Introduction
Medulloblastoma (MB) is the most common invasive
brain tumor of childhood. It arises in the cerebellum, generally
occurs during embryogenesis and is characterized by a massive
expansion of cells from the external granular layer. These events
are thought to originate in response to growth factor secretion
from Purkinje neurons, such as Sonic hedgehog (Shh) and
Wingless-type MMTV integration site family member 3a (Wnt3a)
(1,2). An insidious feature of MB tumors is
their metastatic propensity as evident from MB cell intravasation
through the fourth ventricle into the cerebral spinal fluid,
resulting in dissemination throughout the spinal cord (1,3,4).
Despite advances in therapy, a significant number of patients
succumb to the disease, and survivors often suffer permanent
neurocognitive deficiencies (3–5).
Multiple signaling pathways have been associated
with MB onset and growth, including Shh and Wnt. Shh is a secreted
protein that binds to the transmembrane receptor Ptch1. Upon Shh
binding, Ptch1 inhibition of Smoothened is relieved and Shh signal
is transduced to the Gli family of transcription factors (6,7).
A key feature of both Shh and Wnt3a is their
high-affinity binding to heparan sulfate glycosaminoglycan chains
(HS-GAGs) (8). For example,
treatment of cerebellar sections with bacterial heparinases
abrogates Shh mitogenicity (9).
HS-GAGs are components of HS proteoglycans (HSPGs), ubiquitous
macro-molecules located in the extracellular matrix and on the cell
surface, consisting of a core protein and covalently attached
HS-GAGs (10–14). Cell surface HSPGs, as members of
the syndecan and glypican families, regulate cross-talk between
tumor and host cells by controlling the location and activity of
growth factors, acting as co-receptors. Their unique functions
place HSPGs at the center of cell signaling integration (15).
HSPG expression characteristics have been linked to
tumor cell metastasis (11–14).
Notably, HSPG are targets of heparanase (HPSE), the dominant
mammalian endoglycosidase (endo-β-D-glucuronidase) whose activity
has been proven to be implicated in the angiogenic, tumorigenic and
metastatic abilities of tumor cells (16,17).
HPSE cleaves HS-GAGs at specific intrachain sites resulting in
fragments (10–20 sugar subunits) which are biologically active,
i.e., able to bind growth and angiogenic factors. Although Shh
roles in MB have been demonstrated (2,18,19),
mechanistic links between Shh deregulation and HPSE have yet to be
investigated. Similarly, Wnt pathway activation involves the
interaction of a Wnt ligand with Frizzled receptors, which leads to
inhibition of the β-catenin regulatory complex. Modulation of Wnt
signaling by HPSE has not been addressed, nor the mechanisms or
functional links between Wnt activation and MB (20).
The invasive cell phenotype requires cytoskeletal
dynamics driven by the small GTPases, Rac and Rho (21,22).
As tumor cells invade the surrounding tissue, they establish,
abolish and/or relocate transient focal adhesions (22,23).
Focal adhesion dynamics require Rac and Rho activities (21), and recent evidence supports roles
of syndecan (SDC) cell surface HSPGs (24).
We hypothesized that SDC downstream signaling events
are crucial for driving MB cell proliferation and invasiveness, and
that HPSE modulates HSPG-associated Shh/Wnt3a actions. To this end,
we utilized human MB lines (D721 and D283) that we had previously
characterized, establishing gradients of cell invasiveness and HPSE
functionality (4,25). We identified GEF-H1 as a novel
SDC-associated signal transduction protein in human MB cells. Of
note, GEF-H1 is a Rho guanine nucleotide exchange factor with
microtubule binding ability which also binds Rac1 as an inhibitor
(26). We investigated GEF-H1,
β-catenin and N-Myc protein expression and subcellular
localization, in addition to cell surface HSPG and Gli 1–3 gene
expression, by pre-treating MB cells with human active HPSE,
followed by Shh or Wnt3a exposure. In this study, we demonstrate
the roles of HSPG linking Shh/Wnt3a signaling to GEF-H1, Rac1/RhoA
activities and MB cell proliferative and invasive events.
Furthermore, we provide evidence that HPSE modifies MB cell
responses to HS-binding Shh and Wnt3a.
Materials and methods
Cell culture and treatments
Human MB cell lines of low/high invasive abilities
(D283 and D721) (4) were kindly
provided at low-passage by Dr Darrell Bigner (Duke University,
Durham, NC, USA) and cultured in improved minimal essential media
(IMEM) with Richter's Zn2+ Option (Richter's
modification), 2 mg/l L-glutamine, 2 mg/l L-proline, 50 μg/ml
gentamicin sulfate (Gibco/Invitrogen, Carlsbad, CA, USA)
supplemented with 20% fetal bovine serum (FBS), 100 U/ml penicillin
and 100 μg/ml streptomycin (normal growth medium). The
cultures were maintained at 37°C in 5% CO2. Due to the
inherent difficulty in growing MB cells, special care was taken to
grow cultures in flasks maintained in a canted position and at high
cell density. HPSE treatments were for 12 h (200 ng/ml) using
recombinant human active heparanase (rhHPSE) in IMEM with Richter's
Zn2+ Option (Richter's modification), 2 mg/l
L-glutamine, 2 mg/l L-proline, 50 μg/ml gentamicin sulfate
supplemented with 10 mM HEPES (pH 6.5), 5% FBS, 100 U/ml penicillin
and 100 μg/ml streptomycin. The cells were periodically
tested for Mycoplasma negativity, and only low passage cells were
used in the experiments.
Heparanase, HepIII, Shh and Wnt
treatments
Preparations of highly active rhHPSE were kindly
provided by Dr Israel Vlodavsky (The Bruce Rappaport Medical
Faculty, Technion, Israel) (27).
Heparanase activity was validated by utilizing a heparan
sulfate-degrading enzyme assay kit (Takara Mirus, Madison, WI,
USA). Following the mock or HPSE treatment, the cells were
serum-starved for either 12 or 48 h, as indicated. The growth
factor treatments were for 12 h with either recombinant human Shh
(R&D Systems, Minneapolis, MN, USA) or recombinant human Wnt3a
(R&D Systems) (100 ng/ml each). The removal of HS-GAGs of cell
surface HSPG was accomplished by digestion with Heparitinase III
(HepIII) from Flavobacterium heparinum (Sigma, St. Louis,
MO, USA). The final HepIII concentration was 0.05 U/ml culture
medium. The digestions were carried out for 1 h at 37°C in 5%
CO2.
RT-PCR
Total RNA was isolated from the MB cells using the
RNeasy Plus mini-kit (Qiagen, Valencia, CA, USA) according to the
manufacturer's instructions. The RNA yield was determined using a
NanoDrop ND1000 spectrophotometer (NanoDrop Products, Wilmington,
DE, USA). To ensure a lack of genomic DNA contamination, 1
μg of total RNA was digested with DNase I (Invitrogen) prior
to first-strand synthesis. Inactivated DNase I digestion (2
μl) was used as a template with a first-strand synthesis kit
utilizing SuperScript II reverse transcriptase according to the
manufacturer's instructions (Invitrogen). The first-strand
synthesis reaction was then diluted 1:1 with H2O to be
utilized as a single-strand cDNA template. PCR amplification was
performed in 20-μl reactions consisting of 1X AmpliTaq Gold
buffer (Applied Biosystems, Foster City, CA, USA), 2 mM
MgCl2, 300 μM dNTP mix, 400 nM primer pair, 2
μl single strand cDNA template and 2 units AmpliTaq Gold Taq
polymerase (Applied Biosystems). PCR conditions were: 94°C, 2 min;
40 cycles of 94°C, 20 sec; 58°C, 15 sec; 72°C, 42 sec; 72°C, 30
sec. Gene accession numbers and DNA sequences for the
oligonucleotide primer pairs utilized were: GAPDH, NM_002046.3,
forward TTC CAC CCA TGG CAA ATT CC, reverse TGG CAG GTT TTT CTA GAC
GG; HPSE, NM_006665.3, forward CTG GCA ATC TCA AGT CAA CC, reverse
TCC TAA CCA GAC CTT CTT GC; SDC1, NM_001006946.1, forward TGA CTC
TGA CAA CTT CTC CG, reverse TCT TCT TCA TGC GGT ACA GC; SDC2,
NM_002998.3, forward AAC CAG AAA CTG AAC CTC GG, reverse TCT ACA
TCC TCA TCA GCT CC; SDC3, NM_014654.3, forward AGT GAG AAC TTC GAG
AGA CC, reverse TGT GTG GTC TCT TCT TCT GG; SDC4, NM_002999.2,
forward TAG AAG GCC GAT ACT TCT CC, reverse TCA CGC GTA GAA CTC ATT
GG; GPC1, NM_002081.2, forward AGC CAT GTA TTT CAG GGA CC, reverse
GGA TGC ATG TTT GGA AAA GC; GPC2 NM_152742.1, forward CTC GGT ATT
CAG TTT TCC GG, reverse TTT CCC CAT AGA AGT CTC GC; GPC3,
NM_004484.2, forward GCA AGT ATG TCT CCC TAA GG, reverse CTT GCA
GTG ACT TGG AAA CC; GPC4, NM_001448.2, forward CAA GCT GTC TTT GCT
TCA CG, reverse AAC ATG GCT TCA CAG TCA CG; GPC5, U66033.1, forward
GAC CTG ATC TTC AGG TTT GC, reverse GAA GTG CAG ATA GTC TGT GG;
GPC6, NM_005708.2, forward TCC TCG TTT TGA TTG CAC CG, reverse AAG
TGG TGC GCA CAA AAT GG; Gli1, NM_005269, forward ACC AAT CAG TAG
CTA TGG CG, reverse TAT CAC CTT CCA AGG GTT CC; Gli2, NM_005270.4,
forward ACA TCA ACA ACT CCC GAA GC, reverse TGT CCA GAT CTT CCT TGA
GG; Gli3, NM_000168.5, forward TCT CCA TGA TCT CAG CAA CC, reverse
AGA GTA GGT GAA GCT CAA GG. The PCR reactions were conducted in a
Mastercycler ep gradient thermocycler (Eppendorf North America,
Westbury, NY, USA).
Cloning
For affinity chromatography experiments, the
pGEX-4T-3 vector was used (GE Healthcare, Piscataway, NJ, USA).
RT-PCR was performed to amplify cDNA regions encoding the
intracellular CT domains. Oligonucleotide primer pairs were
designed for insertion into the BamHI/XhoI (New
England Biolabs, Ipswich, MA, USA) restriction sites within the
pGEX vector. The gene accession numbers and DNA sequences for the
oligonucleotide primer pairs utilized were: SDC1, NM_001006946.1,
GST SDCN1 CT forward TTT GGA TCC CGC ATG AAG AAG AAG GAC GA; GST
SDCN1 CT reverse TTT CTC GAG TCA GGC ATA GAA TTC CTC CTG; SDC4,
NM_002999.2, GST SDCN4 CT forward TTT GGA TCC CGT ATG AAG AAG AAG
GAT GAA G; GST SDCN4 CT reverse TTT CTC GAG TCA CGC GTA GAA CTC ATT
GGT. The accuracy of these DNA sequences was confirmed by
sequencing (SeqWright Inc., Houston, TX, USA).
Mass spectrometry
Protein bands excised from the SDS-PAGE were
identified by matrix-assisted laser desorption ionization time of
flight tandem mass spectrometry (MALDI-TOF MS/MS) from the
proteomic core facility at Baylor College of Medicine. Briefly,
unique bands were excised from GSTPD and identified binding
proteins using MALDI-TOF MS/MS. Protein digests were then spotted
on a MALDI target plate, and the analyses were performed using an
ABI/SCIEX 4700 Proteomics Analyzer and TOF/TOF mass spectrometer.
Detected mono-isotopic peptide masses were then analyzed by MS-Fit
(Protein Prospector), and proteins were identified using peptide
mass finger-printing.
Cell lysates
RIPA buffer (Sigma) containing 150 mM NaCl, 1%
IGEPAL CA-630 (Sigma), 0.5% sodium deoxycholate 0.1% SDS, 50 mM
Tris, pH 8.0, supplemented with the protease inhibitors 1 mM PMSF
(Sigma) and Complete-mini (Roche, Mannheim, Germany) was used to
generate whole cell lysates. Nuclear and cytoplasmic lysates were
generated using the NE-PER kit (Thermo, Waltham, MA, USA) according
to the manufacturer's instructions.
GST pulldowns
This procedure was performed as previously described
(28). Briefly, GST-SDC1 or
GST-SDC4 fusion constructs were transformed into the E. coli
strain BL21 (Novagen, Madison, WI, USA). Expression was induced by
the addition of IPTG to LB culture media. Induced fusion proteins
from the bacteria were purified by lysing the cells using B-PER
reagent (Thermo) and passing the cell lysates over columns
containing glutathione beads (Thermo). GST fusion proteins were
eluted from the columns with reduced glutathione. The purified GST
fusion proteins (30 μg) were bound to glutathione bead
columns, and 500 μg of whole cell lysates was passed over
the columns. The columns were then washed three times, and the
bound proteins were eluted with 2X Laemmli buffer with subsequent
heating for 5 min at 95°C. Eluted proteins were detected by
Coomassie blue. Bands of interest were excised from the gels and
identified by mass spectrometry.
GTPase activity assays
Rac1 and RhoA small GTPase activity assays were
performed using respective GLISA kits (BK124, BK125) according to
the manufacturer's instructions (Cytoskeleton, Denver, CO, USA).
Briefly, after determining the cell number to cell lysis buffer
volume ratio that would reproducibly result in protein
concentrations of 0.5 μg/μl, the experimental
treatments were performed. Cell lysates were snap-frozen in liquid
nitrogen to preserve GTP-bound GTPase (Rac1 or RhoA). GTP bound
Rac1 GTPase was bound by Rac1-GTP-binding protein-linked wells of a
96-well plate (Corning). Bound active Rac1 was detected with a
Rac1-specific antibody. The degree of Rac1 activation was
determined by comparison to corresponding values obtained from
lysates derived from untreated serum-starved cells. GTP-bound RhoA
GTPase activity was similarly assayed (GLISA kits; Cytoskeleton).
The experiments were replicated, data were analyzed and
means/standard deviations were calculated.
Western blotting
Cell lysates were quantified by standard
spectrophotometric methods using either Protein Reagent (Bio-Rad,
Hercules, CA, USA) or BCA Reagent (Thermo). Aliquots (20 μg
of protein) of quantified cell lysates were resolved by 10–12%
SDS-PAGE, transferred onto polyvinylidene difluoride (PVDF)
membranes (Bio-Rad) in Tris-buffered saline (TBST) containing 5%
non-fat dry milk and 0.05% Tween-20 (Sigma). Hybridizations were
performed for 16 h at 4°C. The antibodies used were: SDC1 (clone
B-A38, 854.070.000; Cell Sciences, Canton, MA, USA), SDC4 (ab24511;
Abcam, Cambridge, MA, USA), GEF-H1 (55B6; Cell Signaling), N-Myc
(9405; Cell Signaling), β-actin (clone AC-15, sc-69879; Santa Cruz
Biotechnology), β-catenin (9581; Cell Signaling) and Fibrillarin
(clone 38-F3, sc-56676; Santa Cruz Biotechnology). Hybridized
primary antibodies were detected with ECL reagents (Thermo) and
HRP-conjugated secondary goat anti-rabbit (sc-2030) or goat
anti-mouse (sc-2031; Santa Cruz Biotechnology) antibodies.
Stripping of blots was performed with Restore reagent (21059)
(Thermo).
Cell proliferation assays
Cell proliferation was determined by AlamarBlue
staining using the manufacturer's protocol (Sigma). Briefly,
following treatment conditions, cells (3.0×106) were
plated in 96-well plates containing growth media and 10 μl
AlamarBlue, and subsequently cultured for 4 h at 37°C.
Proliferation was determined by measuring OD readings at 570 and
600 nm using a SpectraMax Plus384 spectrophotometer (Molecular
Devices, Sunnyvale, CA, USA). Data were then analyzed for
statistical significance. All assays were performed in
triplicate.
Invasion assays
Transwell membrane inserts (Corning) were coated
with Matrigel™ (BD Biosciences) at a 1:30 dilution in serum-free
media overnight (16 h) before use. The inserts were washed with
serum-free medium, and 1.5×105 cells/insert in IMEM +
0.1% BSA were placed on the top chambers. The bottom chambers
contained IMEM medium with 20% FBS and N-formyl-Met-Leu-Phe (5
μM) as chemoattractant (Sigma). The assays were run for 12 h
and then the inserts were removed, stained with 0.5% crystal violet
solution (400 μl) (Sigma) for 10 min, washed and extracted
with 200 μl of extraction solution (Cell Biolabs, San Diego,
CA, USA) for 10 min. The OD values were read at 560 nm using 100
μl of extracted solution and statistically analyzed.
Statistics
Data are represented as the mean ± standard
deviation. Significance values were obtained by the Student's
paired t-test (P<0.05; P<0.01). P-values of <0.01 were
considered highly significant.
Results
Differential cell surface HSPG and GEF-H1
expression in medulloblastoma
In previous studies, we demonstrated that heparanase
expression of specified MB cell lines correlates with their
invasive phenotype (25). We
considered that SDCs, particularly SDC1/4, in addition to bearing
HS-GAGs as substrates for HPSE enzymatic activity, may have
intracellular CT signaling capabilities that have yet to be
characterized. We hypothesized that HPSE modulates downstream
signaling events that are dependent upon HS/HS-binding growth
factor interactions. To determine the overall cell surface HSPG
expression status in these MB cell lines, semi-quantitative RT-PCR
was performed and gene expression levels for members of the SDC and
glypican families were determined (12). We focused on SDC1/4, considering
their established roles in cell invasion, cell adhesion and
cytoskeletal rearrangement (13,29).
Furthermore, SDC1 has been reported to affect Wnt responsiveness in
other model systems (30), and
SDC4 is a fundamental component of focal adhesions (31). Unique cell surface HSPG gene
expression patterns were observed for each MB cell line analyzed
(Fig. 1A). Notably, SDC1/4 gene
profiling from D283 and D721 cells was detected under normal growth
conditions (Fig. 1A).
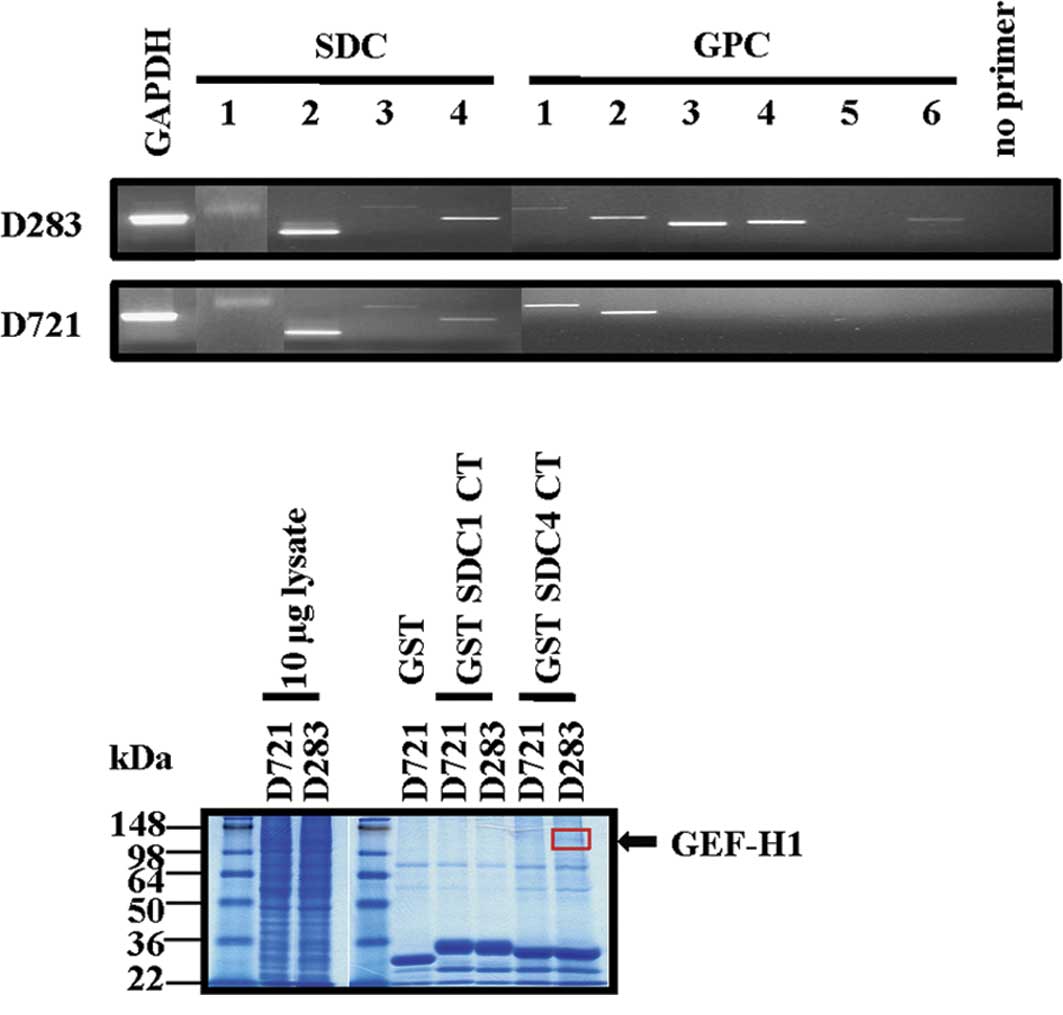 | Figure 1.Medulloblastoma cell lines express
cell surface HSPGs and GEF-H1 is a component of a SDC CT-binding
complex. (A) Semi-quantitative RT-PCR analysis of D283 and D721
cells. GAPDH was used as a positive control. Mastermix with the
primer pair was used as a negative control and a test for
contamination. PCR product sizes: GAPDH, 611 bp; SDC1, 725 bp;
SDC2, 548 bp; SDC3, 824 bp; SDC4 500 bp; GPC1, 849 bp; GPC2, 602
bp; GPC3, 488 bp; GPC4, 517 bp; GPC5, 569 bp; GPC6, 573 bp. (B)
SDS-PAGE of GST pulldown eluates. The arrow indicates the
endogenously-expressed SDC1/4 CT binding protein that was excised
from a Coomassie blue-stained gel of GST pulldown eluates. (C)
GEF-H1 is identified by MALDI-TOF MS/MS as a SDC4 CT-associated
protein from the D283 cell lysate, per B. (D) Representative
Western blotting detecting endogenous SDC1 (core protein), SDC4
(core protein) and GEF-H1 in poorly (D283) and highly (D721)
invasive MB cells. β-actin was used as a control for equal
loading. |
Next, to identify SDC intracellular CT binding
signal transduction components, GST fusion proteins were
constructed containing the intracellular CT domains from human
SDC1/4 and performed GST pulldown assays using cell lysates from
D283 and D721 MB cell lines. Proteins from the GST pull-down
eluates were then separated by SDS-PAGE. A single Coomassie
blue-stained protein band of 110 kDa was eluted preferentially from
the D283 cell lysates compared to the D721 eluted proteins
(Fig. 1B). This band was excised,
subjected to trypsin digestion and the resulting peptides were
identified by MALDI-TOF tandem mass spectrometry. This unique band
was identified as human GEF-H1 (Fig.
1C). Furthermore, SDC1/4 and GEF-H1 protein expression was
visualized in these MB cells under serum-containing conditions by
Western blotting. High GEF-H1/SDC4 protein expression was found in
poorly invasive D283 compared to highly aggressive D721 cells.
Moreover, inverse SDC1/SDC4 profiling was detected according to the
MB cell invasive phenotype (Fig.
1D).
Heparanase modulates GEF-H1, β-catenin
and N-Myc subcellular distribution in medulloblastoma
Recent evidence demonstrated that GEF-H1 functions
to facilitate RhoA activity in the cytoplasm at sites of focal
adhesion turnover, such as in regions of membrane ruffling
(32). Therefore, GEF-H1
localization may influence cell migration and cytoskeletal
dynamics. To characterize endogenous GEF-H1 expression and
subcellular distribution in response to treatment with Shh or
Wnt3a, and subsequent to pre-treatment with exogenous rhHPSE,
nuclear and cytoplasmic fractions were isolated. GEF-H1 antibody
detected both phosphorylated and non-phosphorylated forms of GEF-H1
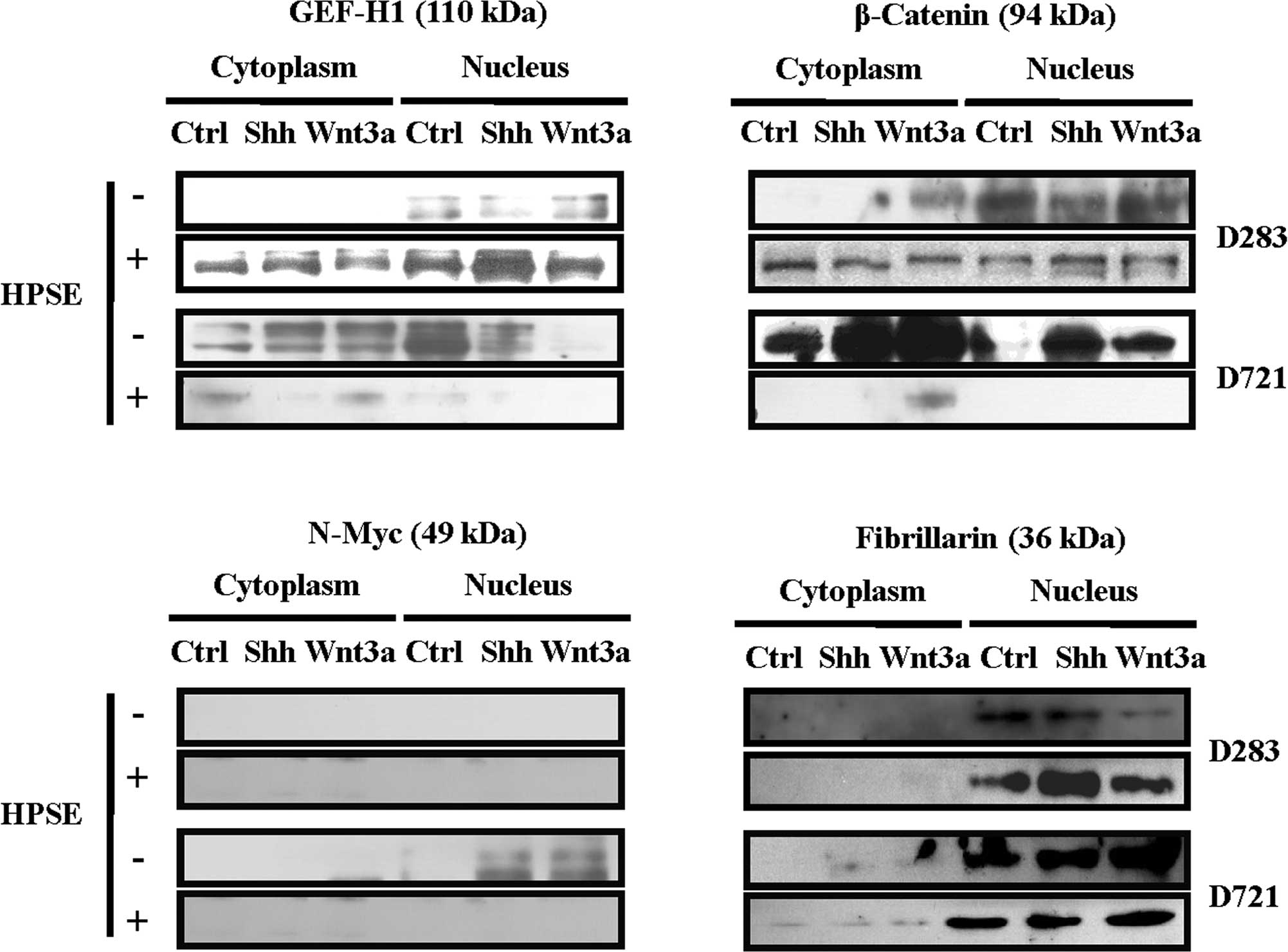
in these subcellular compartments (Fig. 2A). Exposure to rhHPSE altered the
GEF-H1 expression and its subcellular redistribution. The MB cell
lines possessed not only a differential GEF-H1 expression and
subcellular localization by rhHPSE, but also dependency upon Shh or
Wnt3a treatment (Fig. 2A).
Since the expression and subcellular distribution of
β-catenin is a crucial component of adherens junctions and
co-transcription in Wnt signaling (33), signal transduction cross-talk
between Shh and Wnt3a may be involved in MB, as demonstrated in
other systems (34). Exposure to
HPSE altered β-catenin expression and subcellular localization;
differential β-catenin protein expression and subcellular
redistribution were detected following Shh or Wnt3a treatment
(Fig. 2B). D283 cells displayed
primarily nuclear β-catenin, while D721 cells exhibited robust
cytoplasmic β-catenin content, increasing in response to Shh/Wnt3a
(Fig. 2B). To note, the β-catenin
content of D721 cells was drastically reduced subsequent to
pre-treatment with active HPSE (Fig.
2B).
Since a burst of N-Myc expression has been
associated with proliferation of external granular layer neurons of
the developing cerebellum and MB (1), human MB model system was analyzed for
N-Myc expression. Treatment with exogenous active HPSE may modulate
the MB cell response to Shh or Wnt3a treatments. Cytoplasmic and
nuclear fractions were probed for N-Myc protein expression, and
N-Myc modulation by Shh or Wnt3a was distinctly observed (Fig. 2C). No N-Myc expression was detected
in D283. Only exposure with Shh or Wnt3a induced N-Myc in the D721
cells. To note, HPSE abrogated N-Myc expression in the MB cell
lines (Fig. 2C). The purity of the
nuclear fractions was confirmed by probing with fibrillarin
(Fig. 2D).
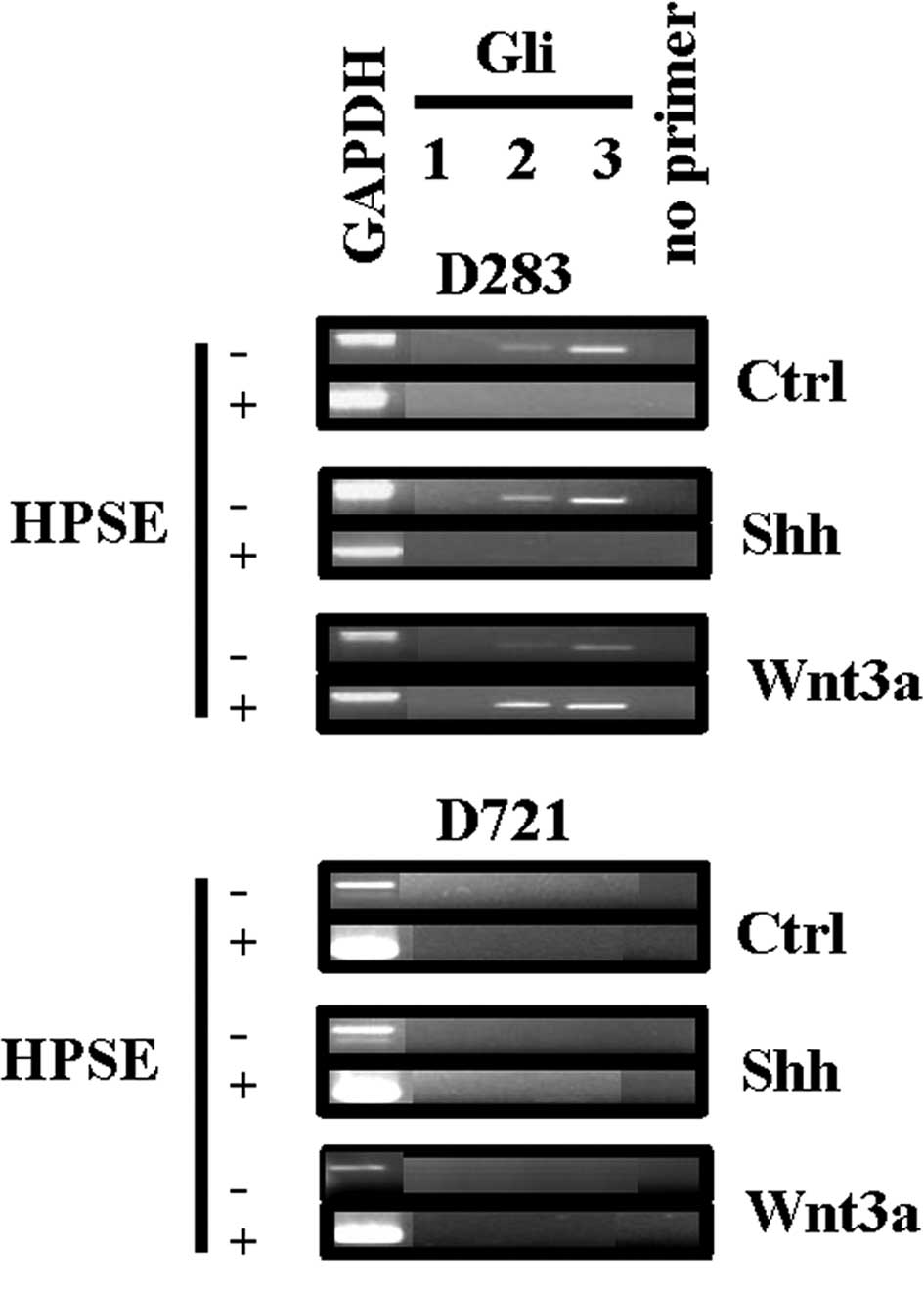
Heparanase modulates Gli gene expression
in medulloblastoma
Regulation of Gli transcription factor genes is a
key downstream read-out of activation of the Shh signal
transduction pathway (1). Since
Gli genes are activating (Gli1) or repressing (Gli2/Gli3) (35), the expression of the Gli genes
subsequent to HPSE exposure was analyzed, followed by treatment
with Shh or Wnt3a. Gli from the D283 cells were expressed under
control conditions and HSPE treatment reduced Gli2/3 gene
expression (Fig. 3). Treatment of
D283 cells with Shh also resulted in Gli2/3 gene expression
(Fig. 3). However, subsequent to
treatment with exogenous HPSE, as without growth factor treatment,
Gli2/3 gene expression was reduced (Fig. 3). The Gli gene expression from the
poorly invasive D283 cells was modulated by treatment with
exogenous HPSE, while not from the highly invasive D721 cells. To
note, signal transduction cross-talk was demonstrated from the D283
cells, as Wnt3a treatment distinctly resulted in Gli2/3 gene
expression (Fig. 3) (34). The Gli gene expression from the
D721 cell line was quiescent in response to Shh or Wnt3a treatment,
an indication that this signal transduction pathway is likely not
involved in the highly invasive phenotype of these cells (Fig. 3).
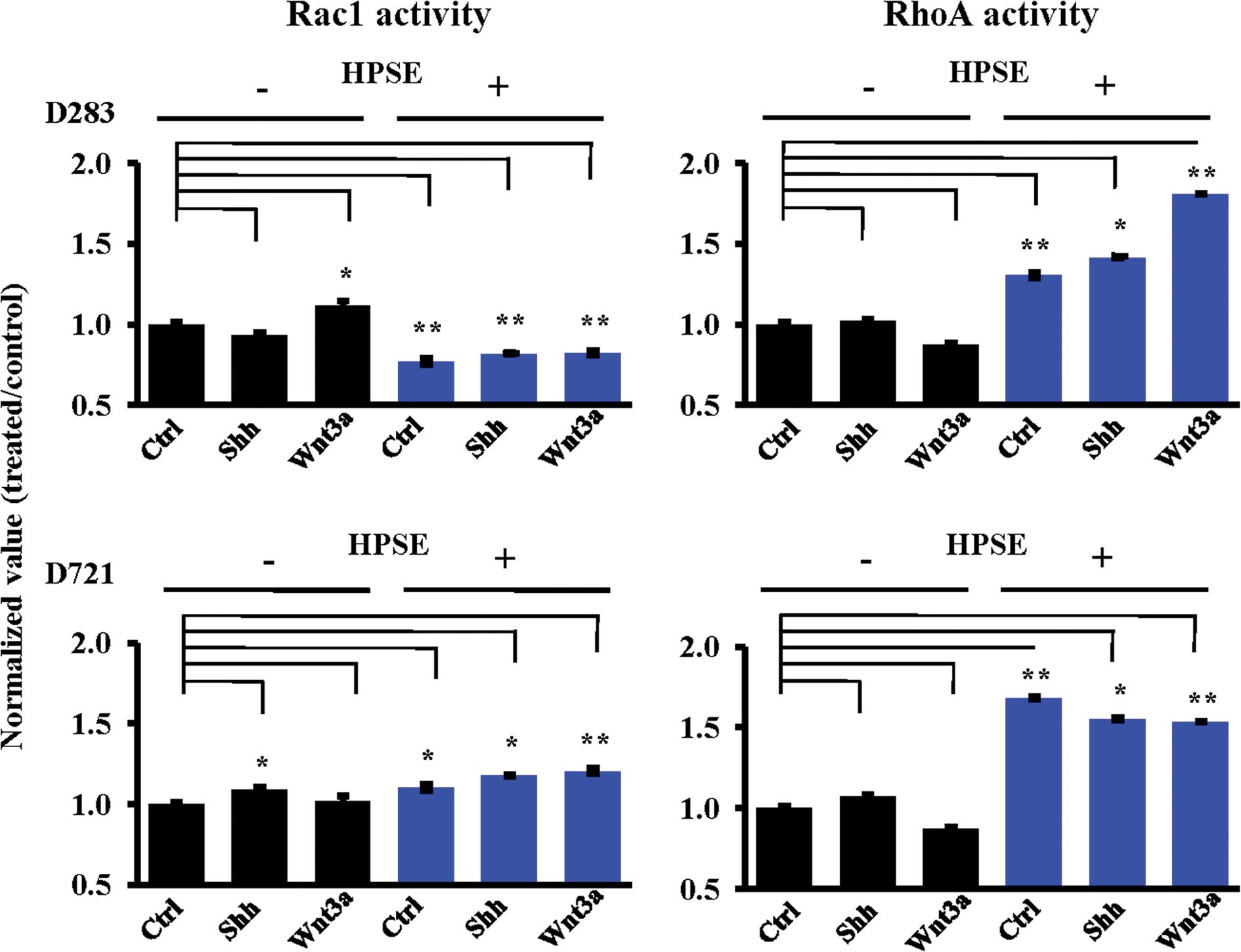
Heparanase modulates Rac1 and RhoA
activities in medulloblastoma
Cell motility requires a dynamic cytoskeleton
(36,37), and small GTPase Rac1 and RhoA
activities are necessary in cytoskeletal dynamics (36,37).
To reveal the mechanisms of cell motility and determine whether
heparanase modulates GTPase activity in medulloblastoma cells, Rac1
and RhoA activities in MB cells were measured following heparanase
treatment. In D283 cells, heparanase pre-treatment significantly
decreased Rac1 activity, while concomitantly increasing RhoA
activity (Fig. 4). In these cells,
HPSE facilitated the RhoA activity response induced by Shh or Wnt3a
(Fig. 4B). Conversely, the D721 MB
cell line displayed increased Rac1 activity as a general response
to HPSE (Fig. 4A). HPSE
pre-treatment resulted in increased Rac1 and RhoA activities in the
D721 cells (Fig. 4). RhoA activity
from the D721 cells decreased following Shh or Wnt3a treatment
subsequent to HPSE pre-treatment, as opposed to the D283 RhoA
activity response which was HPSE-facilitated (Fig. 4B). HPSE pre-treatment modified the
Rac1/RhoA activity response to Shh and Wnt3a under every condition
tested. To note, RhoA activity was increased subsequent to rhHPSE
(Fig. 4B).
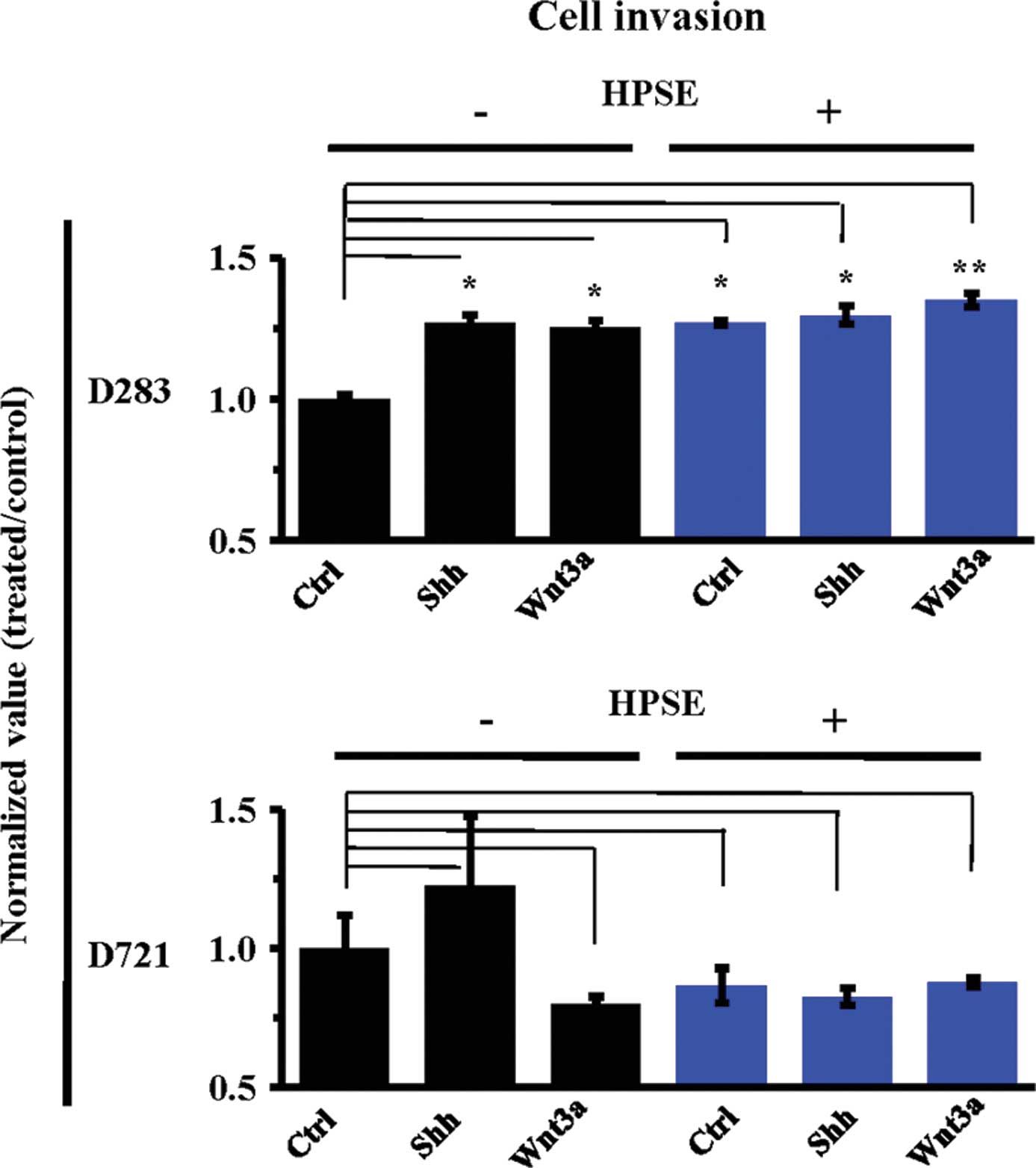
Heparanase modulates Shh and Wnt-induced
medulloblastoma cell invasion
It is known that the invasive phenotype requires a
dynamic cytoskeleton (38).
Considering that cytoskeletal dynamics rely upon the activity of
Rac1 and RhoA small GTPases (38),
and that HPSE regulates Rac1 and RhoA activities, MB cells of
varying invasive characteristics were pre-exposed (4) with HPSE, followed by Shh or Wnt3a
treatment.
HPSE pre-treatment significantly increased the
invasiveness of the D283 MB cells (P<0.05, P<0.01) in the
conditions tested (Fig. 5).
Conversely, highly invasive D721 cells experienced less
invasiveness in response to HPSE. Treatment with Shh or Wnt3a
subsequent to HPSE did not restore D721 cell invasiveness (Fig. 5). Taken together, these data
indicate the differential roles of HPSE in regulating MB cell
invasion in highly invasive vs. non-invasive cell lines.
Heparanase modulates Shh and Wnt-induced
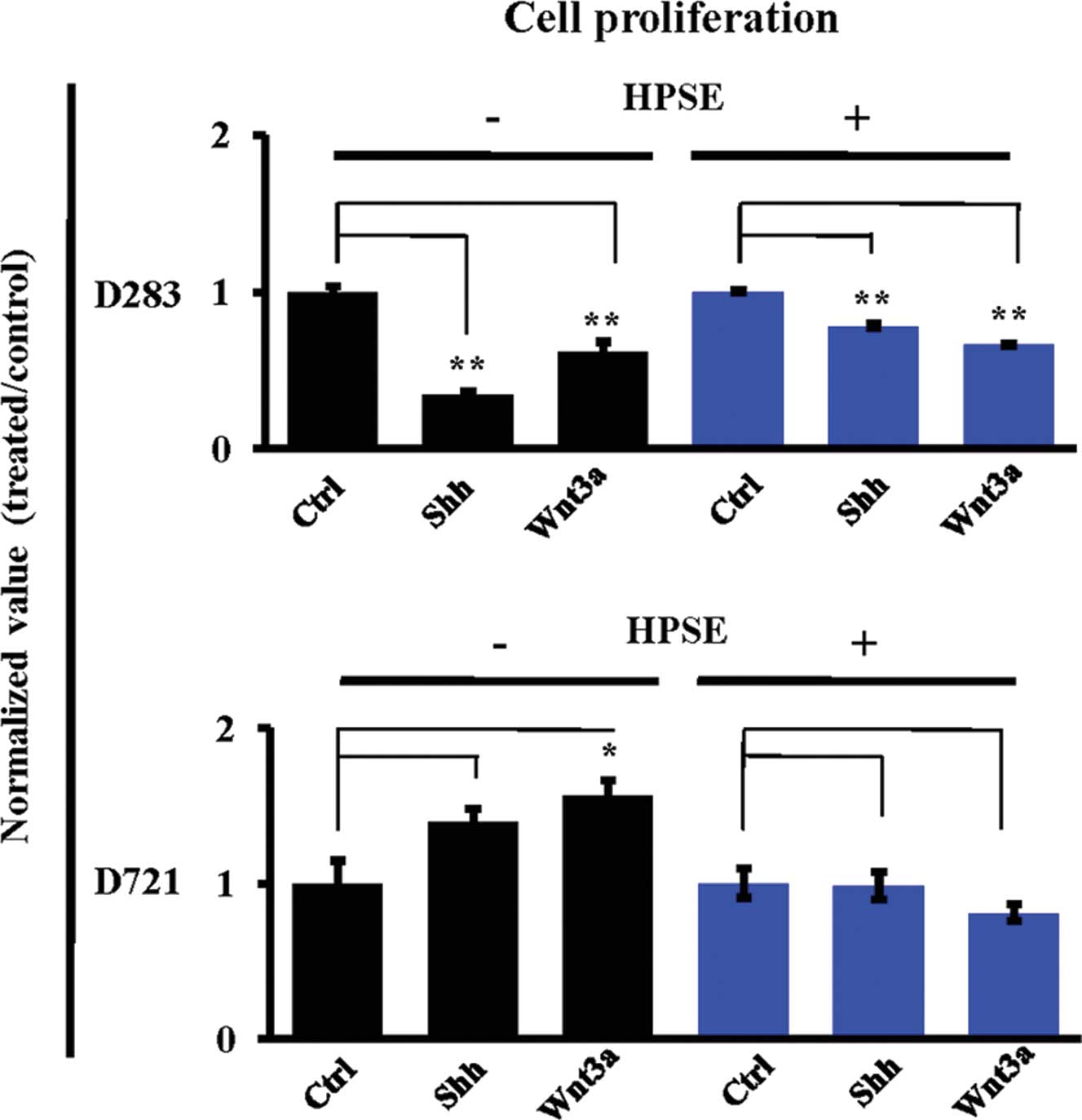
medulloblastoma cell proliferation
MB is marked by a dramatic burst of cell
proliferation from developing external granular layer neurons as a
response to secreted growth factors from Purkinje neurons (1). Accordingly, the role of HPSE
modulating MB cell proliferation was explored. No Shh/Wnt-induced
proliferative responses to these treatments were noted in rapidly
proliferative D283 cells. However, D721 cell proliferation was
augmented in response to treatment with Shh (140%) or Wnt3a (156%,
P<0.05) (Fig. 6). Notably, the
Shh/Wnt-induced proliferative response was not present subsequent
to HPSE treatment (Fig. 6),
indicating that HPSE and Shh/Wnt work in synergy to reduce cell
proliferation in D721 cells.
Discussion
Interest concerning the roles of the HSPGs in MB is
growing and has become a subject of continued investigations. In
this study, we characterized the relevance of the HPSE/HSPG axis on
Shh and Wnt3a signaling in a human MB cell system consisting of two
lines with distinct tumorigenicities. We provide first-time
evidence demonstrating i) a differential cell surface HSPG
expression, notably of SDC1 and 4; ii) the identification of a
guanine nucleotide exchange factor, GEF-H1, associated with SDC CT
domain; iii) the importance of GEF-H1 in HSPG-modulated Shh and
Wnt3a signaling affecting Rac1/RhoA activities and cell
invasiveness; iv) the fact that active HPSE significantly affects
Shh and Wnt3a signaling and alters the expression and localization
of key downstream effectors, namely β-catenin and the Gli family of
transcription factors.
Cell surface SDC1 and 4 are central in cell invasion
and focal adhesion, and small GTPase activity is critical to cell
motility (11,13). We provided evidence that a small
GTPase regulatory protein, GEF-H1, is differentially expressed
under normal (serum-containing) growth conditions among MB lines
previously characterized in regards to HPSE functionality towards
invasiveness (4,25). Recently, Sanz-Moreno et al
indicated that the interchangeable plasticity of Rac1 vs. RhoA
activity is important in tumor cell migration (38). GEF-H1 seems to be uniquely situated
to participate in such interplay, since it binds and regulates both
Rac1 and RhoA (39,40), and thus can be considered a key
regulator of tumor cell migration and phenotypic plasticity. This
is of relevance as it refutes the expectation that a global RhoA
preference throughout a cell is due to the presence of more GEF-H1
protein.
We addressed this issue by comparing RhoA activities
between the D283 and D721 cells. We observed a correlation between
HPSE-induced GEF-H1 expression (Fig.
2A) and decreased Rac1, but increased RhoA activity in the D283
cells, a situation one would anticipate in association with GEF-H1
modulation of Rac1 and RhoA activities. We identified increased
Rac1 and RhoA activities in HPSE-pre-treated D721 cells decreasing
GEF-H1 expression in response to such treatment (Fig. 2A). This indicates that D721 may
rely upon an alternate (or additional) Rho GEF (Fig. 5B) (32).
Similarly, and considering that the β-catenin status
in MB tumors is clinically relevant (20), we observed primarily nuclear
β-catenin in our least invasive D283 cells. By contrast, the more
aggressive D721 cells had increased cytoplasmic localization of
β-catenin augmented without the presence of heparanase.
Furthermore, the Shh/Wnt treatment increased cytoplasmic β-catenin,
and exposure to active HPSE abrogated β-catenin expression and
localization in these cells (Fig.
2B).
Highly proliferative D283 cells did not express
nuclear N-Myc constitutively or in response to growth factor
stimulation (Fig. 2C). The less
proliferative D721 cells proved to be the more growth
factor-inducible and this finding correlated well with N-Myc
expression (Fig. 2C).
Stimulation by Shh and/or Wnt has been demonstrated
in other systems to induce Gli gene expression (41). The expression pattern of the three
Gli genes has been termed the ‘Gli code’ as the resulting
phenotypic outcomes of active or repressive can be predicated by
the combination of Gli gene expression (35). We demonstrated that the less
invasive D283 cells were Gli repressive under conditions without
exogenous active HPSE. Highly invasive D721 cells were Gli inert,
regardless of HPSE treatment. Furthermore, we showed that when a MB
cell responds to Shh or Wnt3a by Gli gene expression, subsequent
exposure to exogenous active HPSE will likely result in repressing
Gli gene expression. Our data also indicated that D283 MB cells
demonstrate Wnt/Gli crosstalk (Fig.
4). HPSE pre-treatment inhibited Rac1 activity, but facilitated
RhoA activity in poorly invasive D283 cells. Thus, the ratios of
Rac1 and RhoA activity in these cells can be modulated to favor
RhoA GTPase activity, in response to exogenous active HPSE, which
represses the Rac1 activity while concomitantly increasing GEF-H1
protein expression. This is particularly valid when GEF-H1 is
highly present in the cytoplasm (Fig.
2A). To note, the greatest increase in Rac1 GTPase activity was
in the D721 cells, and this response was Shh-dependent. Although it
has been shown that Shh down-regulates MB cell growth when cells
are explanted into culture (42),
our study is still relevant to gain insights into the cellular
physiology of MB. For example, we previously demonstrated
modulation of GEF-H1 induced signaling by HPSE in brain metastatic
settings (43). However, it
remains to be demonstrated whether or not GEF-H1 expression is
consistently associated with decreased invasiveness in other tissue
types. HPSE pre-treatment augmented Rac1/RhoA small GTPase
activity, which is indicative of increased cytoskeletal
dynamics.
Furthermore, we demonstrated that HPSE pre-treatment
increased the invasiveness in a RhoA-dependent manner under
conditions of increased GEF-H1 expression which augmented the
invasiveness of D283. Conversely, we found that the highly invasive
D721 cells did not show reduced Rac1 activity to accompany the
increased RhoA activity in response to HPSE pre-treatment and
decreased GEF-H1 expression while increasing RhoA activity.
Therefore, an alternate Rho GEF may facilitate the increased RhoA
activity in these cells. Along with reduced GEF-H1 expression
associated with HPSE pre-treatment, D721 also demonstrated reduced
invasiveness associated with increased RhoA activity.
In summary, we demonstrated a correlation between
cell surface HSPG-induced mechanisms and MB cell invasiveness
linking SDCs to GEF-H1 and Rac1/RhoA activities, the involvement in
Shh/Wnt signaling pathways and their modulation by the presence of
exogenous active heparanase. We also demonstrated roles for
heparanase in regulating MB cell proliferation and adhesion in
addition to its established heparan sulfate chain degrading
activity. These findings provide impetus in further deciphering the
HPSE/HSPG axis to affect gene regulation in MB and to generate
synergies towards the discovery of novel markers for MB clinical
intervention.
Abbreviations:
|
CT
|
carboxy terminal
|
|
DEPC
|
diethylpyrocarbonate
|
|
ECM
|
extracellular matrix
|
|
FBS
|
fetal bovine serum
|
|
GAG
|
glycosaminoglycan
|
|
GEF-H1
|
guanine nucleotide exchange
factor-H1
|
|
GPC
|
glypican
|
|
GPI
|
glycosylphosphotidylinositol
|
|
GST
|
glutathione-S-transferase
|
|
GSTPD
|
GST pulldown
|
|
H2O
|
double-distilled water
|
|
HEPES
|
4-(2-hydroxyethyl)-piperazine-1-ethanesulfonic acid,
N-(2-hydroxyethyl)piperazine-N'-(2-ethanesulfonic acid)
|
|
rhHPSE
|
recombinant human active heparanase
(GS3)
|
|
HS
|
heparan sulfate
|
|
HSPG
|
heparan sulfate proteoglycan
|
|
IPTG
|
isoprythiogalactoside
|
|
MALDI-TOF MS/MS
|
matrix-assisted laser desorption
ionization time of flight tandem mass spectrometry
|
|
MB
|
medulloblastoma
|
|
MEM
|
minimal essential media
|
|
Shh
|
recombinant human Sonic hedgehog
|
|
Wnt3a
|
recombinant human Wingless-type MMTV
integration site family member 3a
|
|
SDC
|
syndecan
|
|
SDS-PAGE
|
sodium dodecyl sulfate-polyacrylamide
gel electrophoresis
|
|
Wnt
|
Wingless-type MMTV integration
site
|
Acknowledgements
We thank Dr Israel Vlodavsky and Dr
Neta Ilan (The Bruce Rappaport Medical Faculty, Technion, Israel)
for kindly providing the rhHPSE. We also thank Dr Richard Cook,
Director of the BCM mass spectrometry core facility for MALDI-TOF
MS/MS analyses, and Dr Anita Chandler (Department of Molecular
Physiology and Biophysics at BCM) for the expert editorial
assistance. This study was supported by NIH grant 2R0-1 CA 086832
(to D. Marchetti).
References
|
1.
|
Guessous F, Li Y and Abounader R:
Signaling pathways in medulloblastoma. J Cell Physiol. 217:577–583.
2008. View Article : Google Scholar
|
|
2.
|
Flora A, Klisch TJ, Schuster G and Zoghbi
HY: Deletion of Atoh1 disrupts Sonic Hedgehog signaling in the
developing cerebellum and prevents medulloblastoma. Science.
326:1424–1427. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Ribi K, Relly C, Landolt MA, Alber FD,
Boltshauser E and Grotzer MA: Outcome of medulloblastoma in
children: long-term complications and quality of life.
Neuropediatrics. 36:357–365. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Marchetti D, Mrak RE, Paulsen DD and
Sinnappah-Kang ND: Neurotrophin receptors and heparanase: a
functional axis in human medulloblastoma invasion. J Exp Clin
Cancer Res. 26:5–23. 2007.PubMed/NCBI
|
|
5.
|
Gottardo NG and Gajjar A: Current therapy
for medulloblastoma. Curr Treat Options Neurol. 8:319–334. 2006.
View Article : Google Scholar
|
|
6.
|
Yauch RL, Dijkgraaf GJ, Alicke B, et al:
Smoothened mutation confers resistance to a hedgehog pathway
inhibitor in medulloblastoma. Science. 326:572–574. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Vaillant C and Monard D: SHH pathway and
cerebellar development. Cerebellum. 8:291–301. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Cool SM and Nurcombe V: Heparan sulfate
regulation of progenitor cell fate. J Cell Biochem. 99:1040–1051.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Rubin JB, Choi Y and Segal RA: Cerebellar
proteoglycans regulate sonic hedgehog responses during development.
Development. 129:2223–2232. 2002.PubMed/NCBI
|
|
10.
|
Mythreye K and Blobe GC: Proteoglycan
signaling co-receptors: roles in cell adhesion, migration and
invasion. Cell Signal. 21:1548–1558. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
O'Connell MP, Fiori JL, Kershner EK, et
al: Heparan sulfate proteoglycan modulation of Wnt5A signal
transduction in meta-static melanoma cells. J Biol Chem.
284:28704–28712. 2009.PubMed/NCBI
|
|
12.
|
Iozzo RV: Heparan sulfate proteoglycans:
intricate molecules with intriguing functions. J Clin Invest.
108:165–167. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Sanderson RD and Yang Y: Syndecan-1: a
dynamic regulator of the myeloma microenvironment. Clin Exp
Metastasis. 25:149–159. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Beauvais DM and Rapraeger AC: Syndecans in
tumor cell adhesion and signaling. Reprod Biol Endocrinol. 2:1–12.
2004. View Article : Google Scholar
|
|
15.
|
Tkachenko E, Rhodes JM and Simons M:
Syndecans: new kids on the signaling block. Circ Res. 96:488–500.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Fux L, Ilan N, Sanderson RD and Vlodavsky
I: Heparanase: busy at the cell surface. Trends Biochem Sci.
34:511–519. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Ilan N, Elkin M and Vlodavsky I:
Regulation, function and clinical significance of heparanase in
cancer metastasis and angiogenesis. Int J Biochem Cell Biol.
38:2018–2039. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Goodrich LV, Milenkovic L, Higgins KM and
Scott MP: Altered neural cell fates and medulloblastoma in mouse
patched mutants. Science. 277:1109–1113. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Berman DM, Karhadkar SS, Hallahan AR, et
al: Medulloblastoma growth inhibition by hedgehog pathway blockade.
Science. 297:1559–1561. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Fattet S, Haberler C, Legoix P, et al:
Beta-catenin status in paediatric medulloblastomas: correlation of
immunohistochemical expression with mutational status, genetic
profiles, and clinical characteristics. J Pathol. 218:86–94. 2009.
View Article : Google Scholar
|
|
21.
|
Symons M and Segall JE: Rac and Rho
driving tumor invasion: who's at the wheel? Genome Biol. 10:1–4.
2009.PubMed/NCBI
|
|
22.
|
Sanz-Moreno V and Marshall CJ: Rho-GTPase
signaling drives melanoma cell plasticity. Cell Cycle. 8:1484–1487.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Ilina O and Friedl P: Mechanisms of
collective cell migration at a glance. J Cell Sci. 122:3203–3208.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Avalos AM, Valdivia AD, Munoz N, et al:
Neuronal Thy-1 induces astrocyte adhesion by engaging syndecan-4 in
a cooperative interaction with alphavbeta3 integrin that activates
PKCalpha and RhoA. J Cell Sci. 122:3462–3471. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Sinnappah-Kang ND, Kaiser AJ, Blust BE,
Mrak RE and Marchetti D: Heparanase, TrkC and p75NTR: their
functional involvement in human medulloblastoma cell invasion. Int
J Oncol. 27:617–626. 2005.PubMed/NCBI
|
|
26.
|
Birkenfeld J, Nalbant P, Yoon SH and
Bokoch GM: Cellular functions of GEF-H1, a microtubule-regulated
Rho-GEF: is altered GEF-H1 activity a crucial determinant of
disease pathogenesis? Trends Cell Biol. 18:210–219. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Nardella C, Lahm A, Pallaoro M, Brunetti
M, Vannini A and Steinkuhler C: Mechanism of activation of human
heparanase investigated by protein engineering. Biochemistry.
43:1862–1873. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Ridgway LD, Kim EY and Dryer SE: MAGI-1
interacts with Slo1 channel proteins and suppresses Slo1 expression
on the cell surface. Am J Physiol Cell Physiol. 297:C55–C65. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Xian X, Gopal S and Couchman JR: Syndecans
as receptors and organizers of the extracellular matrix. Cell
Tissue Res. 339:31–46. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Alexander CM, Reichsman F, Hinkes MT, et
al: Syndecan-1 is required for Wnt-1-induced mammary tumorigenesis
in mice. Nat Genet. 25:329–332. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Dovas A, Yoneda A and Couchman JR:
PKCbeta-dependent activation of RhoA by syndecan-4 during focal
adhesion formation. J Cell Sci. 119:2837–2846. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Nalbant P, Chang YC, Birkenfeld J, Chang
ZF and Bokoch GM: Guanine nucleotide exchange factor-H1 regulates
cell migration via localized activation of RhoA at the leading
edge. Mol Biol Cell. 20:4070–4082. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Hartsock A and Nelson WJ: Adherens and
tight junctions: structure, function and connections to the actin
cytoskeleton. Biochim Biophys Acta. 1778:660–669. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Alvarez-Medina R, Cayuso J, Okubo T,
Takada S and Marti E: Wnt canonical pathway restricts graded
Shh/Gli patterning activity through the regulation of Gli3
expression. Development. 135:237–247. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Ruiz i Altaba A, Mas C and Stecca B: The
Gli code: an information nexus regulating cell fate, stemness and
cancer. Trends Cell Biol. 17:438–447. 2007.PubMed/NCBI
|
|
36.
|
Ridley AJ and Hall A: The small
GTP-binding protein rho regulates the assembly of focal adhesions
and actin stress fibers in response to growth factors. Cell.
70:389–399. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Ridley AJ, Paterson HF, Johnston CL,
Diekmann D and Hall A: The small GTP-binding protein rac regulates
growth factor-induced membrane ruffling. Cell. 70:401–410. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Sanz-Moreno V, Gadea G, Ahn J, et al: Rac
activation and inactivation control plasticity of tumor cell
movement. Cell. 135:510–523. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Ren Y, Li R, Zheng Y and Busch H: Cloning
and characterization of GEF-H1, a microtubule-associated guanine
nucleotide exchange factor for Rac and Rho GTPases. J Biol Chem.
273:34954–34960. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
40.
|
Birkenfeld J, Nalbant P, Bohl BP, Pertz O,
Hahn KM and Bokoch GM: GEF-H1 modulates localized RhoA activation
during cytokinesis under the control of mitotic kinases. Dev Cell.
12:699–712. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Ulloa F and Marti E: Wnt won the war:
antagonistic role of Wnt over Shh controls dorso-ventral patterning
of the vertebrate neural tube. Dev Dyn. 239:69–76. 2009.PubMed/NCBI
|
|
42.
|
Sasai K, Romer JT, Lee Y, et al: Shh
pathway activity is down-regulated in cultured medulloblastoma
cells: implications for preclinical studies. Cancer Res.
66:4215–4222. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Ridgway LD, Wetzel MD and Marchetti D:
Modulation of GEF-H1 induced signaling by heparanase in brain
metastatic melanoma cells. J Cell Biochem. Aug;2010.(E-pub ahead of
print).
|