Introduction
Sodium/potassium pump inhibitors in human plasma are
identical or similar in structure to plant-derived ouabain (EO) and
are synthesized by the adrenal cortex (1). Since Hamlyn et al confirmed
that EO was an endogenous sodium pump inhibitor in 1991 (2), it has been gradually elucidated as an
important factor that participates in the pathogenesis of
hypertension in the neuroendocrine network. The persistent and
specific association of EO with hypertension indicates that 40–50%
of patients with mild to moderate primary hypertension have
elevated EO, with normal dietary salt intakes (3–6).
Evidence indicates that EO does not fulfill the criteria for a
putative natriuretic hormone; however, it is essential in the
adaptation to sodium depletion and sodium loading (7). Several lines of experimental evidence
clearly demonstrate the prohypertensive role of EO, including
induction of hypertension in EO-treated rodents, elevation of EO
levels in hypertensive rats and observations of the central
prohypertensive action of this hormone. Three mechanisms for the
prohypertensive effect, including the ‘adducin paradigm’, the
existence of highly sensitive EO-binding sites in vascular smooth
muscle and central effects, were proposed to link EO to
vasoconstriction in hypertension (8). Chronic EO treatment produces
hypertension, which depends on the activation of central nervous
mechanisms associated with increased sympathetic tone, subsequent
to the activation of the brain renin-angiotensin (9) and endothelin systems (10), as well as peripheral vascular
mechanisms (11). In addition,
vascular endothelial cells, whose functional integrity is crucial
for the maintenance of blood flow and antithrombotic activity, may
be a target for endogenous EO. We previously studied the effect of
EO on human umbilical vein endothelial cells (HUVECs) and
identified that EO stimulates the proliferation of HUVECs at
physiological concentrations (0.3–0.9 nmol/l). However, cell death
was induced at pathological concentrations (0.9–1.8 nmol/l),
including the swelling phenomenon and the appearance of apoptotic
bodies (12). Based on our
previous study, the effects of EO on endothelial dysfunction and
cardiac remodeling were investigated in EO-sensitive (OS)
hypertensive rats.
Materials and methods
Animal model
A total of 22 adult male Sprague-Dawley rats,
weighing 180–250 g, SPF grade, were provided by the Laboratory
Animal Center of Xi’an Jiaotong University College of Medicine. The
local legislation for ethics on experiments on animals and
guidelines for the care and use of laboratory animals (from the
Ethics Committee of Xi’an Jiaotong University) were followed in all
animal procedures. All animals were allowed time to adapt to the
laboratory environment with free access to food and water in
temperature- and humidity-controlled housing with natural
illumination for a week and subsequently fasted for 12 h prior to
the experiment. The animals were randomly divided into two groups:
the EO group (O group, n=12) and the control group (N group, n=10).
The O group was treated with 20 μg/kg/day EO by
intraperitoneal injection for 8 weeks. The N group was treated with
1 ml/kg/day normal saline by intraperitoneal injection for 8 weeks.
Weight and systolic blood pressure (SBP) were recorded three times
for one week and the mean values were calculated. SBP was measured
via an indirect tail cuff method using a blood pressure recorder
for rats (RBP-1B; China-Japan Clinical Medicine Institute, Beijing,
China). After 6 weeks, the O group was divided into two groups
based on their recorded BPs: the OS group (BP increased, n=10) and
the EO-resistant (OR) group (no clear increase in BP, n=2).
Ultrastructural changes in the
myocardium
The rats were anesthetized with a bolus dose of 20%
urethane (6 ml/kg) and fixed on the surgery table. Following the
exposure of the heart through rapid thoracotomy, the middle left
ventricular free wall of the myocardium and the proximal end of the
ascending aorta were collected. The abdominal cavity was opened and
the kidney cortex was removed. The tissue specimens were cut into
pieces (1×1×1 mm) and washed in phosphate buffer (pH 7.6).
Glutaraldehyde fixation solution was used to postfix the tissue,
which was then dehydrated in graded concentrations of acetone. The
tissues were embedded in Araldite, cut into thin sections, stained
with uranyl acetate and lead citrate and examined under a Hitachi
H-600 transmission electron microscope (Hitachi, Tokyo, Japan).
Real-time quantitative reverse
transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from the rat myocardial
tissue using TRIzol reagent according to the manufacturer’s
instructions (Takara Bio Inc., Shiga, Japan). The quality of the
total RNA was determined spec-trophotometrically at 260 and 280 nm.
Following agarose gel electrophoresis, cDNA was synthesized using
M-MLV reverse transcriptase according to the instructions of the
manufacturer (Takara Bio Inc.). All cDNA samples were used
immediately or stored at −20°C until analysis.
Primers and probes were designed and synthesized by
Sangon Biotechnology Co., Ltd. (Shanghai, China).
Glyceraldehyde-3-phosphate dehydrogenase was used as the control.
The primer and probe sequences are listed in Table I.
 | Table I.Primer sequences of Kv4.2
and GAPDH |
Table I.
Primer sequences of Kv4.2
and GAPDH
| Gene | Primer sequence |
|---|
| Kv4.2 | Forward:
5-GCCTTCGTTAGCAAATCTGGATC-3 |
| Reverse:
5-CACTTCCATGCAGCT TTCTTCAA-3 |
| Probe:
5-FAM-CGAGACAACACCACCACCTGCTTCACTA-MRA-3 |
| GAPDH | Forward:
5-TGGTCTACATGTTCCAGTATGACT-3 |
| Reverse:
5-CGTTTGATGTTAGCGGGATCTC-3 |
| Probe:
5-FAM-ACGGCAAGTTCAACGGCACGTCAATA-MRA-3 |
The KV4.2 mRNA expression levels in the
rat myocardial tissue in the different groups were detected by
RT-PCR. The reaction mixture contained 1 μl each forward and
reverse primers, 1 μl fluorescent probe, 1 μl
RT/platinum® Taq mix and 4 μl RNA template.
Diethylpyrocarbonate (DEPC)-treated triple-distilled water was
added to a final volume of 50 μl. The PCR program was as
follows: reverse transcription at 48°C for 45 min; 94°C for 5 min;
then 35 cycles of 94°C for 30 sec, 56°C for 30 sec, 72°C for 30
sec, and a final extension at 72°C for 5 min. The PCR products were
identified by 1.5% agarose gel electrophoresis.
Separation of rat ventricular
myocytes
Before isolation, the solutions were saturated with
O2 for 30 min. The rats were anesthetized with a bolus
dose of 20% urethane (6 ml/kg) and fixed on the surgery table. The
heart was exposed by rapid thoracotomy, removed and slightly
modified in cold (4°C) calcium-free solution. The aorta was fixed
to the heart tube of a Langendorff perfusion device with a surgical
suture. Then, the isolated heart was perfused for 6 min through the
aorta with calcium-free solution and then for 8–12 min with a
calcium-free buffer containing 50 μM Ca2+, 0.33
g/l collagenase and 0.5 mg/ml bovine serum albumin. The digestion
was terminated when the heart became soft and enlarged, followed by
washing for 5 min. The entire process was performed while
maintaining the perfusion pressure at ∼70 cm at 37°C. Following
perfusion, the separated ventricles were cut into small pieces,
incubated in a fresh calcium-free solution and mechanically
dispersed using a large-bore fire-polished Pasteur pipette. Single
intact cells were separated by filtration. Recalcification of the
isolated cells was performed using 0.1 mM Ca2+ per 10
min to a final calcium concentration of 0.8 mM.
Recording action potentials (APs) and
transient outward potassium current (Ito)
Only quiescent rod-shaped cells that presented clear
cross-striations were selected. All recordings were obtained at
room temperature (22–25°C). A small aliquot (∼1.5 ml) of the
solution containing the isolated cells was placed in an open
perfusion chamber mounted on the stage of an inverted microscope.
Cardiac myocytes were allowed to adhere to the bottom of the
chamber for 8 min and were then perfused at 1.5 ml/min with
Tyrode’s solution.
Electrodes were pulled with a microelectrode puller
from borosilicate micropipettes with inner filaments. The
resistance of the recording pipette filled with the pipette
solution was 3–5 mΩ. The capacitance and series resistance
compensation were optimized; 60–80% compensation was usually
obtained. The potential compensation was ∼2 mV. The whole-cell
configuration of the patch-clamp technique was used to record APs
in the current-clamp mode. APs were elicited by stimulation (square
wave, 10 msec duration, 100–120% excitation threshold current, 600
pA and 0 mV constant voltage). Ito was evoked by
depolarizing pulses from a holding potential of −80 mV to voltage
steps from −40 to +60 mV in 10 mV increments for 250 msec. The
calcium current was blocked by adding 0.1 mM CdCl2 to
Tyrode’s solution. A prestep of 30 msec to −40 mV was used to
inactivate sodium currents. The inactivation of Ito was
measured after a 400 msec conditioning prepulse at 0.2 stimulation
frequency from a holding potential of −80 mV to potentials between
−40 and +40 mV (in 10 mV steps), followed by a 250 msec
depolarizing pulse to +40 mV at 0.5 stimulation frequency.
Statistical analysis
SPSS 10.0 (SPSS, Inc., Chicago, IL, USA) was used to
analyze the data. All values are expressed as mean ± standard
deviation (SD). The significance of differences among groups was
determined by single factor analysis of variance (ANOVA). The least
significant difference test was applied for multiple comparisons
between groups. A t-test was used to compare the significant
differences between the pre- and post-experiments. P<0.05 was
considered to indicate a statistically significant difference.
Results
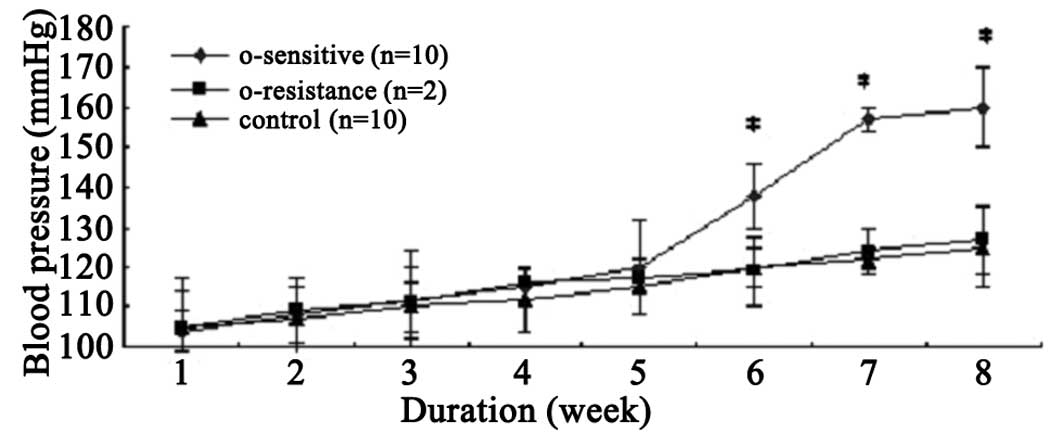
Changes of rat SBP
The effects of the continued infusion of EO on the
SBP in the first 4 weeks were examined. The SBP of all rats
slightly increased and was accompanied by increased weight.
However, no significant differences were observed between the O and
N groups. During the fifth week, the SBP of 10 of the 12 rats in
the O group began to increase. After 6 weeks, the SBP of the 10
rats was significantly higher compared with that in the N group, in
which the rats received normal saline (138.2±8.0 vs. 120.1±5.2
mmHg; P<0.01). The 10 rats were designated as the OS rats. The
remaining two rats in the O group, with normal SBP after 8 weeks of
treatment (126.7±11.4 vs. 125.4±6.9 mmHg; P>0.05) were
designated as the OR rats (Fig.
1).
Ultrastructural changes in the rat
myocardium
The pathological changes in the left ventricular
apical mid-myocardium of the OS rats were observed under electron
microscopy. The cardiac mitochondria were hyperplastic,
hypertrophic and swollen (Fig.
2A). The microvascular membrane permeability in the myocardial
tissue and exudation around the vessels increased (Fig. 2B). Collagen fibers proliferated
among the myocardial cells (Fig.
2C). The connections of the intercalated discs between adjacent
myocardial cells were indistinct (Fig.
2D). There was no significant difference between the
ultrastructural changes in the myocardium of rats in the OR and N
groups (Fig. 2E).
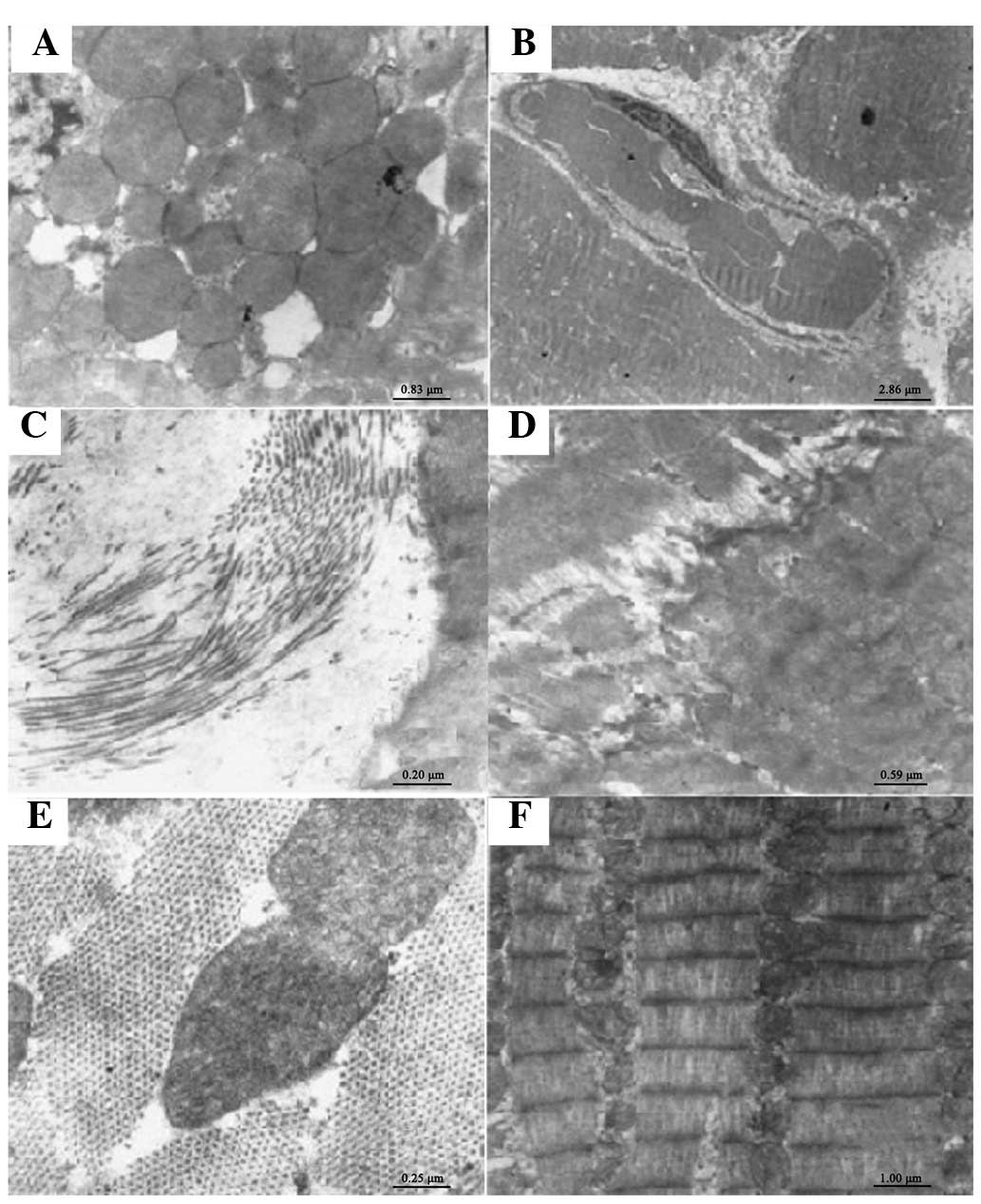 | Figure 2.Ultrastructural changes of the rat
myocardium. (A) Mitochondrial proliferation, hypertrophy and
swelling of the cardiomyocytes of the ouabain-sensitive (OS) rats
(magnification, ×12,000). (B) Microvascular membrane permeability
in myocardial tissue and exudation around the vessels increased in
the OS rats (magnification, ×3,500). (C) Proliferation of collagen
fibers among myocardial cells in the OS rats (magnification,
×50,000). (D) Connections of the intercalated discs between
adjacent myocardial cells were unclear in the OS rats
(magnification, ×17,000). (E) No significant difference was
observed in the ultrastructural changes of the myocardium between
the ouabain-resistant group and the control group (magnification,
×40,000). (F) The morphology of myocardial cells in the control
group was intact. The thick and thin myofilaments were arranged
regularly. The uniformly sized mitochondria were abundant and had a
round or oval shape. Sarcomeres and light-dark bands were clearly
visible. The peri-cellular membrane was uninterrupted and intact
(magnification, ×10,000). |
The left ventricular myocytes in the N group were
regularly arranged. The morphology of the myocardial cells was
intact. The thick and thin myofilaments were regularly arranged.
Uniformly sized, oval mitochondria were abundant. Sarcomeres and
light-dark bands were clearly visible. The pericellular membrane
was uninterrupted and intact. There was a small amount of collagen
fibers among the myocardial cells (Fig. 2F).
KV4.2 expression
There were no significant differences between the
KV4.2 expression levels in the myocardial cells of the
OS (51,345±10,230), OR (53,000±9,880) and control rats
(49,878±12,540) (P>0.05).
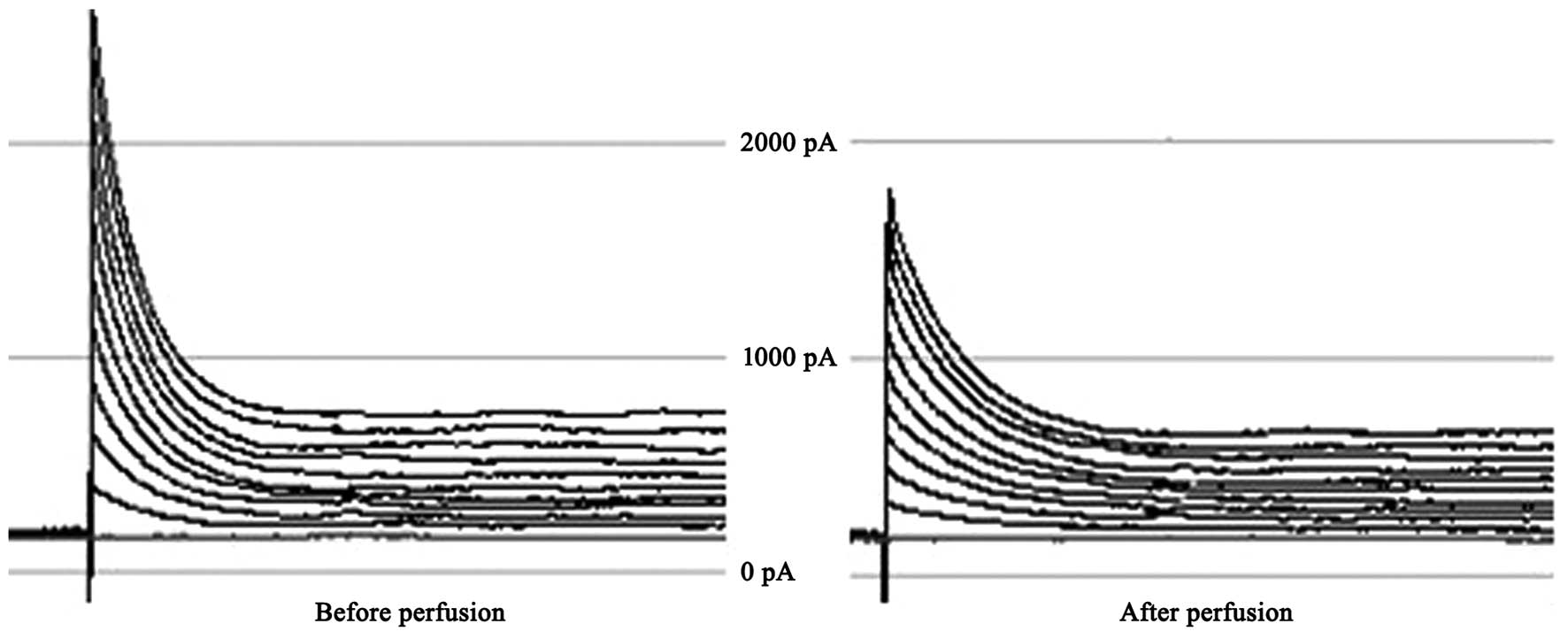
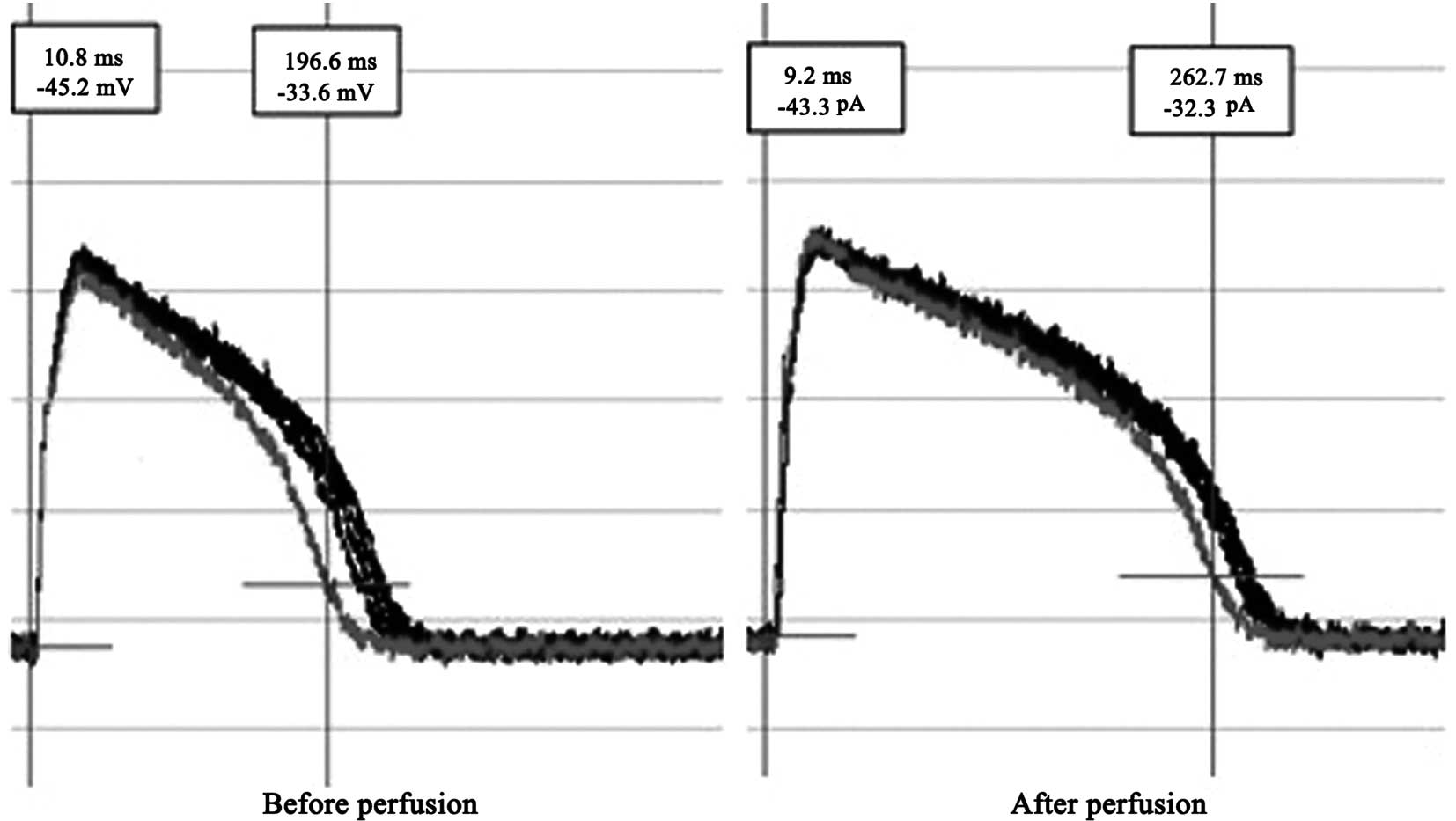
APs and Ito
APs were recorded under the whole-cell current clamp
configuration in rat ventricular myocytes. The AP duration (APD) of
the cardiomyocytes was prolonged after they were perfused for 1 min
with 1.8 nmol/l EO. The difference was statistically significant;
the APD at 50% amplitude (APD50) before and after perfusion was
183±21 and 252±30 msec, respectively (P<0.01). Ito of
the current density significantly decreased (Ito values
before and after perfusion were 2,400±231 and 183±104 pA,
respectively; P<0.01; cell count, n=15; Figs. 3 and 4).
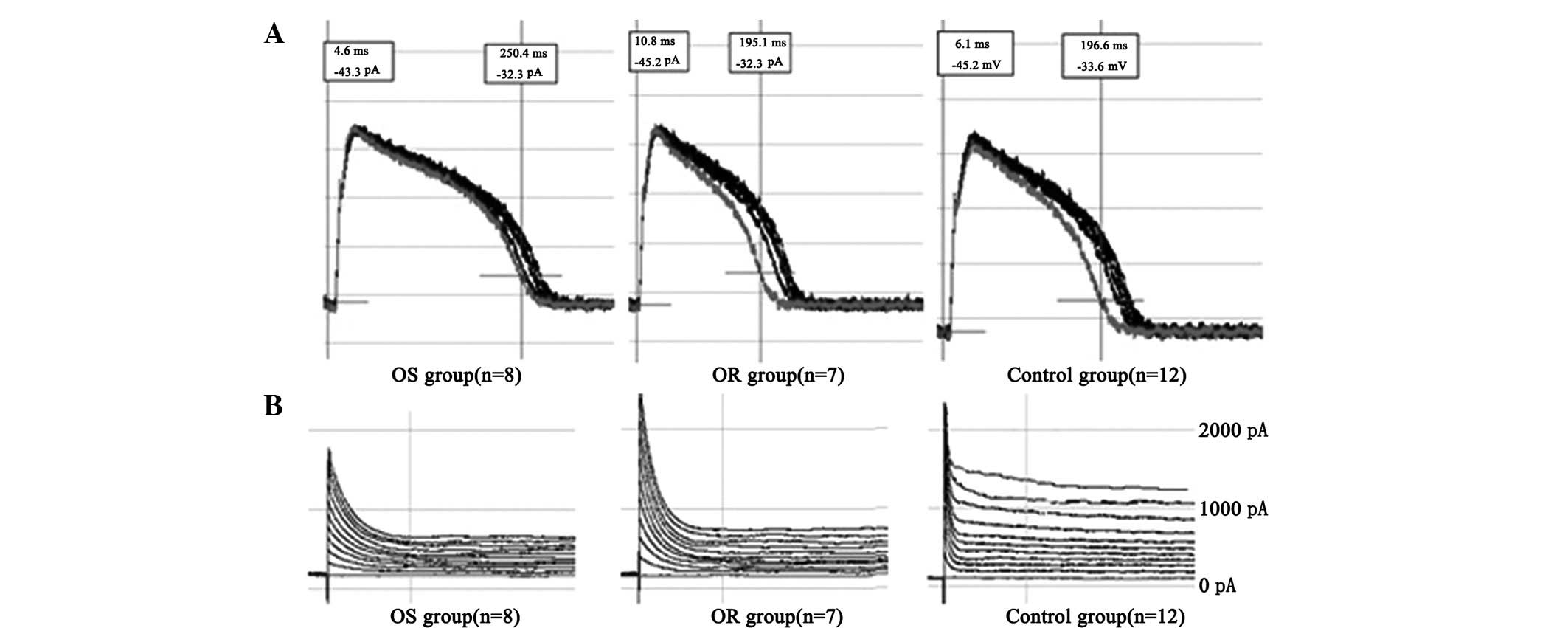
The recordings of the changes in AP and
Ito values of the rat ventricular myocytes in certain
models and the N group indicated significant differences between
the APD90 values of the OS group (242±23 msec; cell
count, n=8) and N group (188±31 msec; cell count, n=12; P<0.01).
No significant differences were observed between the
APD90 values of the OR group (184±19 msec; cell count,
n=7) and N group (188±31 msec; cell count, n=12; P>0.05). As
shown in Fig. 5A and B, the
Ito of rat ventricular myocytes in the OS group
(1,854±192 pA; cell count, n=8) was significantly increased
compared with that in the control group (2,413±331 pA; cell count,
n=12; P<0.01). No significant difference was observed between
the Ito of the OR group (2,580±231 pA; cell count, n=7)
and N group (2,413±331 pA; cell count, n=12; P>0.05).
Discussion
EO, a steroidal hormone, is synthesized by the
hypothalamus and adrenal cortex (1). EO participates in the neuroendocrine
regulation of the cardiovascular system by inhibiting the sodium
pump on the cell membrane. Previous studies indicated that EO
levels are closely related to blood pressure and cardiovascular
complications, including heart failure in rats and humans, during
early hypertension (13–16). EO also affects hemodynamics.
Intravenous application of EO produced an excitatory effect on
baroreceptor nerve activity that was greater in hypertensive rats
(17). Exposure of rats to
nanomolar concentrations of EO for a longer period of time leads to
arterial hypertension (18,19)
and smooth muscle proliferation (20,21).
In patients with essential hypertension, the circulating endogenous
EO concentration correlates directly with the relative wall
thickness of the heart and the total peripheral resistance;
however, it is inversely correlated with the left ventricular
end-diastolic index, stroke index and cardiac index (6).
EO also causes changes in the general morphology of
the left ventricle in patients with essential hypertension
(6,22). In addition, several reports,
including the present study, have also demonstrated that there is a
correlation between EO and endothelial dysfunction (12,23,24).
In the present study, certain significant pathological
ultrastructural changes were also identified in rat apical
mid-myocardium, in accordance with the findings of a previous study
(25). We successfully constructed
an experimental animal model of hypertension using intraperitoneal
EO injection. The blood pressures of the majority of rats (10 out
of 12 rats) increased following injection of EO. The remaining two
rats in the O group that did not have increased SBP after 8 weeks
of treatment were designated as OR rats. Clear pathological changes
were observed in the left ventricular apical mid-myocardium of the
OS rats under electron microscopy. The changes included the rupture
of the aortic intima and outer membranes, as well as endothelial
cell loss. Although no significant loss of the capillary
endothelial cells in the myocardium was observed, microvascular
membrane permeability in the myocardial tissue and exudation around
the vessels increased. These pathological changes in the
endothelial cells from the main artery to capillaries may lead to
the weakening or disappearance of functional membrane barriers,
thereby initiating the remodeling of hypertensive vessels. The
structural integrity of the mitochondria is important for
maintaining the cellular structure. Proliferation and swelling of
the mitochondria in the myocardial cells were observed. The EO
injection also caused structural damage to the intercalated discs
between adjacent myocardial cells. The proliferation of collagen
fibers among myocardial cells was also observed under electron
microscopy. Myocardial fibrosis caused by increased synthesis of
collagen fibers is an important pathological basis for ventricular
remodeling during hypertension, resulting in systolic and diastolic
dysfunction.
Previous in vitro studies suggested that the
function of EO in vascular endothelial cells is related to the
maintenance of reproductive metabolism in normal endothelial cells
under physiological concentrations. EO at pathological
concentrations may induce damage to human vascular endothelial
cells, resulting in direct endothelial injury and the initiation of
hypertensive vascular remodeling (1,12,13).
The consequential target organ damage is also aggravated.
We investigated the effects of EO on myocardial
electrical remodeling in vitro and in vivo. The
results of isolated perfusion with EO and the direct study of EO in
ventricular myocytes in hypertensive rats revealed that EO causes
prolonged APs and reduced Ito density. The results
suggested that EO may cause increased blood pressure and lead to
cardiac remodeling. Consequently, the proliferation of collagen
fibers among myocardial cells, mitochondrial proliferation and
swelling in myocardial cells, as well as the deposition of glycogen
were observed under electron microscopy. However, EO may also lead
to myocardial electrical remodeling due to the direct effect on
myocardial electrophysiology, resulting in prolonged APs and
decreased Ito density. Previous studies demonstrated
that a number of pathological conditions, including myocardial
ischemia, stress overloading, neurohumoral factors and heart
failure, lead to reduced Ito density and cardiac
electrical remodeling (26–29).
In the present study, the results of real-time RT-PCR indicated no
significant difference in KV4.2 expression levels in the
ventricular tissues of the three groups of animal models. This
finding suggests that, although there was no significant difference
among the transcriptional levels of the three groups, the slightly
varied levels may be caused by discrepancies in the translation and
functional protein levels due to certain factors. The specific
mechanism remains unclear.
In view of this, and in line with our previous
findings, it is possible that EO may participate in the cardiac
structural and electrical remodeling of hypertension (30,31).
Therefore, a significant neuroendocrine hormone, such as EO, may be
used as a target for the clinical treatment of hypertension to
reduce blood pressure and myocardial remodeling (16).
References
|
1.
|
Boulanger BR, Lilly MP, Hamlyn JM, Laredo
JM, Shurtleff D and Gann DS: Ouabain is secreted by the adrenal
gland in awake dogs. Am J Physiol. 264:E413–E419. 1993.PubMed/NCBI
|
|
2.
|
Hamlyn JM, Blaustein MP, Bova S, et al:
Identification and characterization of a ouabain-like compound from
human plasma. Proc Natl Acad Sci USA. 88:6259–6263. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Manunta P, Messaggio E, Ballabeni C, et
al: Plasma ouabain-like factor during acute and chronic changes in
sodium balance in essential hypertension. Hypertension. 38:198–203.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Manunta P, Ferrandi M, Bianchi G and
Hamlyn JM: Endogenous ouabain in cardiovascular function and
disease. J Hypertens. 27:9–18. 2009. View Article : Google Scholar
|
|
5.
|
Manunta P, Stella P, Rivera R, et al: Left
ventricular mass, stroke volume, and ouabain-like factor in
essential hypertension. Hypertension. 34:450–456. 1999.PubMed/NCBI
|
|
6.
|
Pierdomenico SD, Bucci A, Manunta P, et
al: Endogenous ouabain and hemodynamic and left ventricular
geometric patterns in essential hypertension. Am J Hypertens.
14:44–50. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Balzan S, Nicolini G, Iervasi A, Dicecco P
and Fommei E: Endogenous ouabain and acute salt loading in
low-renin hypertension. Am J Hypertens. 18:906–909. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Bagrov AY, Shapiro JI and Fedorova OV:
Endogenous cardiotonic steroids: physiology, pharmacology, and
novel therapeutic targets. Pharmacol Rev. 61:9–38. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Huang BS and Leenen FH: Brain
renin-angiotensin system and ouabain-induced sympathetic
hyperactivity and hypertension in Wistar rats. Hypertension.
34:107–112. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Di Filippo C, Filippelli A, Rinaldi B, et
al: Chronic peripheral ouabain treatment affects the brain
endothelin system of rats. J Hypertens. 21:747–753. 2003.PubMed/NCBI
|
|
11.
|
Xavier FE, Yogi A, Callera GE, et al:
Contribution of the endothelin and renin-angiotensin systems to the
vascular changes in rats chronically treated with ouabain. Br J
Pharmocol. 143:794–802. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Ren YP, Huang RW and Lü ZR: Ouabain at
pathological concentration might induce damage in human vascular
endothelial cells. Acta Pharmacol Sin. 27:165–172. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Huang BS, Huang X, Harmsen E and Leenen
FH: Chronic central versus peripheral ouabain, blood pressure, and
sympathetic activity in rats. Hypertension. 23:1087–1090. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Kimura K, Manunta P, Hamilton BP and
Hamlyn JM: Different effects of in vivo ouabain and digoxin on
renal artery function and blood pressure in the rat. Hypertens Res.
23:S67–S76. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Ferrari P: Rostafuroxin: an
ouabain-inhibitor counteracting specific forms of hypertension.
Biochim Biophys Acta. 1802:1254–1258. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Ferrari P, Ferrandi M, Valentini G,
Manunta P and Bianchi G: Targeting ouabain- and adducin-dependent
mechanisms of hypertension and cardiovascular remodeling as a novel
pharmacological approach. Med Hypotheses. 68:1307–1314. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Abreu GR, Futuro Neto HA, Cabral AM and
Vasquez EC: Ouabain produces diverse excitatory effects on afferent
baro-receptor nerve activity in SHR and WKY animals. Clin Exp
Hypertens. 20:85–94. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Manunta P, Hamilton J, Rogowski AC,
Hamilton BP and Hamlyn JM: Chronic hypertension induced by ouabain
but not digoxin in the rat: antihypertensive effect of digoxin and
digitoxin. Hypertens Res. 23:S77–S85. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Yuan CM, Manunta P, Hamlyn JM, et al:
Long-term ouabain administration produces hypertension in rats.
Hypertension. 22:178–187. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Abramowitz J, Dai C, Hirschi KK, et al:
Ouabain- and marinobufagenin-induced proliferation of human
umbilical vein smooth muscle cells and rat vascular smooth muscle
cell line, A7r5. Circulation. 108:3048–3053. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Aydemir-Koksoy A, Abramowitz J and Allen
JC: Ouabain-induced signaling and vascular smooth muscle cell
proliferation. J Biol Chem. 276:46605–46611. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Huang BS and Leenen FH: The brain
renin-angiotensin-aldosterone system: a major mechanism for
sympathetic hyperactivity and left ventricular remodeling and
dysfunction after myocardial infarction. Curr Heart Fail Rep.
6:81–88. 2009. View Article : Google Scholar
|
|
23.
|
Rossoni LV, Salaices M, Miguel M, et al:
Ouabain induced hyper-tension is accompanied by increases in
endothelial vasodilator factors. Am J Physiol Heart Circ Physiol.
283:H2110–H2118. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Aras-López R, Blanco-Rivero J, Hernanz R,
et al: Chronic ouabain treatment increases the contribution of
nitric oxide to endothelium-dependent relaxation. J Physiol
Biochem. 64:115–125. 2008.PubMed/NCBI
|
|
25.
|
Jiang X, Ren YP and Lv ZR: Ouabain induces
cardiac remodeling in rats independent of blood pressure. Acta
Pharmacol Sin. 28:344–352. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Volk T, Nguyen TH, Schultz JH, Faulhaber J
and Ehmke H: Regional alterations of repolarizing K+
currents among the left ventricular free wall of rats with
ascending aortic stenosis. J Physiol. 530:443–455. 2001.PubMed/NCBI
|
|
27.
|
Singarayar S, Singleton C, Tie H, et al:
Effects of components of ischemia on the Kv4.3 current stably
expressed in Chinese hamster ovary cells. J Mol Cell Cardiol.
34:197–207. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Lukas A and Antzelevitch C: Differences in
the electro-physiological response of canine ventricular epicardium
and endocardium to ischemia. Role of the transient outward current.
Circulation. 88:2903–2915. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Verkerk AO, Wilders R, Coronel R,
Ravesloot JH and Verheijck EE: Ionic remodeling of sinoatrial node
cells by heart failure. Circulation. 108:760–766. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Matsumoto S, Yoshida S, Ikeda M, et al:
Effects of acetazolamide on transient K+ currents and
action potentials in nodose ganglion neurons of adult rats. CNS
Neurosci Ther. 17:66–79. 2011.
|
|
31.
|
Matsumoto S, Kitagawa J and Takeda M: The
effects of ouabain on resting membrane potential and
hyperpolarization-activated current in neonatal rat nodose ganglion
neurons. Neurosci Lett. 439:241–244. 2008. View Article : Google Scholar : PubMed/NCBI
|