chen CS, Chen Z, Han HW, Chen S, Sun Q, et al: Effects of moderate
and vigorous exercise on nonalcoholic fatty liver disease: A
randomized clinical trial. JAMA Intern Med. 176:1074–1082.
2016.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Mucinski JM, Salvador AF, Moore MP,
Fordham TM, Anderson JM, Shryack G, Cunningham RP, Lastra G,
Gaballah AH, Diaz-Arias A, et al: Histological improvements
following energy restriction and exercise: The role of insulin
resistance in resolution of MASH. J Hepatol. 81:781–793.
2024.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Houghton D, Thoma C, Hallsworth K, Cassidy
S, Hardy T, Burt AD, Tiniakos D, Hollingsworth KG, Taylor R, Day
CP, et al: Exercise reduces liver lipids and visceral adiposity in
patients with nonalcoholic steatohepatitis in a Randomized
controlled trial. Clin Gastroenterol Hepatol. 15:96–102.e3.
2017.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Charatcharoenwitthaya P, Kuljiratitikal K,
Aksornchanya O, Chaiyasoot K, Bandidniyamanon W and
Charatcharoenwitthaya N: Moderate-intensity aerobic vs resistance
exercise and dietary modification in patients with nonalcoholic
fatty liver disease: A Randomized clinical trial. Clin Transl
Gastroenterol. 12(e00316)2021.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Oh S, So R, Shida T, Matsuo T, Kim B,
Akiyama K, Isobe T, Okamoto Y, Tanaka K and Shoda J: High-intensity
aerobic exercise improves both hepatic fat content and stiffness in
sedentary obese men with nonalcoholic fatty liver disease. Sci Rep.
7(43029)2017.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Ozlu Karahan T, Yilmaz Akyuz E, Yilmaz
Karadag D, Yilmaz Y and Eren F: Effects of intermittent fasting on
liver steatosis and fibrosis, serum FGF-21 and autophagy markers in
metabolic dysfunction-associated fatty liver disease: A Randomized
controlled trial. Life (Basel). 15(696)2025.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Moore MP, Cunningham RP, Meers GM, Johnson
SA, Wheeler AA, Ganga RR, Spencer NM, Pitt JB, Diaz-Arias A, Swi
AIA, et al: Compromised hepatic mitochondrial fatty acid oxidation
and reduced markers of mitochondrial turnover in human NAFLD.
Hepatology. 76:1452–1465. 2022.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Us Altay D, Kaya Y, Mataraci Değirmenci D,
Kocyiğit E, Üner A and Noyan T: Non-alcoholic fatty liver disease:
The importance of physical activity and nutrition education-A
randomized controlled study. J Gastroenterol Hepatol. 39:2723–2734.
2024.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Bhansali S, Bhansali A, Dutta P, Walia R
and Dhawan V: Metformin upregulates mitophagy in patients with
T2DM: A randomized placebo-controlled study. J Cell Mol Med.
24:2832–2846. 2020.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Andreux PA, Blanco-Bose W, Ryu D, Burdet
F, Ibberson M, Aebischer P, Auwerx J, Singh A and Rinsch C: The
mitophagy activator urolithin A is safe and induces a molecular
signature of improved mitochondrial and cellular health in humans.
Nat Metab. 1:595–603. 2019.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Singh A, D'Amico D, Andreux PA, Fouassier
AM, Blanco-Bose W, Evans M, Aebischer P, Auwerx J and Rinsch C:
Urolithin A improves muscle strength, exercise performance, and
biomarkers of mitochondrial health in a randomized trial in
middle-aged adults. Cell Rep Med. 3(100633)2022.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Fang EF, Kassahun H, Croteau DL,
Scheibye-Knudsen M, Marosi K, Lu H, Shamanna RA, Kalyanasundaram S,
Bollineni RC, Wilson MA, et al: NAD+ replenishment improves
lifespan and healthspan in ataxia telangiectasia models via
mitophagy and DNA repair. Cell Metab. 24:566–581. 2016.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Yang X, Lu A, Guan X, Ying T, Pan J, Tan M
and Lu J: An updated review on the mechanisms, pre-clinical and
clinical comparisons of nicotinamide mononucleotide (NMN) and
nicotinamide riboside (NR). Food Front: Nov 17, 2025 (Epub ahead of
print).
|
|
79
|
Tian Y, Wang F, Shen J, Zhang R, Chen Q,
Xu L, Liu D, Fan M, Tian Z, Cen X, et al: Preclinical safety and
toxicokinetic evaluation of TJ0113, a first-in-class mitophagy
inducer for Parkinson's disease. Toxicol Appl Pharmacol.
505(117565)2025.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Fang TZ, Sun Y, Pearce AC, Eleuteri S,
Kemp M, Luckhurst CA, Williams R, Mills R, Almond S, Burzynski L,
et al: Knockout or inhibition of USP30 protects dopaminergic
neurons in a Parkinson's disease mouse model. Nat Commun.
14(7295)2023.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Rusilowicz-Jones EV, Jardine J, Kallinos
A, Pinto-Fernandez A, Guenther F, Giurrandino M, Barone FG,
McCarron K, Burke CJ, Murad A, et al: USP30 sets a trigger
threshold for PINK1-PARKIN amplification of mitochondrial
ubiquitylation. Life Sci Alliance. 3(e202000768)2020.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Jin K, Shi Y, Zhang H, Zhangyuan G, Wang
F, Li S, Chen C, Zhang J, Wang H, Zhang W and Sun B: A
TNFα/Miz1-positive feedback loop inhibits mitophagy in hepatocytes
and propagates non-alcoholic steatohepatitis. J Hepatol.
79:403–416. 2023.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Costa-Machado LF, Garcia-Dominguez E,
McIntyre RL, Lopez-Aceituno JL, Ballesteros-Gonzalez Á,
Tapia-Gonzalez A, Fabregat-Safont D, Eisenberg T, Gomez J, Plaza A,
et al: Peripheral modulation of antidepressant targets MAO-B and
GABAAR by harmol induces mitohormesis and delays aging in
preclinical models. Nat Commun. 14(2779)2023.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Padelli M, Hamelin J, Desterke C, Sebagh
M, Saffroy R, Sanchez CG, Coilly A, Duclos-Vallée JC, Samuel D and
Lemoine A: Analysis of the mitochondrial dynamics in NAFLD: Drp1 as
a marker of inflammation and fibrosis. Int J Mol Sci.
26(7373)2025.PubMed/NCBI View Article : Google Scholar
|














