Introduction
It has been estimated that up to 15.2% of the adult
population exhibits a certain degree of chronic kidney disease
(CKD) (1), while the prevalence of
patients with end-stage renal disease (ESRD) has shown a global
increase. CKD leading to ESRD is associated with tubulointerstitial
fibrosis, regardless of the underlying causes (2). Interstitial renal fibrosis is
characterized by tubular atrophy, lumen dilation, accumulation of
fibroblasts and increased interstitial matrix deposition (3). While the understanding of the
molecular mechanisms of fibrogenesis has improved, only a limited
number of antifibrotic therapies are currently used in the clinic
(4). Therefore, further
investigations into specific treatments to target fibrosis are
required.
Although most types of resident and bone
marrow-derived cells are involved in fibrogenesis, fibroblasts are
considered to be the key mediators of fibrosis in the kidney and in
other organs (5). Once activated,
fibroblasts are designated to be myofibroblasts. Myofibroblasts are
the predominant type of interstitial cell in the fibrotic kidney
(6) and have been suggested to be
the major source of the extracellular matrix (ECM) components that
accumulate during renal fibrosis. Myofibroblasts appear de
novo in areas of fibrosis in response to stimuli such as
transforming growth factor (TGF)-β1 (7).
TGF-β1 is a ubiquitous cytokine belonging to the
TGF-β superfamily (8). TGF-β1
transduces signaling through a transmembrane receptor
serine/threonine complex that comprises the type I and type II
receptor kinases. Once TGF-β1 binds to the constitutively active
type II receptor, the type I receptor kinase, activin receptor-like
kinase, is subsequently recruited and activated by TGF-β type II
receptor-mediated phosphorylation. Phosphorylation of
serine/threonine residues in the type I receptor kinase
subsequently phosphorylates the major downstream signaling mediator
proteins, Smad2 and Smad3. Phosphorylated Smad2 (pSmad2) and Smad3
(pSmad3) form a complex with Smad4. This complex translocates into
the nucleus and regulates the transcription of fibrosis-associated
genes (9). Deregulation of TGF-β 1
has been implicated in the pathogenesis of various diseases,
including fibrosis, atherosclerosis and cancer (10). It has been indicated that TGF-β1
acts as a potent fibrogenic cytokine, evoking pathological fibrosis
in various organs, including the kidney (11).
Asiatic acid (AA) is one of the triterpenoid
compounds present in Centella asiatica. A number of studies
have shown that AA demonstrates a variety of antitumor (12) and neuroprotective (13) pharmacological effects. In
particular, AA has been shown to combat liver fibrosis (14); however, whether AA is able to
inhibit renal fibrosis has not yet been elucidated. Therefore, the
aim of the present study was to investigate whether AA has an
antifibrotic effect in the kidneys of mice with ureteral
obstruction, a well-established model of interstitial fibrosis, and
to explore any underlying mechanisms.
Materials and methods
Animals
Male C57BL6 mice (weight, 18–20 g) were housed at
the Experimental Animal Center of Wuhan University (Wuhan, China),
at a constant temperature and with a regular light-dark cycle. Food
and water were provided ad libitum. All surgical and
experimental procedures were approved by the Institutional Animal
Care and Use Committee of Wuhan University. Efforts were made to
minimize any animal suffering and the number of animals used
throughout the experiment.
Agents and antibodies
The purified natural product AA (96%) was obtained
from Shanghai Yuanye Biotechnology Co., Ltd. (Shanghai, China) and
was used as described in the following sections. The goat
polyclonal anti-fibronectin (sc-6952) and anti-p-Smad2/3 (sc-11769)
antibodies, and the rabbit polyclonal anti-TGF-β1 (sc-146) antibody
was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA,
USA). The rabbit polyclonal anti-collagen III (BA0326) and the
mouse monoclonal anti-α-smooth muscle actin (SMA) antibodies
(BM0002) were purchased from Boshide Bioengineering Co., Ltd.
(Wuhan, China).
Experimental groups and treatments
Twenty-five male C57BL6 mice were randomly assigned
into five groups, with five mice per group, as follows: (i)
sham-surgery (Sh); (ii) unilateral ureteral obstruction plus
vehicle (UUO+V); (iii) UUO plus 1 mg/kg body weight AA (UUO+A1);
(iv) UUO plus 4 mg/kg body weight AA (UUO+A2); and (v) UUO plus 16
mg/kg body weight AA (UUO+A3). The AA dosages used in this study
were chosen on the basis of a previous study (15) and the UUO was conducted using an
established procedure (16).
Briefly, under sodium pentobarbital (40–60 mg/kg body
weight)-induced anesthesia, complete ureteral obstruction was
performed by ligating the left ureter at the level of lower pole of
the left kidney using 4-0 sutures, following a left abdominal
incision. Sham-surgery mice had their ureters exposed and
manipulated, but not ligated. For mice receiving the AA treatment,
escalating doses of AA (1, 4, and 16 mg/kg body weight), suspended
in 1.2% methyl cellulose (MC) were administered daily by oral
gavage from the day subsequent to the surgery for six days. Mice
receiving the vehicle treatment were administered 300 μl
phosphate-buffered saline (PBS) in 1.2% MC. Mice were sacrificed on
the seventh day subsequent to surgery, and the obstructed kidneys
were harvested. A sample of the kidney was fixed in 4% buffered
paraformaldehyde and was embedded in paraffin for histological and
immunohistochemical studies. The remaining kidneys were snap-frozen
in liquid nitrogen and stored at −80°C for protein extraction.
Histological and immunohistochemical
examination
Kidney sections from the paraffin-embedded tissues
were prepared at a 5-μm thickness, using an established procedure
(17). Sections were stained with
hematoxylin and eosin (H&E) and Periodic acid-Schiff (PAS)
reagents to assess the grade of tubular injury. A further set of
sections was stained using the Masson’s trichrome method for
identifying interstitial collagen.
Collagen III and fibronectin expression was examined
with individual antibodies, while the presence of interstitial
myofibroblasts was measured with anti-α-SMA antibody.
Paraffin-embedded sections (5 μm) were mounted onto glass slides.
The sections were dewaxed in xylene three times for 15 min each,
rehydrated in decreasing concentrations of ethanol for 5 min and
washed twice in PBS for 10 min. Endogenous peroxidase activity was
quenched for 10 min with 3% hydrogen peroxide. Once the sections
had been washed in PBS, a blocking step was included, using 5%
bovine serum albumin (BSA) in PBS for a total of 30 min. Primary
antibodies against specific antigens, as described previously, were
then incubated overnight at 4°C in a humidified chamber. Subsequent
to returning to room temperature, the sections were washed in PBS
for 10 min and the biotin-coupled secondary antibodies were
incubated for 1 h at room temperature. The sections were washed in
PBS for 10 min, prior to antigen-specific positive cells being
visualized by horseradish peroxidase-coupled streptavidin and
diaminobenzidine reagents, provided by Boshide Biotechnology Co.,
Ltd. (Wuhan, China). Brown staining represented a positive result.
Ten random, nonoverlapping, high-power (original magnification,
×400) fields of each slide were selected for evaluation and tubular
injury was scored on a scale from 0 to 3, as previously described
by Fujiu et al: 0, absent; 1, mild; 2, moderate and 3,
severe (18).
Western blot analysis
Western blot analysis was performed as described in
a previous study (19). In brief,
the kidney tissues were homogenized with a polytron homogenizer
(IKA GmbH, Königswinter, Germany) in a lysis buffer containing 20
mM Tris (pH 7.5), 150 mM NaCl, 1% Triton X-100, sodium
pyrophosphate, β-glycerophosphate, EDTA,
Na3VO4 and leupeptin (Biyuntian, Wuhan,
China) on ice. The lysates were centrifuged at 12,000 × g at 4°C
for 20 min and the protein concentration was determined using a
bicinchoninic acid (BCA) protein assay kit (cat. no. p0012s;
Biyuntian). Following this, the lysates were mixed with 5X sodium
dodecyl sulfate (SDS) loading buffer (125 mM Tris-HCl, 4% SDS, 20%
glycerol, 100 mM dithiothreitol (DTT) and 0.2% bromophenol blue).
Samples were heated at 95°C for ~5 min prior to loading and the
supernatants (40 μg protein/lane) were subsequently separated by
SDS-polyacrylamide gel electrophoresis (PAGE) on a 12% acrylamide
gel. The proteins were then electrotransferred to a nitrocellulose
membrane (Millipore, Billerica, MA, USA) in transfer buffer
containing 48 mM Tris-HCl, 39 mM glycine, 0.037% SDS and 20%
methanol at 4°C for 1 h. Nonspecific binding to the membrane was
blocked for 1 h at room temperature with 5% non-fat milk in
Tris-buffered saline (TBS) buffer (20 mM Tris-HCl, 150 mM NaCl and
0.1% Tween 20) or 5% BSA. The membranes were then incubated
overnight at 4°C with various primary antibodies in a blocking
buffer containing 5% milk, at the dilutions specified by the
manufacturers. Following this, the membranes were incubated with
horseradish peroxidase-conjugated secondary antibodies, developed
with the enhanced chemiluminescence plus detection system
(Amersham, Buckinghamshire, UK) (20).
Statistical analysis
Data are presented as the mean ± standard deviation.
One-way analysis of variance (ANOVA) and the Student-Newman-Keuls
test were used for quantitative data, while Kruskal-Wallis ANOVA
was used for abnormally distributed data. Statistical analyses of
the data were performed using the Graphpad Prism®
software package, version 5.0 (Graphpad Software, Inc., La Jolla,
CA, USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
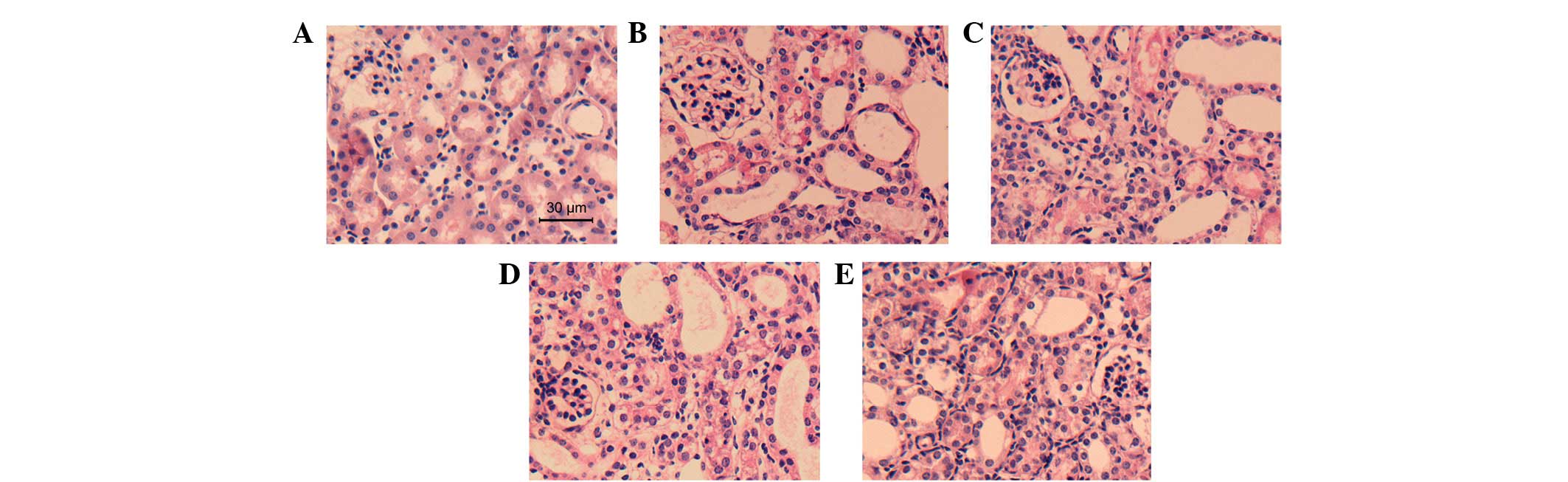
AA attenuates tubular injury
Seven days subsequent to the ureteral obstruction,
the effect of AA on the suppression of tubular injury was examined.
H&E- and PAS-stained micrographs of the kidney are shown in
Figs. 1 and 2, respectively. A significantly greater
amount of tubular damage was observed in the UUO+V group compared
with the Sh group (2.86±0.35 versus 0.10±0.30, respectively;
P<0.05); however, this damage was alleviated in the UUO+A3 group
(1.44±0.64 versus 2.86±0.35, P<0.05). The tubular damage was not
significantly alleviated in the UUO+A1 (2.62±0.53 versus 2.86±0.35,
P>0.05) or UUO+A2 (2.42±0.67 versus 2.86±0.35, P>0.05)
groups.
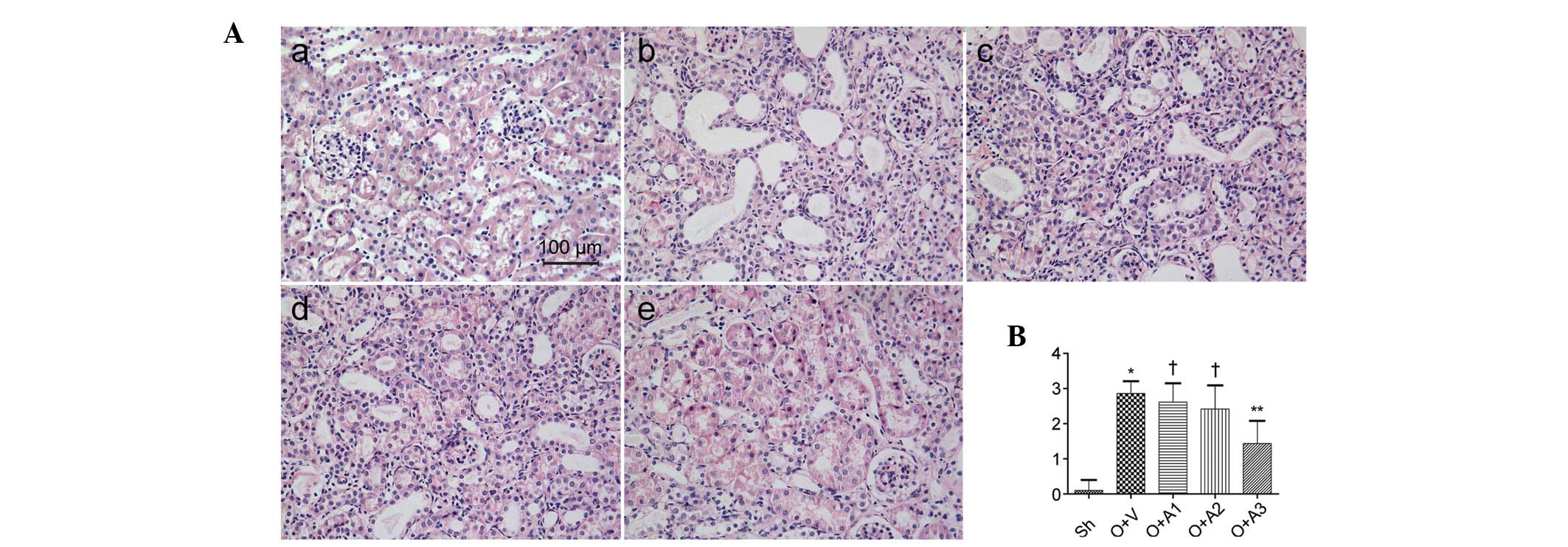
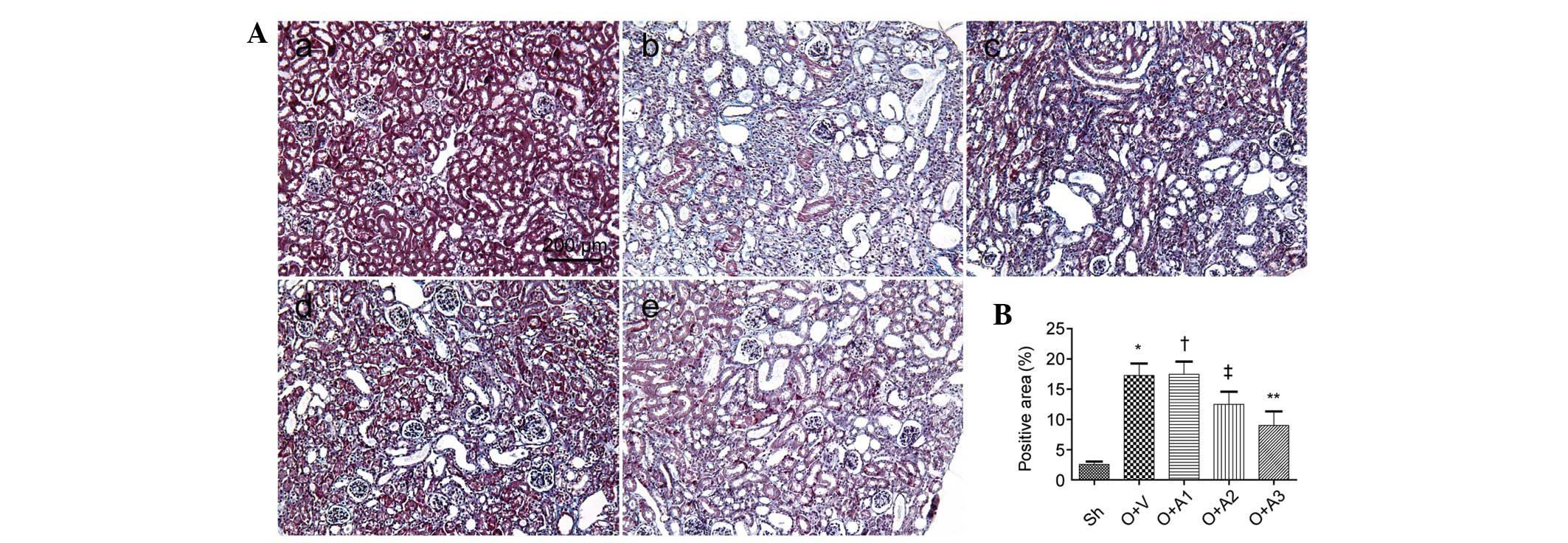
AA attenuates interstitial fibrosis
Seven days subsequent to the ureteral obstruction,
the effect of AA on the suppression of interstitial fibrosis was
examined. Masson’s trichrome-stained micrographs of the kidney are
shown in Fig. 3. A significantly
greater amount of interstitial fibrosis was observed in the UUO+V
group compared with the Sh group (17.28±1.98 versus 2.56±0.46,
respectively; P<0.05); however, this fibrosis was alleviated in
the UUO+A2 (12.51±2.09 versus 17.28±1.98, P<0.05) and UUO+A3
(9.03±2.31 versus 17.28±1.98, P<0.05) groups. Interstital
fibrosis was not alleviated in the UUO+A1 group (17.48±2.08 versus
17.28±1.98, P>0.05). The anti-fibrotic effect of AA was also
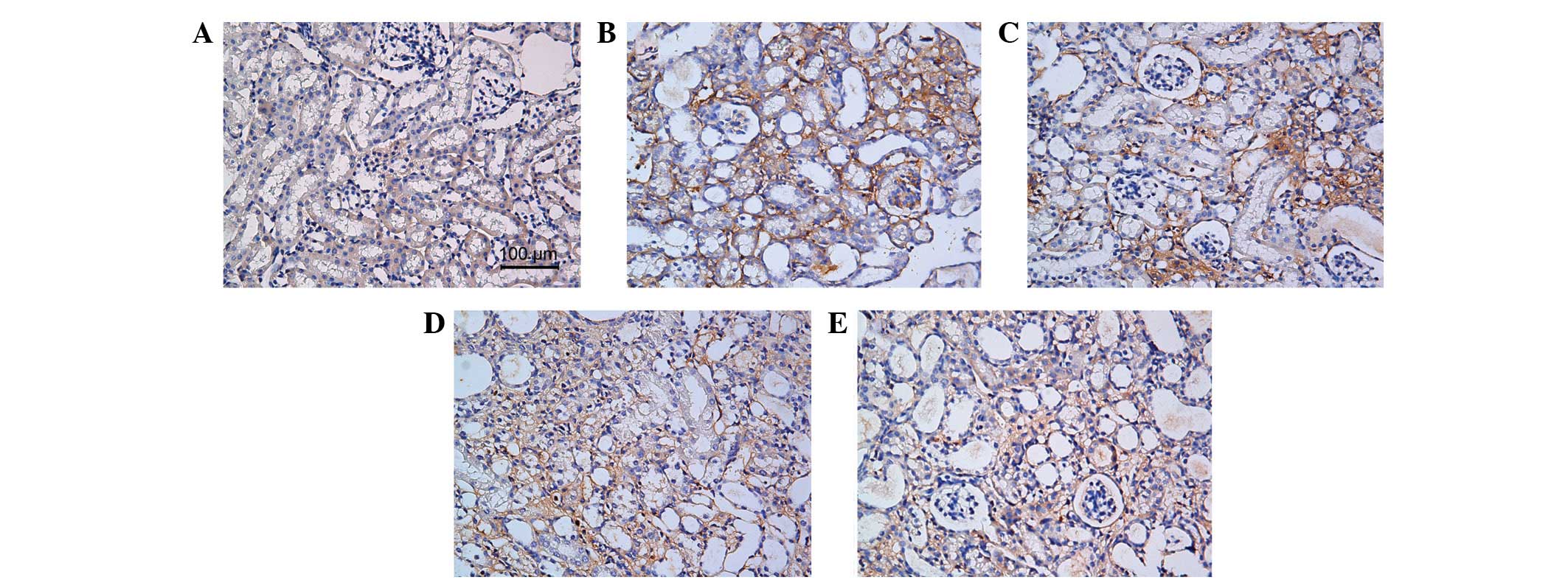
shown by histopathological staining of collagen III and fibronectin
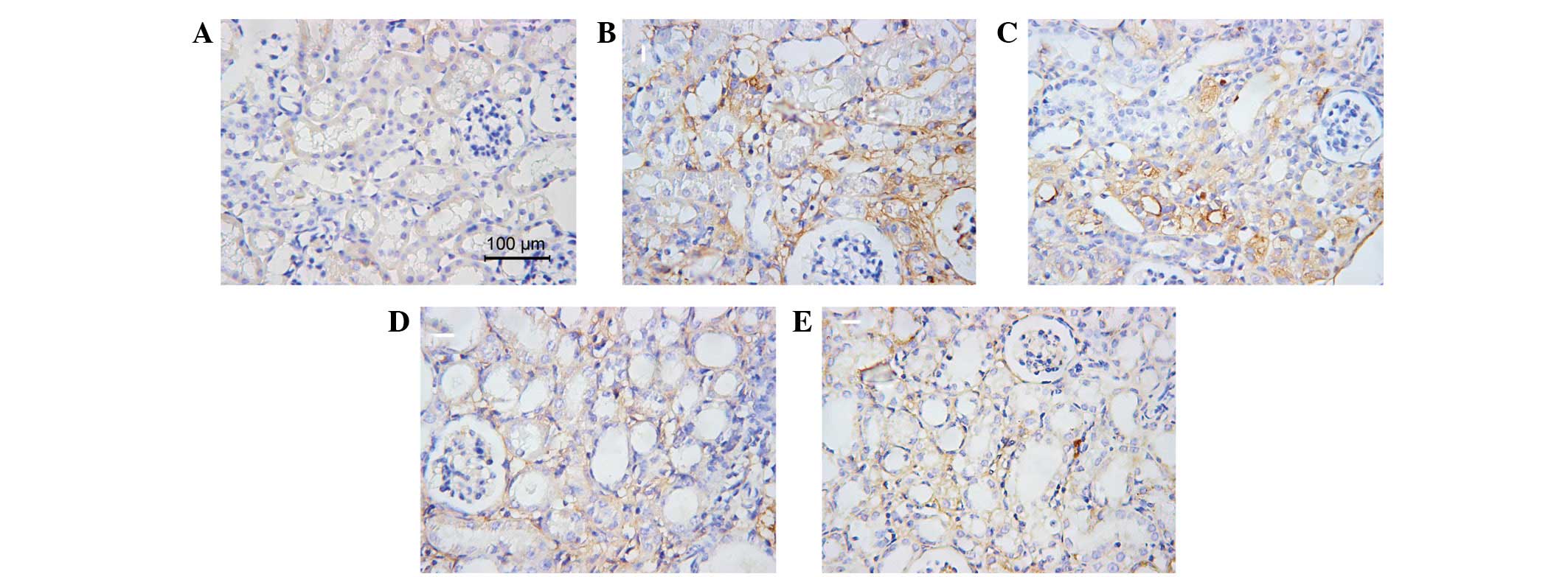
(Figs. 4 and 5).
AA decreases the accumulation of
myofibroblasts
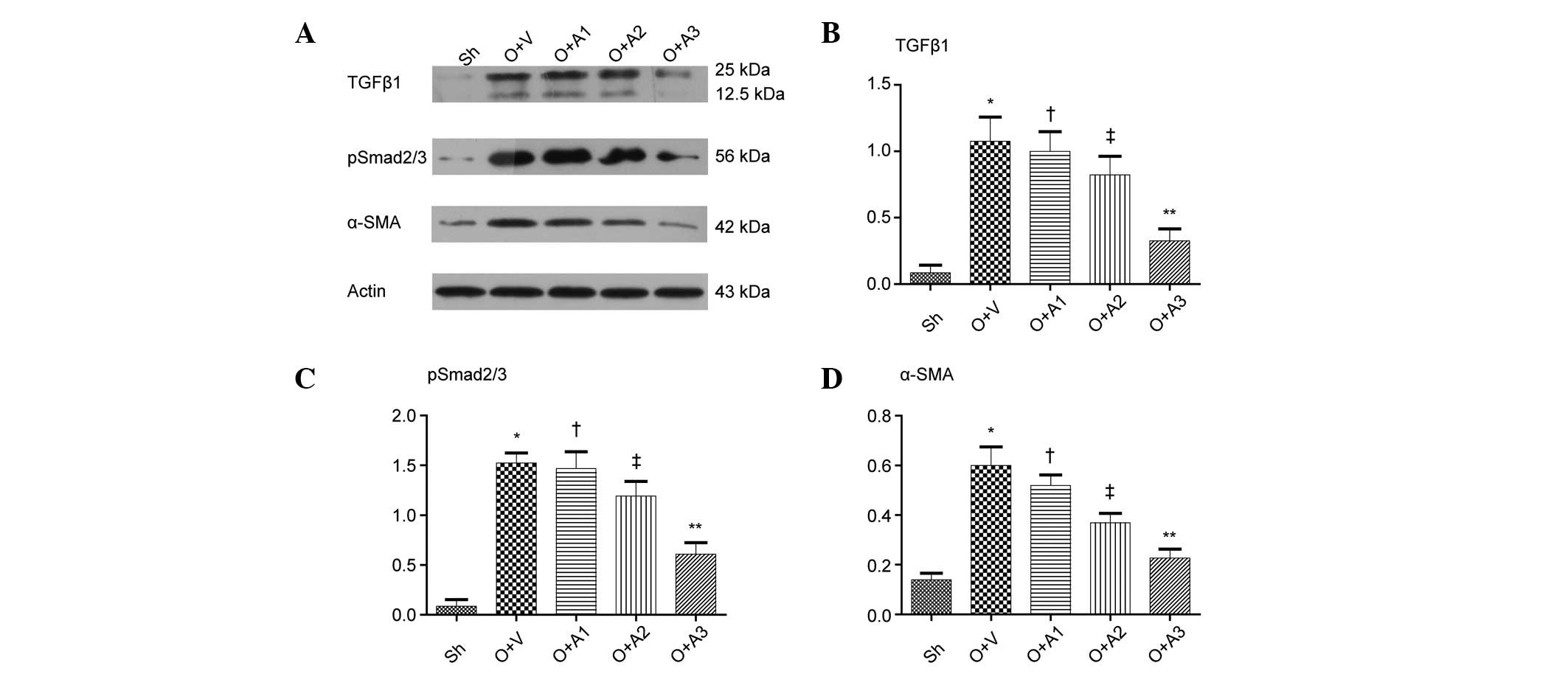
As shown in Fig. 6,
western blot analysis demonstrated that ureteral obstruction
resulted in a marked increase in α-SMA expression in the UUO+V
group mice as compared with the Sh group mice (0.60±0.07 versus
0.14±0.03, respectively; P<0.05). Compared with the UUO+V group,
the UUO+A2 and UUO+A3 groups demonstrated significant reductions in
α-SMA expression in renal tissue (0.37±0.04 and 0.23±0.04,
respectively, versus 0.60±0.07, P<0.05). There was no
significant difference between the UUO+A1 and the UUO+V groups
(0.52±0.04 versus 0.60±0.07, respectively; P>0.05).
AA inhibits the TGFβ1 pathway
As shown in Fig. 6,
western blot analysis demonstrated that ureteral obstruction
resulted in a marked increase in the level of TGF-β1 in the UUO+V
group as compared with Sh group (1.08±0.18 versus 0.09±0.06,
respectively; P<0.05). Compared with the UUO+V group, the UUO+A2
and UUO+A3 groups demonstrated significant reductions in TGF-β1
expression in renal tissue (0.82±0.14 and 0.33±0.09, respectively,
versus 1.08±0.18; P<0.05); however, there was no significant
difference in the TGF-β1 expression between the UUO+A1 and UUO+V
groups (1.00±0.15 versus 1.08±0.18, respectively; P>0.05).
Ureteral obstruction also led to a significant increase in
p-Smad2/3 levels in UUO+V mice as compared with the Sh group mice
(1.52±0.10 versus 0.09±0.07, respectively; P<0.05). The UUO+A2
and UUO+A3 groups demonstrated significant reductions in p-Smad2/3
levels in the renal tissue compared with the UUO+V group,
(1.19±0.15 and 0.61±0.11 versus 1.52±0.10, respectively;
P<0.05). However, there was no significant difference between
the UUO+A1 and UUO+V groups (1.47±0.17 versus 1.52±0.10,
P>0.05).
Discussion
In CKD, progressive renal tissue injury leads to an
irreversible reduction in renal function. At present, there are no
specific treatments for CKD, which ultimately progresses into ESRD.
Renal replacement therapy (RRT) is the only therapeutic option
available. The main pathological feature of CKD is interstitial
fibrosis, and this is positively correlated with the prognosis of
the patient; therefore, the attenuation of renal interstitial
fibrosis presents a therapeutic option for patients with CKD. A
previous study indicated that AA inhibited liver fibrosis induced
by CCl4(14), while the
present study demonstrated that AA was able to attenuate renal
fibrosis in mice with UUO.
Tubular epithelial cells are the predominant
component of renal parenchyma and are the primary target in a
variety of injuries. Depending on the severity and duration of the
injury, tubular cells exhibit a wide range of responses (3). In patients with CKD, the
histopathological presentation of tubular damage is often
characterized by tubular atrophy, most likely as a consequence of
apoptosis and epithelial-mesenchymal transition (EMT) (8). While activated and injured tubules
compromise renal function, those tubules may also be involved in
the process of interstitial fibrosis, by producing profibrotic and
proinflammatory cytokines (21).
The present study revealed that high-dosage AA alleviated tubular
injury, although similar effects were not observed with lower and
intermediate dosages. AA may, therefore, preserve tubular function
and halt the progression of interstitial fibrosis.
The ECM is mainly composed of collagen and
proteoglycans (22). In the
glomerular and tubulointerstitial compartments, there is a small
amount of ECM, which constitutes the framework structure of the
renal interstitium. Abnormal deposition of ECM is one of the main
pathological changes observed following UUO, and leads to a
decrease in the diffusion of oxygen (23). It also contributes to abnormal
intercellular signaling, leading to a vicious cycle of ECM
deposition (24,25). In the present study, Masson’s
trichrome staining revealed that small quantities of ECM were
present in the normal kidneys, predominantly in the glomerular
basement membrane and the peritubular blood vessels. By contrast,
UUO induced the accumulation of a large volume of ECM in the
interstitium. Whereas low-dosage AA did not decrease the levels of
ECM, intermediate and high dosages of AA resulted in marked
reductions. Similar results were observed by the
immunohistochemical examination of collagen III and fibronectin.
This indicated that AA decreased the volume of ECM, which rescued
normal tubulointerstitial architecture and function.
During fibrosis, interstitial myofibroblasts are the
main cells producing ECM (26).
Although the source of the myofibroblasts remains debated, a
strategy that reduces myofibroblast recruitment and inhibits their
proliferation and activation is a therapeutic option. Mature
myofibroblasts are tissue-contracting cells. It has been shown that
biomechanical forces are crucial for cell differentiation and are
major triggers for the induction of a myofibroblast phenotype
(27). α-SMA is a biomarker of
activated myofibroblasts and α-SMA levels represent and correlate
with the progression of CKD. The majority of antifibrotic
interventions have been shown to be correlated with a reduction in
the number of α-SMA-expressing cells (7). The present study showed that
intermediate and high doses of AA decreased the expression of
α-SMA, although low-dosage AA failed to produce a significant
effect. This indicated that AA was able to alleviate renal fibrosis
by inhibiting myofibroblast recruitment and activation.
TGF-β1 has been shown to be a central factor in many
pathological events associated with CKD progression (28), and contributes to tubular loss,
fibroblast recruitment, proliferation and activation and excessive
ECM accumulation (2,29). It has been shown that targeting
TGF-β1 signaling produces encouraging results (29). Therefore, the present study
examined whether AA was able to influence this pathway. The results
showed that intermediate and high doses of AA decreased TGF-β1
levels and reduced the phosphorylation of Smad2/3, a downstream
meditator of TGF-β1. This finding indicated that AA decreased
interstitial fibrosis by affecting the Smad-dependent TGF-β1 signal
transduction.
In conclusion, the results of the present study
provided in vivo evidence that AA was able to attenuate
renal tubulointerstitial fibrosis in a dose-dependent manner. The
effects of AA may have been mediated by the inhibition of
Smad-dependent TGF-β1 signaling which, in turn, attenuated tubular
injury and fibroblast recruitment, proliferation and
activation.
References
|
1
|
Collins AJ, Foley RN, Chavers B, et al:
United States Renal Data System 2011 Annual Data Report: Atlas of
chronic kidney disease & end-stage renal disease in the United
States. Am J Kidney Dis. 59(Suppl 1): A7e1–e420. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yanagita M: Inhibitors/antagonists of
TGF-beta system in kidney fibrosis. Nephrol Dial Transplant.
27:3686–3691. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hodgkins KS and Schnaper HW:
Tubulointerstitial injury and the progression of chronic kidney
disease. Pediatr Nephrol. 27:901–909. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Formentini I, Bobadilla M, Haefliger C, et
al: Current drug development challenges in chronic kidney disease
(CKD) - identification of individualized determinants of renal
progression and premature cardiovascular disease (CVD). Nephrol
Dial Transplant. 27(Suppl 3): iii81–iii88. 2012.
|
|
5
|
Barnes JL and Glass WF II: Renal
interstitial fibrosis: a critical evaluation of the origin of
myofibroblasts. Contrib Nephrol. 169:73–93. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Grande MT and López-Novoa JM: Fibroblast
activation and myofibroblast generation in obstructive nephropathy.
Nat Rev Nephrol. 5:319–328. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Strutz F and Zeisberg M: Renal fibroblasts
and myofibroblasts in chronic kidney disease. J Am Soc Nephrol.
17:2992–2998. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
López-Hernández FJ and López-Novoa JM:
Role of TGF-beta in chronic kidney disease: an integration of
tubular, glomerular and vascular effects. Cell Tissue Res.
347:141–154. 2012.PubMed/NCBI
|
|
9
|
Verrecchia F and Mauviel A: Transforming
growth factor-beta signaling through the Smad pathway: role in
extracellular matrix gene expression and regulation. J Invest
Dermatol. 118:211–215. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Santibañez JF, Quintanilla M and Bernabeu
C: TGF-beta/TGF-beta receptor system and its role in physiological
and pathological conditions. Clin Sci (Lond). 121:233–251.
2011.PubMed/NCBI
|
|
11
|
Lan HY: Diverse roles of TGF-β/Smads in
renal fibrosis and inflammation. Int J Biol Sci. 7:1056–1067.
2011.
|
|
12
|
Kavitha CV, Agarwal C, Agarwal R and Deep
G: Asiatic acid inhibits pro-angiogenic effects of VEGF and human
gliomas in endothelial cell culture models. PLoS One. 6:e227452011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lee KY, Bae ON, Serfozo K, et al: Asiatic
acid attenuates infarct volume, mitochondrial dysfunction, and
matrix metalloproteinase-9 induction after focal cerebral ischemia.
Stroke. 43:1632–1638. 2012.PubMed/NCBI
|
|
14
|
Tang LX, He RH, Yang G, et al: Asiatic
acid inhibits liver fibrosis by blocking TGF-beta/Smad signaling in
vivo and in vitro. PLoS One. 7:e313502012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang X, Wu J, Dou Y, et al: Asiatic acid
protects primary neurons against C2-ceramide-induced apoptosis. Eur
J Pharmacol. 679:51–59. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Eddy AA, López-Guisa JM, Okamura DM and
Yamaguchi I: Investigating mechanisms of chronic kidney disease in
mouse models. Pediatr Nephrol. 27:1233–1247. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Grande MT, Fuentes-Calvo I, Arévalo M, et
al: Deletion of H-Ras decreases renal fibrosis and myofibroblast
activation following ureteral obstruction in mice. Kidney Int.
77:509–518. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fujiu K, Manabe I and Nagai R: Renal
collecting duct epithelial cells regulate inflammation in
tubulointerstitial damage in mice. J Clin Invest. 121:3425–3441.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sörensen I, Susnik N, Inhester T, et al:
Fibrinogen, acting as a mitogen for tubulointerstitial fibroblasts,
promotes renal fibrosis. Kidney Int. 80:1035–1044. 2011.PubMed/NCBI
|
|
20
|
Barnes JL and Gorin Y: Myofibroblast
differentiation during fibrosis: role of NAD(P)H oxidases. Kidney
Int. 79:944–956. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Prunotto M, Budd DC, Gabbiani G, et al:
Epithelial-mesenchymal crosstalk alteration in kidney fibrosis. J
Pathol. 228:131–147. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zeisberg M and Neilson EG: Mechanisms of
tubulointerstitial fibrosis. J Am Soc Nephrol. 21:1819–1834. 2010.
View Article : Google Scholar
|
|
23
|
Mimura I and Nangaku M: The suffocating
kidney: tubulointerstitial hypoxia in end-stage renal disease. Nat
Rev Nephrol. 6:667–678. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Loeffler I, Liebisch M and Wolf G:
Collagen VIII influences epithelial phenotypic changes in
experimental diabetic nephropathy. Am J Physiol Renal Physiol.
303:F733–F745. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Schaefer L: Extracellular matrix
molecules: endogenous danger signals as new drug targets in kidney
diseases. Curr Opin Pharmacol. 10:185–190. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Boor P and Floege J: The renal
(myo-)fibroblast: a heterogeneous group of cells. Nephrol Dial
Transplant. 27:3027–3036. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Boor P, Ostendorf T and Floege J: Renal
fibrosis: novel insights into mechanisms and therapeutic targets.
Nat Rev Nephrol. 6:643–656. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gewin L and Zent R: How does TGF-beta
mediate tubulointerstitial fibrosis? Semin Nephrol. 32:228–235.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Meng XM, Chung AC and Lan HY: Role of the
TGF-beta/BMP-7/Smad pathways in renal diseases. Clin Sci (Lond).
124:243–254. 2013. View Article : Google Scholar : PubMed/NCBI
|