Introduction
Cancer-directed immune-based therapies focus on
eliciting a cytotoxic T cell (CTL) response due to the fact that
CTLs directly kill tumor cells. However, most of these strategies
have been occluded by strong immunosuppression and immune-escape
mechanisms that become established in tumor-bearing hosts (1,2).
Tumor-induced immune suppression is caused by numerous mechanisms,
many of which involve the accumulation of immune-suppressive
infiltrates into the tumor microenvironment. One of the most potent
and well-studied suppressive phenotypes found in the tumor
microenvironment is the regulatory T cell (Treg). Tumors may
differentiate, expand, recruit and activate Tregs via multiple
mechanisms, and potentially abrogate antitumor immunity (3). Tregs have been shown to increase in
the tumor and the peripheral blood of patients with cancer
(4–12), and to be inversely related to the
outcome of several types of human malignancies (10,12).
Consistent with these findings, previous studies have demonstrated
that deletion of Tregs results in the enhancement of effective
tumor immune responses via the removal of such strong
immunosuppression (13–18). Therefore, blocking the migration of
Tregs or their function by means of immunotherapeutic approaches
may prove beneficial in cancer therapy.
Recently, it has been reported that CD4+
T helper (Th) cells are crucial for the induction of effective
antitumor immunity. Th cells are primarily responsible for
enhancing and sustaining CD8+ T cell responses (19,20).
Apart from the key role of CD4+ cells in controlling
CD8+ T cell reactivity to antigens, which emphasizes the distinct
roles of Th1 and Th2 cells in tumor eradication, it has been
demonstrated that Th cells have direct cytolytic effects via
multiple pathways (21,22) and activate antigen-presenting cells
(23). In addition, Zhang et
al demonstrated in an animal model that Th1 cell therapy
significantly inhibits the accumulation of Tregs in tumor-draining
lymph nodes and that this abrogation is associated with interferon
(IFN)-γ secreted from Th1 cells (24). By introducing Th1-dominant immunity
and reducing the number of Tregs, overcoming immunosuppression and
inducing fully activated tumor-specific CTLs becomes critically
important. Several potential mechanisms by which Th cell therapy
modulates the tumor microenvironment include enhancing the
production of inflammatory cytokines and reducing the number of
Tregs, thus supporting the elaborate tumor-specific immune
response. Despite this encouraging data, clinical use of Th cells
remains limited because of technical difficulties in acquiring and
expanding Th cells to the numbers deemed necessary for adoptive
cellular therapy (ACT) to be effective (25).
Anti-CD3 stimulated lymphokine-activated killer
(CD3-LAK) cells used in this study have been reported to be
effective in therapeutic animal models (26,27),
whereby CD3-LAK cells are already being used in clinical
applications. Although CD3-LAK therapy was found to be only
minimally effective in the treatment of large tumor burdens,
adjuvant use of CD3-LAK therapy is reported to contribute to
significant risk reduction of tumor recurrence (28,29).
There are many reports that CD3-LAK cells display non-specific
lytic activity in a broad spectrum of tumors (30). However, it remains to be clarified
whether CD3-LAK therapy in patients with advanced cancer, in whom
strong immunosuppression and immune-escape mechanisms are
established, has the potential to alter cytokine secretion from
blood cells and affect the number of Tregs. To resolve these
issues, the secretion ability of cytokines in whole blood and the
number of peripheral blood Tregs before and after ACT using CD3-LAK
cells were examined in advanced cancer patients. The relationship
of the immunological response with clinical efficacy after ACT
treatment was also assessed.
Patients and methods
Patients and blood samples
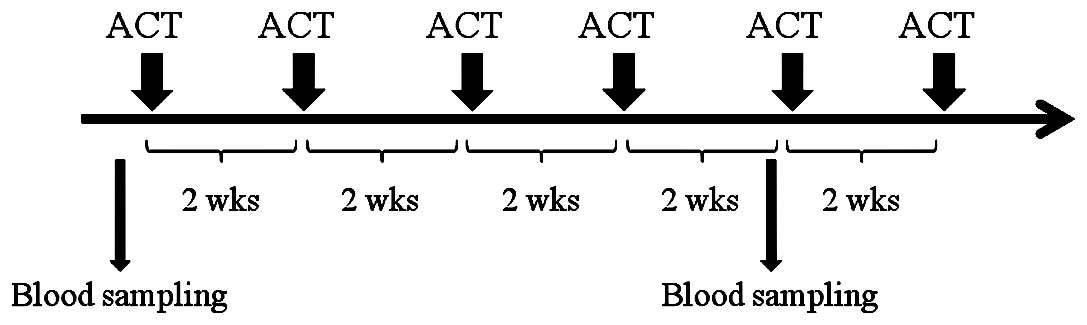
Blood samples from 109 consecutive cancer patients
who were scheduled to receive ACT with CD3-LAK cells were collected
intravenously at Hyakumanben Clinic between October 2008 and
December 2009. Among the 109 patients, 3 received ACT as
postoperative adjuvant therapy for the prevention of disease
recurrence, and 106 received this therapy for advanced or recurring
diseases. The patients received ACT at 2-week intervals and this
continued until they were unable to receive further treatment due
to either poor general condition or if they refused any further
treatment. For testing immune function, venous blood was obtained
from patients before starting the therapy and during the follow-up,
which was after 4 cycles of ACT. Of the 109 patients, 76 received
ACT four times or more. First follow-up blood sampling was carried
out immediately before the 5th ACT (2 weeks after the 4th ACT;
Fig. 1). All 109 blood samples at
baseline and 76 first follow-up samples were available.
Healthy individuals were selected as controls for
comparison with our cancer patients. Of the 49 healthy subjects, 41
were men and 8 were women. These subjects were selected from
individuals receiving medical examinations at the clinic who had no
acute or chronic disease. The median age of the healthy controls
was 54 years (range 40–78).
All subjects gave written informed consent. The
institutional review board approved the present study.
Lymphokine-activated killer cell
preparation
Peripheral blood (30–40 ml) was taken from the
patients. Mononuclear cells were separated and resuspended in a
CultiLife215 bag (Takara Bio, Otsu, Japan) pre-coated with anti-CD3
(Janssen Pharmaceutical, Tokyo, Japan) and then cultured in
serum-free media, GT-T505 (Takara Bio), supplemented with 1%
heat-inactivated plasma and 1,000 U/ml recombinant interleukin
(IL)-2 (Nipro, Osaka, Japan) for 2 days. At day 7, the cells were
transferred to a CultiLifeEva bag (Takara Bio), and GT-T503 media
was added. At day 9, IL-2 and 1% heat-inactivated plasma were
added. At day 14, the cells were harvested and resuspended in 100
ml saline with 1% human albumin, as the final cell product. These
cell products were assessed for viability and checked for
contamination with bacteria, fungi and endotoxins.
Cytokine assay
Methods for quantifying IFN-γ production in whole
human blood have been described previously (31). Briefly, heparinized peripheral
blood was cultured with Sendai virus (HVJ, 500 HA/ml) within 8 h
after the withdrawal of blood. The blood-virus mixture was
incubated at 37°C for 20 h, and IFN-γ activity in the supernatants
was quantified by bioassay. Other cytokines were measured according
to the following procedure: heparinized whole blood was diluted
4-fold with Eagle’s minimal essential medium (MEM; Nissui
Pharmaceutical Co., Ltd., Tokyo, Japan) and stimulated with
phytohemagglutinin (PHA; 25 μg/ml). After 48 h of incubation at
37°C, supernatants were harvested by centrifugation at 800 x g for
10 min and stored at −80°C until analysis. Cytokine levels in the
samples were measured using a multiplex cytokine array system
(Bio-Plex; Bio-Rad Laboratories, Hercules, CA, USA) according to
the manufacturer’s instructions. The Multiplex Th1/Th2 bead kit
(Bio-Rad) included various cytokines [IL-2, IL-4, IL-5, IL-10,
IL-12(p70), IL-13, TNF-α, IFN-γ and granulocyte-monocyte
colony-stimulating factor (GM-CSF)]. Data acquisition and analysis
were carried out using the Bio-Plex Manager Software, version
5.0.
Flow cytometric analysis
To determine the regulatory cell phenotype, flow
cytometry of whole blood was performed using the following
antibodies: PE-Cy™5 mouse anti-human CD4; PE mouse anti-human CD25,
Alexa Fluor® 488 mouse anti-human Foxp3 and Alexa
Fluor® 488 mouse IgG1κ isotype control (all purchased
from BD Biosciences Pharmingen, San Diego, CA, USA). For whole
blood staining, 500 μl of whole blood was incubated with
appropriate amounts of fluorochrome-labeled antibodies in the dark
at room temperature for 30 min. After lysing red blood cells using
BD FACS™ lysing solution, cells were treated with human FoxP3
buffer A and buffer B as described in the manufucturer’s
recommended procedure for the Human Foxp3 Buffer set (BD
Biosciences Pharmingen). After permealization, anti-Foxp3 was added
for 30 min at room temperature and washed once. Flow cytometry was
performed on a Becton Dickinson FACSCalibur, and CellQuest Software
(Becton Dickinson, Oxford, UK) was used for the analysis. The
phenotype of the Tregs was defined as positive for CD4, CD25 and
Foxp3.
Statistical analysis
Variables were compared by means of a non-parametric
test (Mann-Whitney) for intergroup comparisons. A paired t-test was
used to compare results before and after treatment. Overall
survival was calculated from the first day of ACT until the last
follow-up. Overall survival rates were analyzed using Kaplan-Meier
curves and the related log-rank test. P-values of <0.05 were
considered significant. All statistical analyses were performed
using GraphPad Prism (version 5.0) for Windows (GraphPad Software,
Inc., La Jolla, CA, USA).
Results
Patient characteristics
Patient characteristics are summarized in Table I. The median age was 63 years
(range 23-89). The types of cancer varied, as shown in Table I. Only 3 patients were in a post
completely resected state for their cancers, and all remaining
patients had inoperable advanced or recurrent cancers. There was a
number of patients with defective general status and 51 patients
(46.8%) with an Eastern Cooperative Oncology Group (ECOG)
performance score of ≥2. Approximately 90% of the patients had
already received some treatment and most of them continued their
treatment (e.g., chemotherapy, radiation therapy and hyperthermia)
after starting ACT. The median frequency of ACT that patients had
received was 6 times (range 0–27), and the total number of
transferred LAK cells was 25 (range, 0–116.8) × 109.
 | Table I.Clinical characteristics of the
cancer patients. |
Table I.
Clinical characteristics of the
cancer patients.
| Characteristics
(n=109) | |
|---|
| Age (years) | |
| Median
(range) | 63 (23–89) |
| Gender | |
| Male/female | 53/56 |
| Disease site | |
| Gastric
cancer | 18 |
| Esophageal
cancer | 6 |
| Colorectal
cancer | 12a |
| Pancreatic
cancer | 17a |
| Cholangioductal
cancer | 6 |
| Hepatic cell
carcinoma | 4 |
| Head and neck
cancer | 8 |
| Lung cancer | 12 |
| Breast
cancer | 4 |
| Uterine cancer,
sarcoma | 6 |
| Ovarian
cancer | 7 |
| Other | 10 |
| Disease status | |
| Post-operative
state | 3 |
| Advanced
inoperable state | 71 |
| Recurrent | 35 |
| ECOG performance
status | |
| 0/1/2/3/4 | 24/34/26/22/3 |
| Prior
treatment | |
| None | 11 |
| Surgery | 50 |
| Chemotherapy | 81 |
| Radiation
therapy | 13 |
| Other | 10 |
| Combined
treatment | |
| Chemotherapy | 82 |
| Radiation
therapy | 2 |
| Other | 37 |
| Adoptive immune
transfer | |
| Frequency of
transfer | 6 (0–27)b |
| Total number of
transferred cells (×109) | 25
(0–116.8)b |
Comparison of cytokine production and
absolute number, and proportion of Tregs in the peripheral blood
between healthy controls and cancer patients
As shown in Table
II, the level of IFN-α secreted (P=0.0005) in whole blood was
significantly lower in cancer patients compared to the healthy
controls, while IFN-γ (P=0.0007) secretion was significantly
higher. The levels of secreted TNF-α, IL-2, IL-4, IL-5, IL-10,
IL-12(p70), IL-13 and GM-CSF in cancer patients and controls did
not differ. Even though white blood cell counts in the peripheral
blood did not differ between the two groups, the lymphocyte count
in cancer patients was significantly lower compared to the healthy
controls (P<0.0001). The proportion of Tregs to CD4+
lymphocytes (Treg/CD4) did not differ between the two groups.
| Table II.Cytokine production and absolute
number and proportion of Tregs in healthy controls and cancer
patients. |
Table II.
Cytokine production and absolute
number and proportion of Tregs in healthy controls and cancer
patients.
| Healthy controls
(n=49) | Cancer patients
(n=109) | P-value |
|---|
| IFN-α | 6,974±3199 | 5,102±3974 | 0.0005 |
| IFN-γ (pg/ml) | 418.0±623.6 | 1,447±2413 | 0.0007 |
| TNF-α (pg/ml) | 537.5±416.1 | 722.7±814.0 | NS |
| IL-2 (pg/ml) | 71.5±47.9 | 123.9±146.3 | NS |
| IL-4 (pg/ml) | 5.44±7.07 | 7.11±8.71 | NS |
| IL-5 (pg/ml) | 37.6±52.8 | 69.6±122.8 | NS |
| IL-10 (pg/ml) | 52.8±31.1 | 65.7±76.2 | NS |
| IL-12(p70)
(pg/ml) | 2.07±2.95 | 6.38±31.7 | NS |
| IL-13 (pg/ml) | 248.1±175.0 | 367.9±409.3 | NS |
| GM-CSF (pg/ml) | 9.58±12.62 | 16.77±29.98 | NS |
| WBC
(/mm3) | 6,275±1,454 | 6,102±4150 | NS |
| Lymphocyte
(/mm3) | 2,427±690 | 1,230±572 | <0.0001 |
| Treg/CD4 | 4.72±1.94 | 4.28±2.21 | NS |
Effect of ACT on cytokine production
The levels of many cytokines secreted after ACT
treatment were substantially altered (Table III). The value for IFN-α did not
change after ACT treatment when compared to the value before
treatment. As for Th1 cytokines, both IFN-γ and TNF-α were markedly
increased after ACT, while IL-12 (p70) was decreased and IL-2 did
not change. As for Th2 cytokines, IL-4, IL-5, IL-13 and GM-CSF were
significantly increased after ACT, while IL-10 remained unchanged.
The proportions of IFN-γ to IL-4 (IFN-γ/IL-4) and IFN-γ to IL-10
(IFN-γ/IL-10) were both significantly increased after ACT.
| Table III.Cytokine production in the cancer
patients before and after ACT. |
Table III.
Cytokine production in the cancer
patients before and after ACT.
| Cytokine | ACT
| P-value |
|---|
| Pre | Post |
|---|
| IFN-α | 5,102±3,974 | 5,941±6,584 | NS |
| IFN-γ (pg/ml) | 1,447±2,413 | 4,127±6,535 | 0.0003 |
| TNF-α (pg/ml) | 722.7±814.0 | 1,246±1,272 | 0.0005 |
| IL-2 (pg/ml) | 123.9±146.3 | 163.6±304.4 | NS |
| IL-4 (pg/ml) | 7.111±8.711 | 14.70±22.04 | 0.0001 |
| IL-5 (pg/ml) | 69.37±123.8 | 235.1±320.1 | <0.0001 |
| IL-10 (pg/ml) | 65.65±76.15 | 69.14±73.17 | NS |
| IL-12 (p70)
(pg/ml) | 6.378±31.69 | 2.855±17.38 | 0.0400 |
| IL-13 (pg/ml) | 367.9±409.3 | 879.4±1,230 | <0.0001 |
| GM-CSF (pg/ml) | 16.77±29.98 | 30.14±42.11 | 0.0200 |
| IFN-γ/IL-4 | 257.3±416.4 | 388.4±694.2 | 0.0200 |
| IFN-γ/IL-10 | 26.40±39.24 | 67.07±115.3 | 0.0020 |
Effect of ACT on the absolute number and
proportion of Tregs
As shown in Table
IV, the number of lymphocytes in the peripheral blood after ACT
was slightly elevated compared to pre-treatment levels, but the
difference was not significant (P=0.085). The number and the
proportion of Tregs, however, were significantly lower compared to
pre-treatment values.
| Table IV.Number and proportion of Tregs in
cancer patients before and after ACT. |
Table IV.
Number and proportion of Tregs in
cancer patients before and after ACT.
| ACT
| P-value |
|---|
| Pre | Post |
|---|
| No. of lymphocytes
(/mm3) | 1,234±576 | 1,333±548 | NS |
| No. of Tregs
(/mm3) | 20.4±14.2 | 18.0±11.6 | <0.005 |
| Treg/CD4 | 4.281±2.211 | 3.656±2.04 | <0.005 |
Relationship between immunological
parameters and clinical outcome
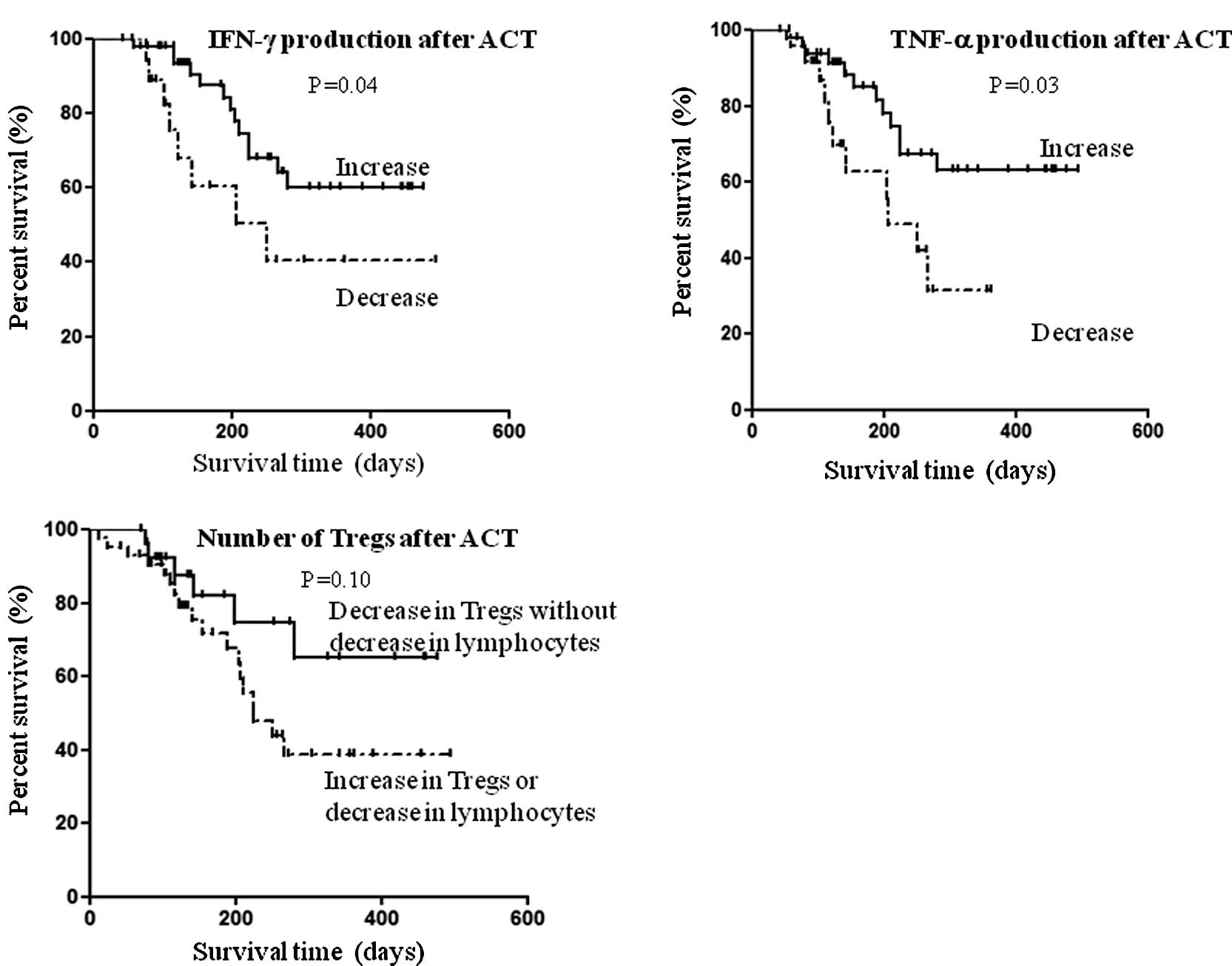
The relationship between overall survival and
immunological variables, which was altered significantly after ACT,
was assessed in 76 patients whose blood samples at baseline and
after four cycles of treatment were available. The median length of
follow-up was 155 days (range 43–494). According to Kaplan-Meier
analysis, there was a significantly longer overall survival in
patients who had increased IFN-γ secretion after ACT (log-rank test
P=0.04; Fig. 2A). Similarly, the
overall survival was significantly longer in patients with
increased TNF-α secretion after ACT (log-rank test P=0.03; Fig. 2B). As for the Tregs, overall
survival was evaluated based on the number of lymphocytes present.
Thus, after ACT the overall survival tended to be longer in
patients with decreased Tregs, but with normal levels of
lymphocytes compared to the other patients; yet this was not
statistically significant (log-rank test P=0.10; Fig. 2C).
Discussion
It remains to be determined whether ACT in patients
with advanced cancer, in whom strong immunosuppression and
immune-escape mechanisms are established, may potentially alter the
undesirable immunological conditions noted in cancer. In the
present study, we demonstrated that in advanced cancer patients ACT
had the strong potential to enhance the Th response and to shift
the balance of cytokine production to Th1. Moreover, we confirmed
that ACT induced a decrease in the number of Tregs in the
peripheral blood of patients with advanced cancer. In the present
study, cytokine secretion and Tregs in the peripheral blood of 109
patients with various types of cancers and in 76 patients after ACT
were extensively examined. To the best of our knowledge, there has
been no prior clinical study reporting the effects of ACT on
specific immunological parameters in patients with cancer.
When certain immunological parameters were examined
in cancer patients before ACT, we found that IFN-α production in
whole blood was significantly low compared to the healthy controls.
These results agree with previous findings in patients with lung
cancer and hepatocellular carcinoma (31,32).
Secretion of IFN-γ in cancer patients was significantly high when
compared to the healthy controls, while Th2 cytokines, such as
IL-4, IL-5, IL-10 and IL-13, did not differ between the cancer
patients and healthy controls. This suggests that peripheral Th
cells in patients with cancer differentiate towards Th1, yet they
are not active enough against cancer to result in cancer rejection.
Evidence from a number of laboratories indicates that a higher
amount of Tregs is observed in the blood of patients with cancer
compared to healthy subjects (4–11).
In disagreement with these studies, we did not find a higher amount
of Tregs in patients with cancer compared to healthy subjects. This
may be explained by the different conditions of the subjects
between the studies. In the present study, most of the patients had
previously received several courses of chemotherapy or radiation
therapy, and their general condition had deteriorated more than
that of patients in the other studies who had not received any
treatment.
We demonstrated that, after four cycles of
treatment, ACT enhanced the secretion of both Th1 cytokines (IFN-γ
and TNF-α) and Th2 cytokines (IL-4, IL-5, IL-13 and GM-CSF) from
blood cells. ACT also had the potential to shift the balance of Th
cytokines to the Th1 type in advanced cancer patients as the
proportion of IFN-γ to IL-4 was significantly increased after ACT.
It is generally believed that the predominant tumoricidal effector
mechanism is the cytotoxic killing effect of CD8+ T
cells. However, increasing attention is being focused on the
stimulation of CD4+ (Th) cells in their response to
cancer immunotherapy. Th cells produce many factors that are able
to overcome immunosuppression and influence the tumor
antigen-specific CTL response (33,34).
Th1 cells, characterized by their secretion of IFN-γ and TNF-α, are
primarily responsible for activating and regulating the development
and persistence of CTLs (19,20).
Several studies found that Th cells have direct cytolytic effects
via multiple pathways involving the Fas/FasL and TNF-related
apoptosis-inducing ligand (TRAIL) pathways (22). In addition, Th1 cells have been
found to activate antigen-presenting cells and contribute to
antigen presentation to CTLs (23). Th2 cells recruit and activate
eosinophils that are able to produce antitumor factors (35). Thus, Th cells mediate antitumor
effects via a variety of mechanisms, by enhancing and supporting
the immune environment via cytokine secretion, directly stimulating
the recruitment of CTLs, directly inducing cytotoxic effects to the
tumor itself and orchestrating the recruitment and activation of
innate immune cells.
Romagnani et al reported that cell-mediated
immunity is preferentially activated by Th1 cytokines, whereas Th2
cytokines have a suppressive action on cell-mediated immunity
(36). However, it has been
clearly shown that not only Th1, but also Th2 cells initiate a
CTL-based immune response in some murine tumor models. Fallarino
et al showed that by infusing Th1 and Th2 clones into
tumor-bearing mice a CTL-mediated antitumor response is activated,
resulting in quantitative rather than qualitative differences in
antitumor activity between Th1 and Th2 cells (37). Hung et al demonstrated that
protection against B16 melanoma challenge in vaccination-challenge
experiments was completely eliminated in IFN-γ−/− mice,
whereas in IL-4−/− mice, protection was reduced by
approximately 50% relative to vaccinated wild-type controls
(35). These findings indicate
that Th1 and Th2 effector mechanisms actually collaborate with each
other in directing an effective antitumor response rather than
antagonizing each other. These mechanisms have important
implications for the development of cancer immunotherapy.
In the present study, a significant decrease in the
number and proportion of Tregs in the peripheral blood was also
observed. A number of studies in mice have revealed that depletion
of Tregs elicits effector responses that lead to tumor eradication
(17,18). Therefore, manipulation of Tregs,
including their depletion, blocking their trafficking into tumors
or reducing their differentiation and suppressive mechanisms,
represent novel strategies in cancer treatment. Previous clinical
studies have demonstrated that Treg depletion alone or in
combination with active immunotherapy increased effector T cell
activation (14–16). In the present study, it is
suggested that ACT has a strong potential to reduce the number of
Tregs. Nishikawa et al (38) showed that IFN-γ abrogated the
generation/activation of CD4+CD25+ Tregs. In
addition, it has been shown that the amount of Tregs in
tumor-bearing mice was significantly decreased by Th1 cell therapy,
in which tumor-specific Th1 cells were intradermally injected with
a tumor antigen near tumor-draining lymph nodes (24). The authors of that study
demonstrated that the observed blockade of Treg accumulation did
not occur in tumor-bearing IFN-γR−/− mice. Thus, it is
possible that the decrease in Tregs in the present study after ACT
administration was IFN-γ-dependent as the secretion of IFN-γ from
peripheral blood cells was increased significantly after ACT. It
will be necessary to examine the exact mechanism by which ACT
reduces the number of Tregs in peripheral blood in more detail.
CD3-LAK cells used in this study are defined as
lymphoid cells activated in vitro by exposure to IL-2 and
monoclonal antibodies that bind to the CD3 complex (26,27).
It is generally believed that the antitumor effect of CD3-LAK cell
depends on its direct lytic activity against tumors (30). Unlike CTL, which are characterized
by their MHC-restricted immunoreactivity to tumor-associated
antigens, CD3-LAK cells display non-specific immunoreactivity. In
this study, we demonstrated that CD3-LAK therapy has a strong
potential to enhance the production of both Th1 and Th2 cytokines
and to reduce the number of Tregs. Furthermore, we revealed that,
among the immunological parameters examined in this study, only the
increase in secretion of IFN-γ and TNF-α after CD3-LAK therapy
correlated with survival. There were no significant differences in
age, gender or performance status at the start of CD3-LAK therapy
between the patients with an increase in IFN-γ or TNF-α secretion
and the patients without these changes (data not shown). Therefore,
longer survival in the patients with increased IFN-γ or TNF-α
secretion can be regarded as the direct effect of CD3-LAK therapy.
We provide the first evidence that CD3-LAK therapy supports the
immune environment via cytokine secretion and the reduction of
Tregs. Thus, it is necessary to reevaluate the potential role of
CD3-LAK in cancer immunotherapy from these aspects.
In conclusion, we demonstrated that ACT has the
potential to enhance the Th response and reduce the number of Tregs
in advanced cancer patients who are in a strong immunosuppressive
state. Overall survival is significantly longer in patients who
have increased IFN-γ or TNF-α secretion after receiving ACT. These
findings suggest a novel potential therapeutic role of ACT in
cancer immunotherapy. We believe that novel strategies of cancer
immunotherapy based on these antitumor effects of ACT will succeed
in the near future.
Abbreviations:
|
ACT,
|
adoptive cellular therapy;
|
|
CTLs,
|
cytotoxic T cells;
|
|
TGF,
|
transforming growth factor;
|
|
IL,
|
interleukin;
|
|
TNF-α,
|
tumor necrosis factor-α;
|
|
IFN,
|
interferon;
|
|
Tregs,
|
regulatory T cells;
|
|
Th,
|
CD4+ T helper;
|
|
CD3-LAK,
|
anti-CD3 stimulated
lymphokine-activated killer;
|
|
PHA,
|
phytohemagglutinin;
|
|
ECOG,
|
Eastern Cooperative Oncology Group
|
Acknowledgements
This study was partially supported by
Grant-in-Aid for Young Scientists (B) from the Ministry of
Education, Culture, Sports, Science and Technology of Japan
(21790638).
References
|
1.
|
Ribas A, Butterfield LH, Glaspy JA and
Economou JS: Current developments in cancer vaccines and cellular
immunotherapy. J Clin Oncol. 21:2415–2432. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Rosenberg SA, Yang JC and Restifo NP:
Cancer immunotherapy: moving beyond current vaccines. Nat Med.
10:909–915. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Liu Z, Kim JH, Falo LD II and You Z: Tumor
regulatory T cells potently abrogate antitumor immunity. J Immunol.
182:6160–6167. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Bluestone JA and Abbas AK: Natural versus
adaptive regulatory T cells. Nat Rev Immunol. 3:253–257. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Ling KL, Pratap SE, Bates GJ, et al:
Increased frequency of regulatory T cells in peripheral blood and
tumour infiltrating lymphocytes in colorectal cancer patients.
Cancer Immun. 7:72007.PubMed/NCBI
|
|
6.
|
Wolf AM, Wolf D, Steurer M, Gastl G,
Gunsilius E and Grubeck-Loebenstein B: Increase of regulatory T
cells in the peripheral blood of cancer patients. Clin Cancer Res.
9:606–612. 2003.PubMed/NCBI
|
|
7.
|
Sasada T, Kimura M, Yoshida Y, Kanai M and
Takabayashi A: CD4+CD25+ regulatory T cells
in patients with gastrointestinal malignancies: possible
involvement of regulatory T cells in disease progression. Cancer.
98:1089–1099. 2003.
|
|
8.
|
Kono K, Kawaida H, Takahashi A, et al:
CD4(+)CD25 high regulatory T cells increase with tumor stage in
patients with gastric and esophageal cancers. Cancer Immunol
Immunother. 55:1064–1071. 2006.
|
|
9.
|
Gray CP, Arosio P and Hersey P:
Association of increased levels of heavy-chain ferritin with
increased CD4+ CD25+ regulatory T-cell levels
in patients with melanoma. Clin Cancer Res. 9:2551–2559.
2003.PubMed/NCBI
|
|
10.
|
Liyanage UK, Moore TT, Joo HG, et al:
Prevalence of regulatory T cells is increased in peripheral blood
and tumor microenvironment of patients with pancreas or breast
adenocarcinoma. J Immunol. 169:2756–2761. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Schaefer C, Kim GG, Albers A, Hoermann K,
Myers EN and Whiteside TL: Characteristics of
CD4+CD25+ regulatory T cells in the
peripheral circulation of patients with head and neck cancer. Br J
Cancer. 92:913–920. 2005.
|
|
12.
|
Curiel TJ, Coukos G, Zou L, et al:
Specific recruitment of regulatory T cells in ovarian carcinoma
fosters immune privilege and predicts reduced survival. Nat Med.
10:942–949. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Nicholl M, Lodge A, Brown I, Sugg SL and
Shilyansky J: Restored immune response to an MHC-II-restricted
antigen in tumor-bearing hosts after elimination of regulatory T
cells. J Pediatr Surg. 39:941–946. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Zou W: Regulatory T cells, tumour immunity
and immunotherapy. Nat Rev Immunol. 6:295–307. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Barnett B, Kryczek I, Cheng P, Zou W and
Curiel TJ: Regulatory T cells in ovarian cancer: biology and
therapeutic potential. Am J Reprod Immunol. 54:369–377. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Dannull J, Su Z, Rizzieri D, et al:
Enhancement of vaccine-mediated antitumor immunity in cancer
patients after depletion of regulatory T cells. J Clin Invest.
115:3623–3633. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Shimizu J, Yamazaki S and Sakaguchi S:
Induction of tumor immunity by removing
CD25+CD4+ T cells: a common basis between
tumor immunity and autoimmunity. J Immunol. 163:5211–5218.
1999.PubMed/NCBI
|
|
18.
|
Onizuka S, Tawara I, Shimizu J, Sakaguchi
S, Fujita T and Nakayama E: Tumor rejection by in vivo
administration of anti-CD25 (interleukin-2 receptor alpha)
monoclonal antibody. Cancer Res. 59:3128–3133. 1999.PubMed/NCBI
|
|
19.
|
Sun JC, Williams MA and Bevan MJ:
CD4+ T cells are required for the maintenance, not
programming, of memory CD8+ T cells after acute
infection. Nat Immunol. 5:927–933. 2004.
|
|
20.
|
Pardoll DM and Topalian SL: The role of
CD4+ T cell responses in antitumor immunity. Curr Opin
Immunol. 10:588–594. 1998.
|
|
21.
|
Schattner EJ, Mascarenhas J, Bishop J, et
al: CD4+ T-cell induction of Fas-mediated apoptosis in
Burkitt’s lymphoma B cells. Blood. 88:1375–1382. 1996.
|
|
22.
|
Thomas WD and Hersey P: TNF-related
apoptosis-inducing ligand (TRAIL) induces apoptosis in Fas
ligand-resistant melanoma cells and mediates CD4 T cell killing of
target cells. J Immunol. 161:2195–2200. 1998.PubMed/NCBI
|
|
23.
|
Fruh K and Yang Y: Antigen presentation by
MHC class I and its regulation by interferon gamma. Curr Opin
Immunol. 11:76–81. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Zhang Y, Wakita D, Chamoto K, et al: Th1
cell adjuvant therapy combined with tumor vaccination: a novel
strategy for promoting CTL responses while avoiding the
accumulation of Tregs. Int Immunol. 19:151–161. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Muranski P and Restifo NP: Adoptive
immunotherapy of cancer using CD4(+) T cells. Curr Opin Immunol.
21:200–208. 2009.
|
|
26.
|
Ting CC, Hargrove ME and Yun YS:
Augmentation by anti-T3 antibody of the lymphokine-activated killer
cell-mediated cytotoxicity. J Immunol. 141:741–748. 1988.PubMed/NCBI
|
|
27.
|
Anderson PM, Blazar BR, Bach FH and Ochoa
AC: Anti-CD3 + IL-2-stimulated murine killer cells. In vitro
generation and in vivo antitumor activity. J Immunol.
142:1383–1394. 1989.
|
|
28.
|
Takayama T, Sekine T, Makuuchi M, et al:
Adoptive immunotherapy to lower postsurgical recurrence rates of
hepatocellular carcinoma: a randomised trial. Lancet. 356:802–807.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Kimura H and Yamaguchi Y: A phase III
randomized study of interleukin-2 lymphokine-activated killer cell
immunotherapy combined with chemotherapy or radiotherapy after
curative or noncurative resection of primary lung carcinoma.
Cancer. 80:42–49. 1997. View Article : Google Scholar
|
|
30.
|
Chang AE and Shu S: Current status of
adoptive immunotherapy of cancer. Crit Rev Oncol Hematol.
22:213–228. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Uno K, Nakano K, Maruo N, et al:
Determination of interferon-alpha-producing capacity in whole blood
cultures from patients with various diseases and from healthy
persons. J Interferon Cytokine Res. 16:911–918. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
32.
|
Uno K, Hirosaki M, Kakimi K, et al:
Impaired IFN-alpha production and the risk of cancer development. J
Interferon Cytokine Res. 27:1013–1017. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Nishimura T, Iwakabe K, Sekimoto M, et al:
Distinct role of antigen-specific T helper type 1 (Th1) and Th2
cells in tumor eradication in vivo. J Exp Med. 190:617–627. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Trinchieri G: Interleukin-12 and the
regulation of innate resistance and adaptive immunity. Nat Rev
Immunol. 3:133–146. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Hung K, Hayashi R, Lafond-Walker A,
Lowenstein C, Pardoll D and Levitsky H: The central role of CD4(+)
T cells in the antitumor immune response. J Exp Med. 188:2357–2368.
1998.
|
|
36.
|
Romagnani S: Human TH1 and TH2 subsets:
doubt no more. Immunol Today. 12:256–257. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Fallarino F, Grohmann U, Bianchi R, Vacca
C, Fioretti MC and Puccetti P: Th1 and Th2 cell clones to a poorly
immunogenic tumor antigen initiate CD8+ T cell-dependent
tumor eradication in vivo. J Immunol. 165:5495–5501. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
38.
|
Nishikawa H, Kato T, Tawara I, et al:
IFN-gamma controls the generation/activation of
CD4+CD25+ regulatory T cells in antitumor
immune response. J Immunol. 175:4433–4440. 2005.PubMed/NCBI
|