Introduction
Coronary thrombotic microembolism (CME) may be
induced by spontaneous plaque rupture and disrupted plaque
components produced during coronary interventions. CME has been
shown to lead to myocardial microinfarcts, as reflected by elevated
creatine kinase (CK) and troponin I (TnI) (1–7),
which may partly contribute to impaired microvascular perfusion and
result in the ‘slow’ or ‘no-reflow’ phenomenon (6–10). A
number of efforts, including intravascular thrombolysis drug
application and thrombosis suction, have been attempted for the
prevention and treatment of CME and CME-induced myocardial
impairment (11–16). Previous studies have demonstrated
that calcium antagonists are capable of relieving microvascular
spasm (17,18) and that the intravascular
application of diltiazem may attenuate coronary artery spasms in
patients with microvascular angina or acute myocardial infarction
with ‘no-reflow’ phenomenon (19,20).
In our previous experimental study, a rat CME model was established
by the aortic injection of automicrothrombotic particulates
(9). In the present study, we
examined whether this model is suitable for testing and reflecting
the therapeutic effects of drugs that may be capable of attenuating
coronary microembolization. It was hypothesized that intravenous
diltiazem, an agent with proven clinical effectiveness in reducing
coronary microembolization, may attenuate the cardiac dysfunction
and pathological changes in the rat CME model.
Materials and methods
Animals
A total of 152 adult male Sprague Dawley rats
(weight, 250–350 g; age, 12 weeks) were used in the present study.
Thirty-two rats were used for the dose-finding pilot study. Forty
rats were subjected to an aortic saline injection (sham group,
n=40) and 80 rats underwent procedures to induce CME, as described
previously (9). Briefly, blood
(0.5 ml) was obtained from the ventral tail artery of all rats 1
day prior to surgery (21). The
blood was left to clot for 1 h and then dried overnight at 37°C
(9,22). The dried blood was fragmented into
thrombotic particulates with a Sabi Crush Easy Pill Smasher (SABI
Co., Palo Alto, CA, USA) for 5 min. Subsequently, 5 mg thrombotic
particulates were dissolved in 0.2 ml saline and filtered through a
45-μm metal filter. The filtrates were collected and the
mixed solution (1 μl) was utilized for a particulate number
count and size determination using a TS-M2 micrometer (OPLENIC
Manufacturer, HangZhou, China). Examination revealed that the
number of particulates with a diameter of 10–45 μm was
∼600,000, whereas the majority of particulates (>90%,
∼16,000,000) had a diameter <10 μm. The remaining 0.199
ml filtrates were injected into sodium pentobarbital-anesthetized
(50 mg/kg, i.p.) rats. The second intercostal space was exposed
through parasternotomy and a microretractor was used to separate
the second and third rib in order to adequately expose the
operating region under strictly aseptic conditions. Following the
removal of the pericardium, the ascending aorta was visualized. The
ascending aorta was temporarily clamped by a microvascular clip and
0.199 ml filtrate or 0.2 ml saline was injected into the aorta over
10 sec using a 28-gauge tuberculin syringe. The mean quantity of
hemorrhage ranged from 0.5 to 1.0 ml following injection into
animals without massive hemorrhage. During surgery, the animals
were monitored with electrocardiograms using standard lead II
through subcutaneous needle electrodes. Penicillin (800,000 IU,
i.p.) was administered daily for 7 days to animals studied at 7 and
28 days after the aortic injection of automicrothrombotic
particulates or saline. Four rats died immediately after the
injection of automicrothrombotic particulates (three due to large
hemorrhage and one due to malignant arrhythmia). The CME model rats
that survived were randomly assigned to the untreated (CME group,
n=38) or diltiazem-treated CME groups (CME+DIL group, n=38). In the
latter group, diltiazem (1 mg/ml) was intravenously injected at an
infusion rate of 50 μg/min/kg using an infusion pump
(Perfusor®, TCI-II; Guangxi Veryark Technology Co., Ltd., Guangxi,
China) through the tail vein for 175 min, 5 min following the
injection of automicrothrombotic particulates. The dose of drug
used in the main study was selected based on the results of the
pilot study (as described in Results). The rats were examined and
sacrificed at 3 h, 24 h, 7 days and 28 days postoperatively (n=8–10
at each time point).
Experiments were approved by the Tongji Medical
College Council on the Animal Care Committee of Huazhong University
of Science and Technology (Wuhan, China). Animals were maintained
in accordance with the Guide for the Care and Use of Laboratory
Animals published by the US National Institute of Health (NIH
Publication No.85–23, revised 1996).
Transthoracic echocardiography
Prior to and 28 days following surgery, rats in the
28 days group were anesthetized (sodium pentobarbital, 50 mg/kg,
i.p.) and lightly secured to a warming pad in the supine position,
and their precordium was shaved. Transthoracic echocardiography was
performed using a cardiac ultrasound machine with a 11.2 MHz
transducer (Vivid 7; GE Healthcare, Fairfield, CT, USA). The heart
was first imaged in the two-dimensional mode in the parasternal
long-axis and parasternal short-axis views. Left ventricular (LV)
areas were measured from the transverse sections and the LV
short-axis lengths were used to calculate LV end-systolic diameters
(LVESDs) or volumes (LVESVs) and end-diastolic diameters (LVEDDs)
or volumes (LVEDVs) using the modified Simpson’s rule formula. The
inner endocardial margin defined the LV lumen and the LVEF was
derived using the following formula: LVEF = (LVEDV – LVESV)/LVEDV ×
100. LV thickness was measured from the M-mode recording at the
mid-papillary level. The results from three different cardiac
cycles were averaged.
Hemodynamic measurements
Hemodynamic measurements were performed in the 3 h
group. LV systolic pressure (LVSP) and end-diastolic pressure
(LVEDP), the maximum rate of increase of LV systolic pressure
(dp/dtmax) and heart rate (HR) were measured using a 1% heparinized
short segment of a saline-filled PE 50 catheter connected to a
solid-state miniature pressure transducer and the Philips apparatus
(Integris Allura 12; Philips Healthcare, Heide, The Netherlands)
(23).
Serum c-troponin I (c-TnI)
measurement
Prior to sacrifice, blood (1.0 ml) was obtained from
the femoral vein of each rat in the 24 h group at 6 and 24 h
postoperatively. c-TnI was measured by immunofluorescence methods
and an immunofluorescence assay (OPUS, Dade Behring, Inc., Mariani
Cupertino, CA, USA).
Determination of von Willebrand factor
(vWF) and endothelin-1 (ET-1)
Blood (2.0 ml) was obtained from the femoral vein of
each rat in all 4 groups (3 h, 24 h, 7 days and 24 days) prior to
sacrifice. Blood was centrifuged at 2,000 x g and the serum vWF and
ET-1 levels were determined using antibodies in an antigen-based
sandwich ELISA assay (von Willebrand Factor Elisa kit, Helena
Laboratories, Beaumont, TX, USA; Endothelin-1 Quantikine ELISA kit,
ELISAs More Quality Reagents from R&D Systems, Minneapolis, MN,
USA).
Evaluating areas of no-flow
Three hours postinjection, a single bolus of
thioflavin-S (1 ml/kg of 4%; Sigma, St. Louis, MO, USA) was
injected via the jugular vein to stain the vascular endothelium
in vivo (24). One min
later, the heart was stopped in the diastolic phase by an
intravenous injection of potassium chloride (1 ml, 10%) and removed
immediately. The atrium and right ventricle were separated from the
left ventricle. The left ventricle was then placed into a −20°C
freezer for 5 min and sectioned into 10 or 11 cross-sectional
slices (1 mm in thickness) along the long axis using a cutting
apparatus (JP40; Shanghai Shiyuan Scientific Equipment Co., Ltd.,
Shanghai, China). Even slices (2,4,6,8 and 10) were fixed in
formalin solution and used for light microscopic and
immunohistochemical analyses. Uneven slices (1,3,5,7,9 and 11) were
irradiated with ultraviolet light (wave length, 365 nm) to
determine the no-flow zone (NF, area not perfused by thioflavin-S).
The NF was traced and analyzed using NIH imaging software
(http://rsb.info.nih.gov/nih-image/).
The ratios of NF/LV area (NF/LV) from all examined slices were
calculated and averaged.
Light microscopic analysis
LV tissue samples from the 3 h group were fixed in
10% buffered formalin solution for 24 h, embedded in paraffin, cut
into 4-μm sections and stained with hematoxylin and eosin
(H&E), Carstair’s (25) and
hematoxylin basic fuchsin picric acid (HBFP) (26), respectively. An Olympus-BX41TF
microscope (Olympus, Tokyo, Japan) incorporated with Image-Pro Plus
4 (Media Cybernetics, Inc., Rockville, MD, USA) was employed to
observe the micro-thrombosis in the coronary arteriole in
H&E-stained slices. One hundred coronary arterioles with a
diameter <100 μm were randomly observed under a
microscope (magnification, ×200) and the percentage of
microthrombosis in these arterioles was determined. For animals
studied 28 days postoperatively, hearts were embedded in paraffin,
sectioned at 5-μm intervals and stained with Masson’s
trichrome (27). Myocardial
leukocyte infiltration was observed in rats 24 h and 7 days
postoperatively in H&E-stained sections. Leukocytes were
counted in 10 randomly selected visual fields in each section and
five sections per rat were examined.
Immunohistochemical analysis
LV tissue samples from the 3 h group were fixed in
10% buffered formalin solution for 24 h, embedded in paraffin and
cut into 4-μm sections for immunohistochemical analysis.
Tissue sections were blocked with 10% normal serum for 1 h at 27°C.
The blocker was removed and the primary antibody, vascular smooth
muscle α-actin (VSMA-α) antibody (mouse monoclonal anti-VSMA; Santa
Cruz Biotechnology Inc., Santa Cruz, CA, USA) was added. The tissue
sections were incubated for 15 h at 4°C and washed with PBS, then
the peroxidase block was performed with 0.5%
H2O2 in methanol for 30 min. The respective
biotinylated secondary antibody (rabbit anti mouse IgG, Santa Cruz
Biotechnology Inc.) was added and incubated for 1 h at 27°C. After
washing with PBS, the streptavidin peroxidase label (Zymed, San
Diego, CA, USA) was added and incubated for 10 min at 27°C. The
sections were washed with PBS and incubated with AEC color
development substrate (Zymed) for a further 10 min at 27°C.
Sections were washed in water and counterstained with Mayer’s
hematoxylin. The vascular smooth muscle was dyed claybank and the
arterioles (10–50 μm) were counted in 10 random fields of
vision in each section, and five sections per rat were examined
under a light microscope (magnification, ×200). Arteriolar density
(AD) was calculated using the following formula: AD = total
arteriole number/10 x 5/ the area of one vision
(mm2).
Western blot analysis
At 24 h, 7 days and 28 days after injection, the
apex of the left ventricle was used for western blot analysis.
Tissues were homogenized in PBS and centrifuged at 10,000 × g for
10 min at 4°C, then 70 μg supernatant was lysed in
electrophoresis buffer, boiled for 10 min and subsequently
subjected to electrophoresis on a SDS-polyacrylamide gel. The
separated blots were transferred to nitrocellulose membranes and
blocked for 1 h in TTBS buffer containing 5% nonfat milk. The
membranes were incubated overnight with primary anti-TNF-α or IL-6
polyclonal antibodies (TNF-α, 1:500 dilution; IL-6, 1:200 dilution)
and then with horseradish peroxidase (HRP)-conjugated rabbit
anti-goat IgG antibody (1:1,000 dilution) for 2 h at 37°C. Blots
were detected by chemiluminescence and relative protein expression
was quantified by scanning densitometry.
Statistical analyses
The data were analyzed using SPSS 13.0 (SPSS, Inc.,
Chicago, IL, USA). Data are presented as the mean ± SD. Comparisons
among groups and different experimental stages were first carried
out using a test of homogeneity of variances and then by two-way
ANOVA, Tukey’s test (homogeneity of variance) and the Games-Howell
test (heterogeneity of variance). A P-value of 0.05 was considered
to indicate a statistically significant difference.
Results
Pilot study
Prior to the main study, a pilot study was performed
to observe the blood pressure and heart rate changes induced by
various concentrations of diltiazem. In our previous study
(9), thrombosis was observed at 1
h, peaked at 3 h and decreased at 12 h, while heart rate decreased
significantly at 1 min and returned to baseline level at 5 min
after the injection of automicrothrombotic particulates. Therefore,
diltiazem was infused at doses of 10, 50, 100 or 200
μg/min/kg (n=8 for each dose) through the tail vein for 175
min with an infuser at 5 min after the automicrothrombotic
particulate injection. The 200 μg/min/kg dose of diltiazem
significantly reduced blood pressure (<90 mmHg) and heart rate
(<200 beats per minute), while the 100 μg/min/kg dose did
not affect the blood pressure but significantly reduced the heart
rate (<250 beats per minute). Blood pressure and heart rate were
not significantly affected by the 10 and 50 μg/min/kg doses
of diltiazem, and the beneficial effects of reducing the NF/LV area
were greater using 50 μg/min/kg diltiazem compared with 10
μm/min/kg diltiazem (5% vs. 2%). As a result, diltiazem at a
dose of 50 μg/min/kg was selected for the main study.
Mortality
In the sham group, 2 rats died during the
perioperative period (1 due to excess anesthesia, 1 due to massive
hemorrhage). The remaining 38 rats were examined at 3 h (n=9), 24 h
(n=10), 7 days (n=10) and 28 days (n=9). In the CME group, 3
animals died at 10 h, 26 h and 4 days postinjection, respectively.
This was due to cardiac failure based on the postmortem
pathological findings (pulmonary venous pleonaemia and pleural
effusion). The remaining 35 rats were examined at 3 h (n=9), 24 h
(n=9), 7 days (n=9) and 28 days (n=8). In the CME+DIL group, no
rats died and 38 rats were examined at 3 h (n=10), 24 h (n=9), 7
days (n=10) and 28 days (n=9).
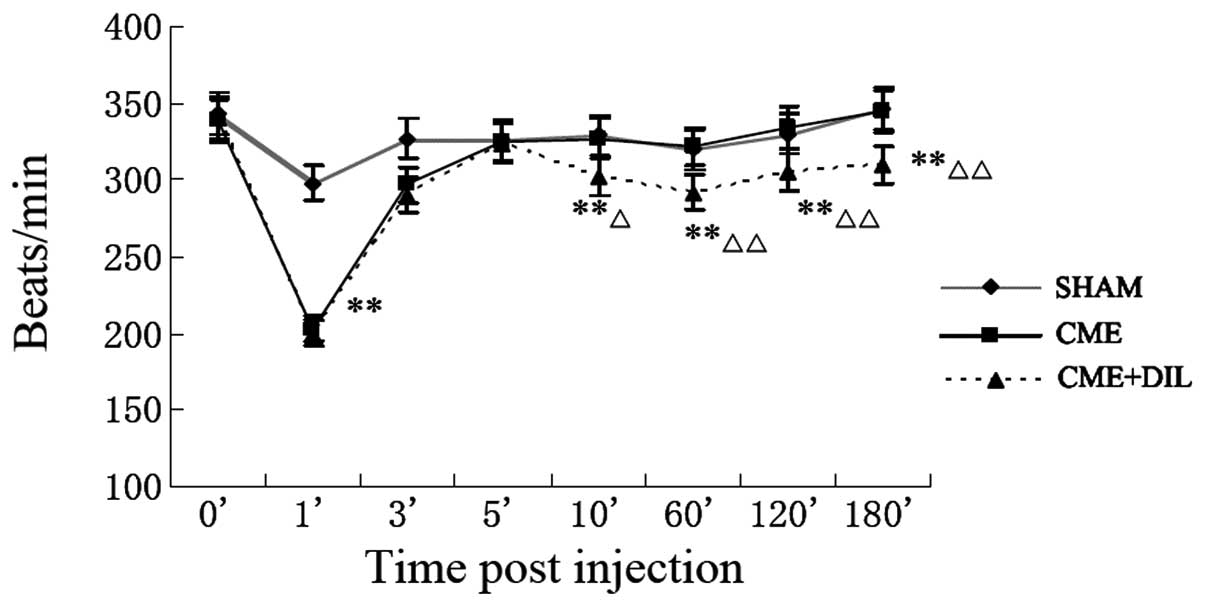
Heart rate changes
Following the injection of automicrothrombotic
particulates, the heart rate decreased significantly at 1 min and
returned to baseline level 5 min postinjection in the CME and
CME+DIL groups, while it remained unchanged in the sham group
(Fig. 1).
Transthoracic echocardiography
Preoperative values were comparable between the CME
and CME+DIL groups. Four weeks postoperatively, the LVEDV was
significantly lower, while the FS and LVEF were significantly
higher, in the CME+DIL group compared with the CME group (Table I).
 | Table I.Two-dimensional mode transthoracic
echocardiography results. |
Table I.
Two-dimensional mode transthoracic
echocardiography results.
| Variable | Status | Sham group | CME group | CME+DIL group |
|---|
| LVEDD (mm) | Pre-op | 5.66±0.59
(n=10) | 5.84±0.47
(n=10) | 5.88±0.39
(n=10) |
| Post-op | 5.77±0.42
(n=8) |
6.65±0.61a,b (n=8) | 6.20±0.44
(n=9) |
| LVESD (mm) | Pre-op | 2.88±0.24
(n=10) | 2.95±0.21
(n=10) | 2.90±0.19
(n=10) |
| Post-op | 2.92±0.13
(n=8) |
4.58±0.45a,b (n=8) |
3.73±0.43a–c (n=9) |
| LVEDV (ml) | Pre-op | 0.47±0.11
(n=10) | 0.48±0.10
(n=10) | 0.49±0.09
(n=10) |
| Post-op | 0.47±0.05
(n=8) |
0.92±0.10a,b (n=8) |
0.71±0.10a–c (n=9) |
| LVESV(ml) | Pre-op | 0.09±0.02
(n=10) | 0.09±0.01
(n=10) | 0.09±0.01
(n=10) |
| Post-op | 0.09±0.02
(n=8) |
0.38±0.04a,b (n=8) |
0.25±0.05a–c (n=9) |
| FS (%) | Pre-op | 48.9±1.7
(n=10) | 49.4±1.2
(n=10) | 50.7±2.7
(n=10) |
| Post-op | 49.3±1.9 (n=8) |
31.2±2.1a,b (n=8) |
40.4±4.3a–c (n=9) |
| LVEF (%) | Pre-op | 81.3±1.6
(n=10) | 80.3±2.4
(n=10) | 80.9±2.6
(n=10) |
| Post-op | 80.0±3.7 (n=8) |
58.2±6.8a,b (n=8) |
64.5±9.8a,b (n=9) |
Hemodynamic measurements
In the 3 h group, dp/dtmax and-dp/dtmax were
significantly higher, while LVEDP was significantly lower, in the
CME+DIL group than in the CME group (Table II).
| Table II.Hemodynamic measurements. |
Table II.
Hemodynamic measurements.
| Group | n | LVSP (mmHg) | LVEDP (mmHg) | dp/dtmax
(mmHg/s) | –dp/dtmax
(mmHg/s) |
|---|
| Sham | 9 | 150.1±9.8 | 5.4±2.9 | 5605±693 | −5034±394 |
| CME | 9 | 104.3±9.6a | 15.3±2.8a | 2986±236a | −2612±239a |
| CME+DIL | 10 | 106.0±7.3a |
10.8±2.0a,c |
3407±359a,b | −2905±316a |
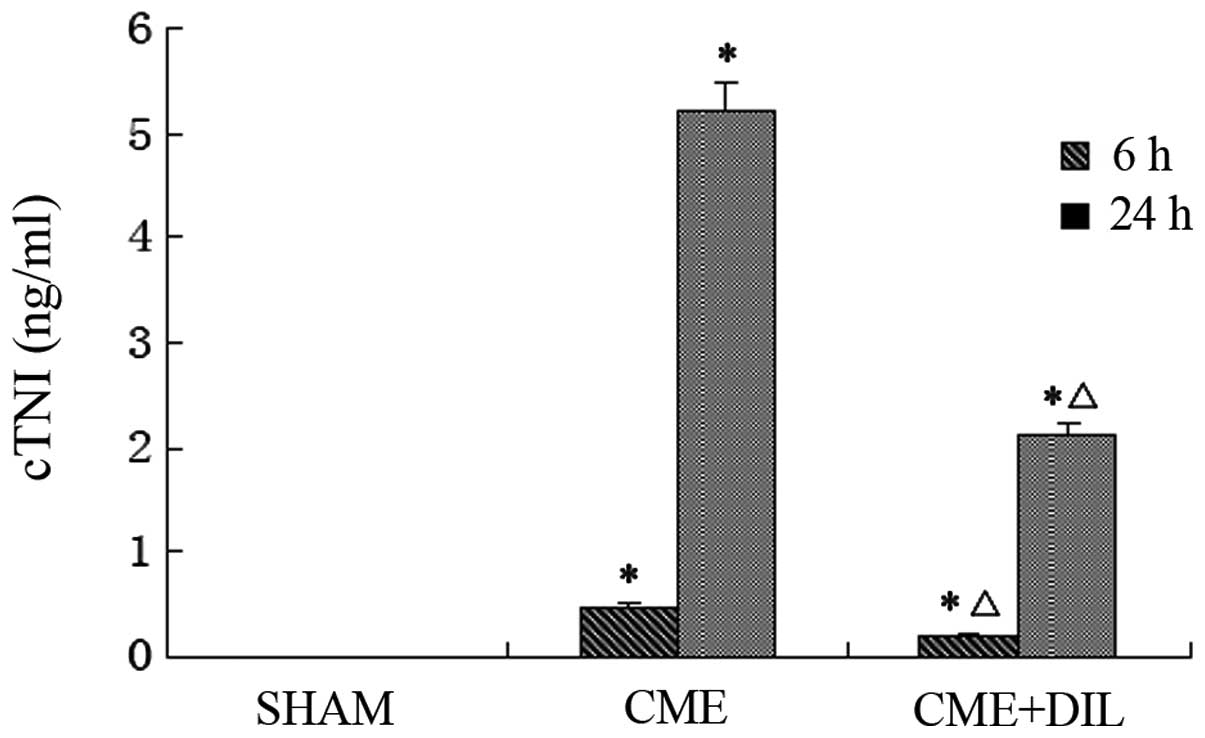
Levels of c-TNI at 6 and 24 h
postinjection
c-TNI levels were significantly reduced in the
CME+DIL group compared with those in the CME group at 6 and 24 h
post-automicrothrombotic particulate injection (Fig. 2).
Levels of plasma vWF and ET-1
postinjection
Plasma vWF is regarded as a good indicator of
endothelial dysfunction and has been shown to contribute to the
activation of the coagulation cascade (28,29).
Plasma vWF levels were significantly lower at 3 h following the
injection of post-automicrothrombotic particulates in the CME+DIL
group than in the CME group (Table
III). Levels of ET-1, the endothelium-derived vasoconstrictor
peptide, increase in response to myocardial ischemia and infarction
(30,31). Plasma ET-1 levels at 3 h, 24 h and
7 days after the injection of post-automicrothrombotic particulates
were also significantly lower in the CME+DIL group than in the CME
group (Table III).
| Table III.Plasma vWF and ET-1 levels
postinjection. |
Table III.
Plasma vWF and ET-1 levels
postinjection.
| Group | Variable | Time
postinjection |
|---|
|
|---|
| 3 h | 24 h | 7 days | 28 days |
|---|
| Sham | vWF (ng/ml) | 6.08±0.29
(n=9) | 5.98±0.25
(n=9) | 5.94±0.26
(n=9) | 6.02±0.29
(n=8) |
| ET-1 (pg/ml) | 50.2±0.24
(n=9) | 52.2±0.26
(n=9) | 51.2±0.23
(n=9) | 52.2±0.29
(n=8) |
| CME | vWF (ng/ml) | 7.80±0.58a (n=9) | 6.61±0.46a (n=10) | 6.10±0.41
(n=10) | 6.23±0.35
(n=9) |
| ET-1 (pg/ml) | 93.6±1.24a (n=9) | 154.2±2.46a (n=10) | 114.8±2.46a (n=10) | 84.0±1.35a (n=9) |
| CME+DIL | vWF (ng/ml) |
6.95±0.59a,b (n=10) | 6.31±0.49
(n=9) | 6.06±0.36
(n=10) | 6.13±0.39
(n=9) |
| ET-1 (pg/ml) |
73.2±0.48a,b (n=10) |
100.2±0.49a,b (n=9) |
78.6±0.76a,b (n=10) | 74.8±1.68a (n=9) |
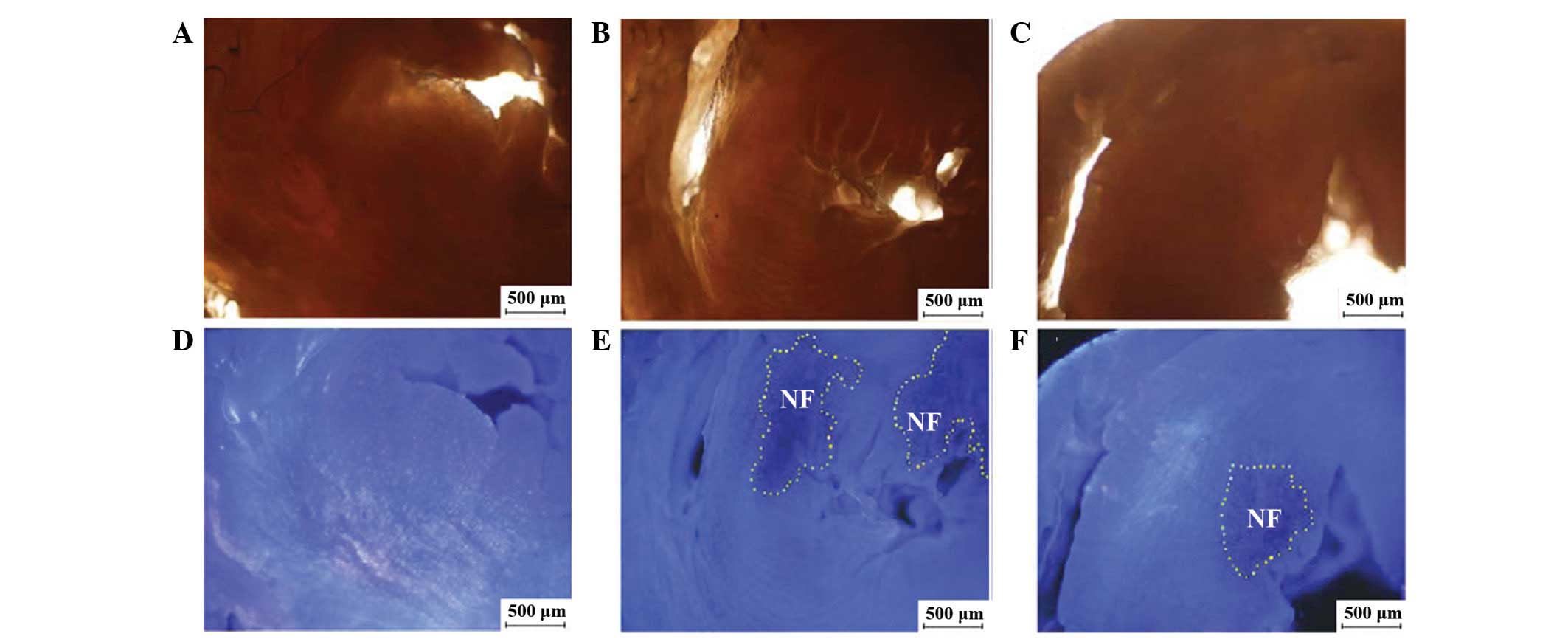
NF evaluation at 3 h
postinjection
The NF was evaluated by thioflavin S (blue
fluorescence represented the perfused zone and non-fluorescent
areas represented the NF when examined under ultraviolet light at a
365-nm wavelength). The NF/LV ratio was significantly lower in the
CME+DIL group than in the CME group (5.6±2.5 vs. 11.2±2.7%,
P<0.01; Fig. 3).
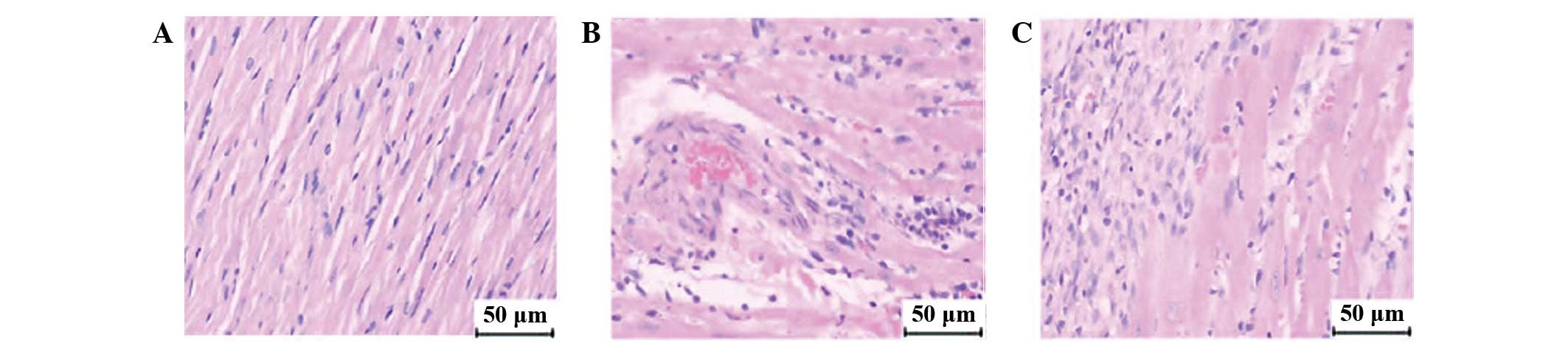
Light microscope analyses
H&E staining at 3 h
postinjection
Three hours postinjection, red thrombi were
identified in 18.2±4.5% of coronary arterioles with diameters
<100 μm in the CME group, compared with 10.4±2.5% in the
CME+DIL group (P<0.01; Fig. 4B and
C).
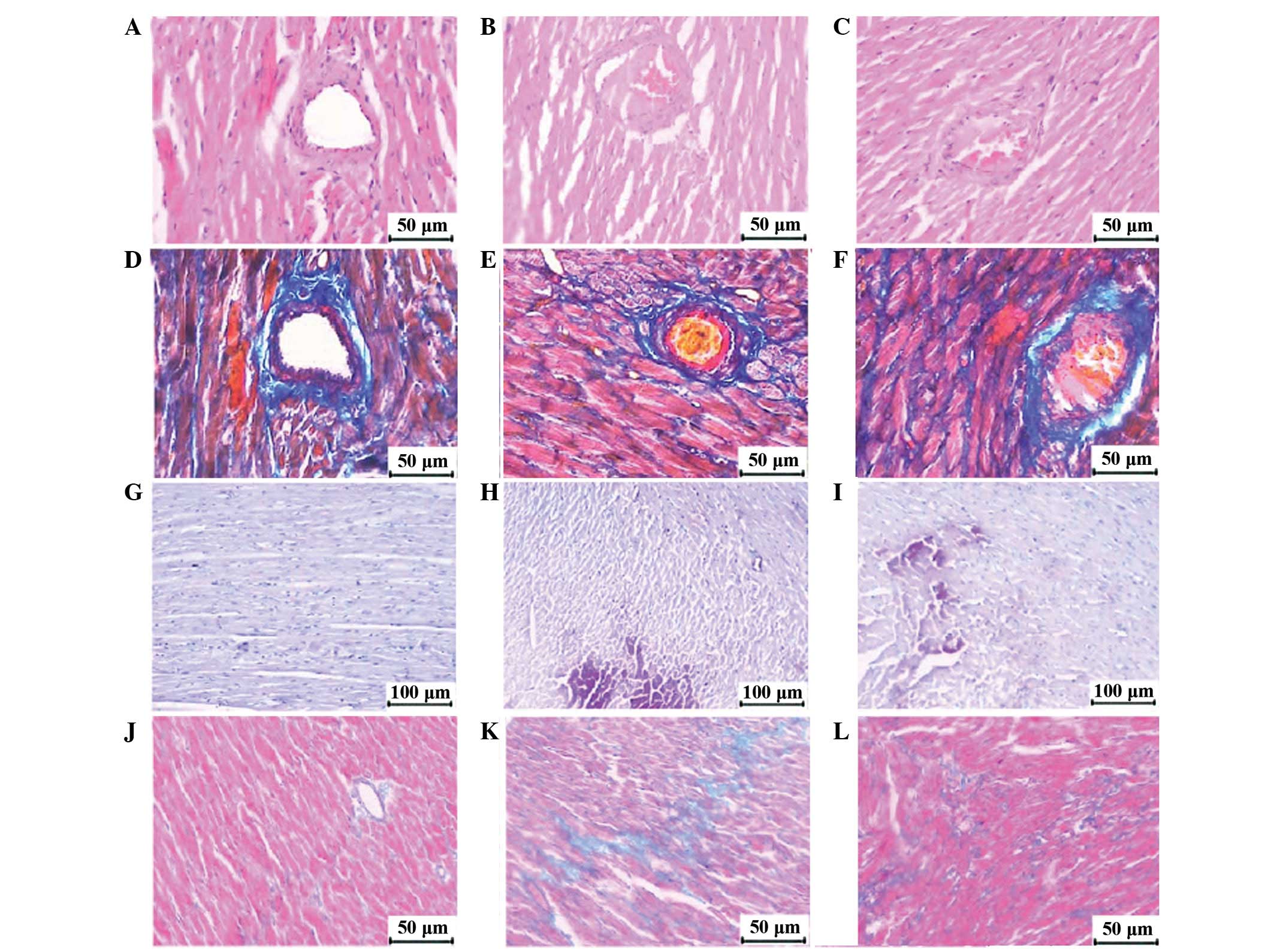 | Figure 4.Light microscopic analyses. In
H&E-stained slices (magnification, ×200), red thrombosis was
not observed in arterioles of (A) the sham group, but was observed
in the (B) CME and (C) CME+DIL groups. (D–F) In the sham, CME and
CME+DIL groups, respectively, Carstair’s staining (magnification,
×200) showed that the major components of thrombosis in the CME
group were fibrins (bright red), platelets (gray-blue to navy blue)
and red cells (yellow). (G-I) In the sham, CME and CME+DIL groups,
respectively, HBFP staining (magnification, ×100) showed cardinal
red regions 3 h postinjection in the CME and CME+DIL groups. (J–L)
In the sham, CME and CME+DIL groups, respectively, Masson staining
at 4 weeks postinjection (magnification, ×200) showed increased
collagen in the CME group and decreased collagen deposition in the
CME+DIL group. H&E, hematoxylin and eosin; CME, coronary
thrombotic microembolism; DIL, diltiazem; HBFP, hematoxylin basic
fuchsin picric acid. |
Carstair’s staining at 3 h
postinjection
Different colors in Carstair’s staining represented
different components of the thrombosis in arterioles; bright red
for fibrin, gray-blue to navy blue for platelets, bright blue for
collagen, red for muscle and clear yellow for red blood cells.
Three hours postinjection, evidence of thrombosis was observed in
the CME and CME+DIL groups. The major components of thrombosis were
fibrins and platelets, and there was also red cell accumulation in
the vascular lumen (Fig. 4E and
F).
HBFP staining at 3 h post
injection
HBFP staining was used to detect early myocardial
ischemia or infarct regions. The normal myocardium was stained
yellow or yellow-brown, and the ischemic or necrotic myocardial
tissue was stained cardinal red. The ischemic area (IA) was
calculated using the following formula: IA (%) = IA/area of field
of vision x 100. The IA was 6.3±1.2% in the CME group and reduced
to 3.3±1.2% in the CME+DIL group (P<0.01; Fig. 4H and I).
Masson staining 28 days
postoperatively
Masson staining was carried out in the 28 days
postinjection group. Cardiomyocytes were stained red and collagen
stained blue. The collagen volume fraction (CVF = area of
collagen/area of the field of vision × 100%) was measured. The CVF
was significantly lower in the CME+DIL group (Fig. 4L) than in the CME group (Fig. 4K; 5.38±1.46 vs. 2.60±1.07%,
P<0.01).
Inflammatory cell infiltration
At 24 h postinjection, coagulative necrosis occurred
in the microinfarct zone and polymorphonuclear leukocyte
infiltration was observed around the blocked vessel in the CME
group (Fig. 5B). Seven days
postinjection, the majority of infiltrated leukocytes were
macrophages and leukomonocytes in the CME+DIL group (Fig. 5C). The leukocyte counts were
significantly lower in the CME+DIL group than in the CME group 24 h
and 7 days postoperatively (Table
IV).
| Table IV.Leukocyte infiltration postinjection
(leukocytes/mm2). |
Table IV.
Leukocyte infiltration postinjection
(leukocytes/mm2).
| Group | Time
postinjection |
|---|
|
|---|
| 24 h | 7 days | 28 days |
|---|
| Sham | 158±42 (n=9) | 160±42 (n=9) | 157±22 (n=8) |
| CME | 930±126a (n=10) | 836±105a (n=10) | 160±24 (n=9) |
| CME+DIL |
652±112a,b
(n=9) | 322±66a,b (n=10) | 159±22 (n=9) |
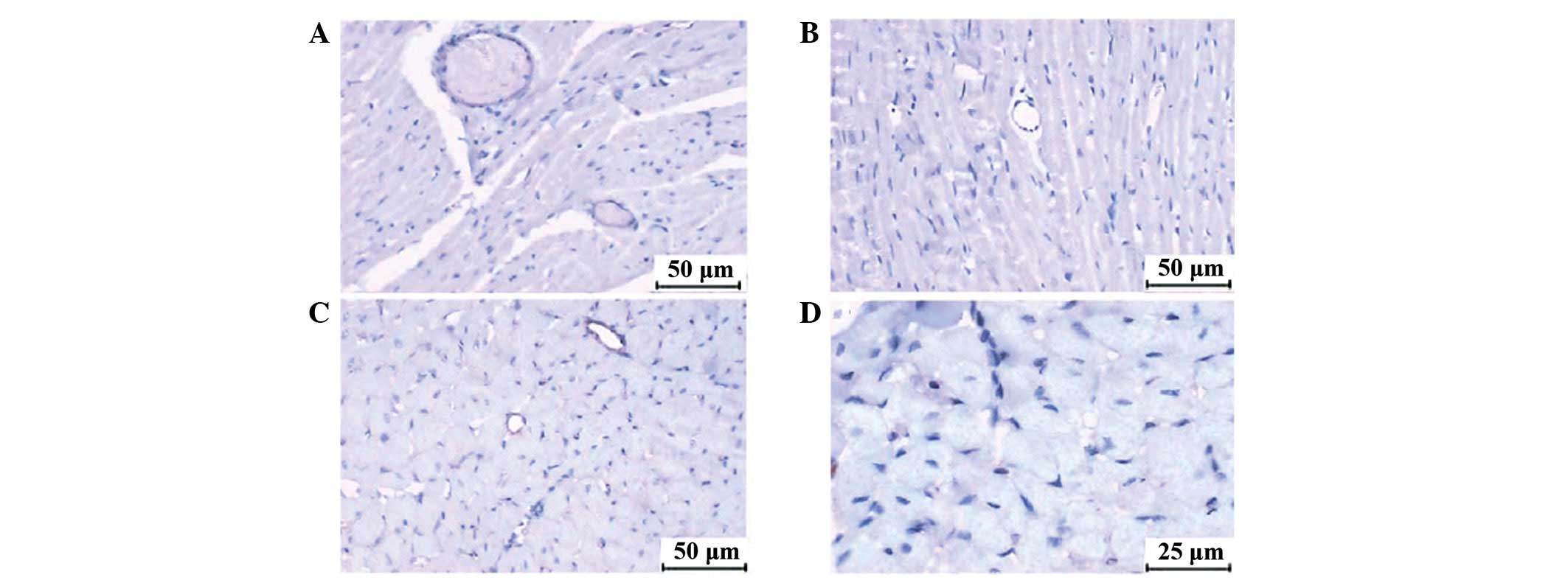
Immunohistochemical staining
At 3 h postinjection, VSMA-α was expressed in
vascular smooth muscle cells and the vascular smooth muscle was
dyed claybank in order to count the arterioles (10–100 μm).
Fig. 6 shows arterioles with
different diameters in the CME and CME+DIL groups.
Immunohistochemical staining analysis indicated that the number of
arterioles with a diameter in the range of 10–50 μm,
particularly arterioles with diameters of 20–50 μm, was
significantly higher in the CME+DIL group than in the CME group at
3 h postinjection (Table V).
| Table V.Number of arterioles at 3 h
postinjection (s/mm2). |
Table V.
Number of arterioles at 3 h
postinjection (s/mm2).
| Arteriole
diameter | Group |
|---|
|
|---|
| Sham | CME | CME+DIL |
|---|
| 10–20
μm | 2.61±0.18
(n=9) | 2.15±0.26b (n=9) | 2.4±0.19c (n=10) |
| 20–50
μm | 0.70±0.06
(n=9) | 0.32±0.10b (n=9) | 0.59±0.14d (n=10) |
| 50–100
μm | 0.30±0.03
(n=9) | 0.28±0.05
(n=9) | 0.32±0.04
(n=10) |
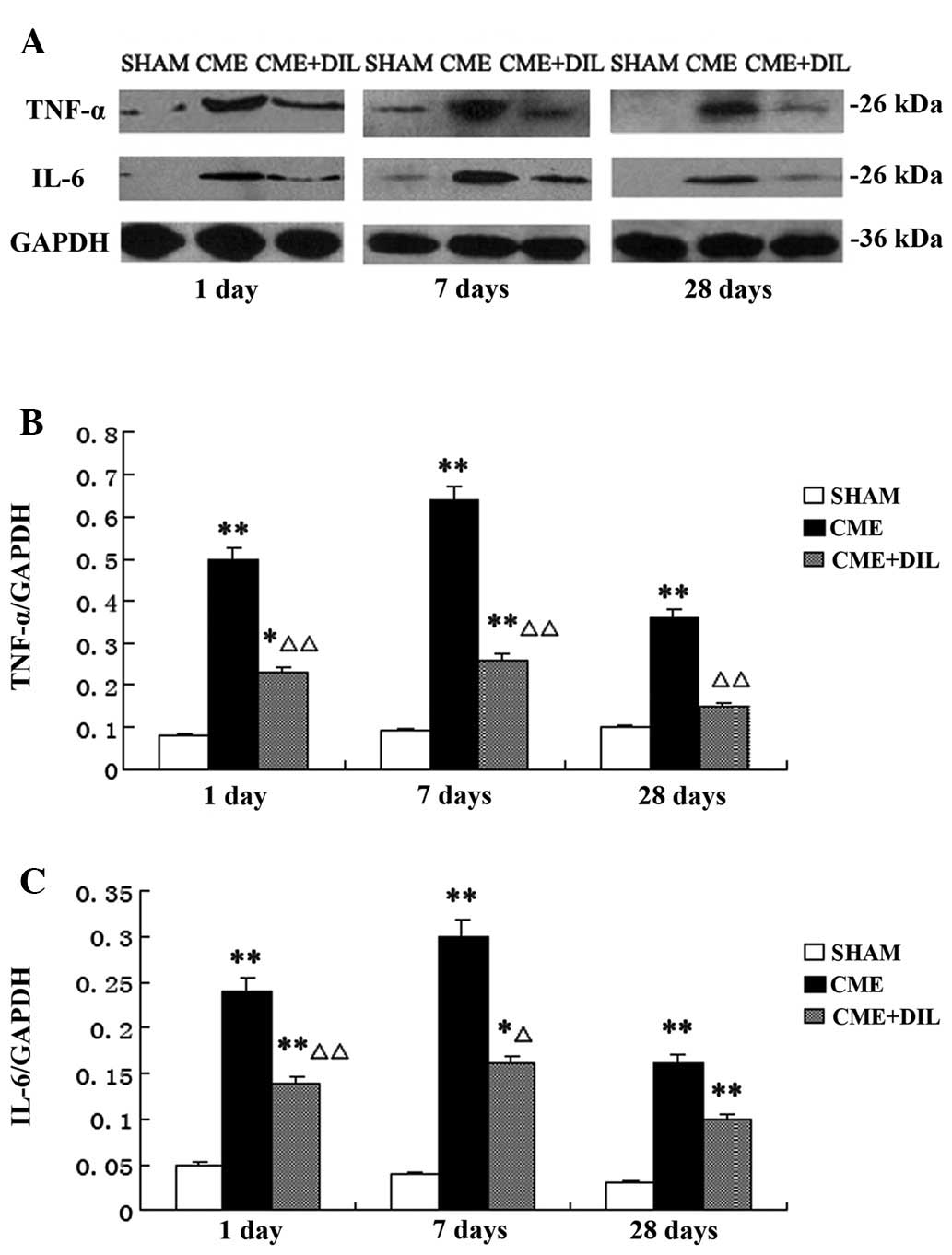
Western blot analysis
The myocardial protein expression levels of TNF-α
and IL-6 were significantly downregulated in the CME+DIL group
compared with those in the CME group at various time-points
postinjection (Fig. 7).
Discussion
The present study demonstrated that the injection of
auto-microthrombotic particulates into the aorta of male Sprague
Dawley rats successfully induced CME, arteriolar thrombosis,
histologically-confirmed ischemic regions and NF. This confirmed
the pathological changes that were shown in this model in a
previous study (9).
Results from this model demonstrated that the
components of the thrombosis in coronary arterioles were fibrin,
aggregated platelets and red blood cells, indicating the presence
of vessels that were obstructed by automicrothrombotic particulates
and newly formed thrombosis in situ. Increased vWF and ET-1
levels, indicators of endothelial function (32), at 3 h postinjection in CME rats
indicated that microthrombotic particulates may also induce
microvascular endothelial injury. Acute myocardial injury was
demonstrated by increased c-TnI levels at 6 and 24 h and myocardial
infarctlets in HBFP-stained myocardium at 3 h
post-automicrothrombotic particulate injection in this model.
Immunohistochemical staining analysis indicated that the number of
10–50 μm diameter arterioles (particularly 20–50 μm)
was significantly reduced in the CME group at 3 h postinjection
compared with the number in the sham rats. Therefore, the injection
of automicrothrombotic particulates induced not only arteriolar
thrombosis, but also arteriolar spasm. Moreover, increased serum
c-TnI levels and myocardial leukocyte infiltration at early
time-points postinjection and prolonged inflammatory responses
indicated by increased myocardial TNF-α and IL 6 expression
resembled typical inflammatory responses post-ischemia (33). This finding is in line with
previous reports showing that an inflammatory reaction was the most
significant mechanism resulting in systolic heart failure in CME
and that the inflammatory mediator TNF-α may be causal in
contractile dysfunction following CME (34–38).
The interactions between microembolism, plaque fissuring,
arrhythmia and dysfunction have been summarized previously
(5,9,12).
Thus, there may be a correlation between microembolism and the NF
phenomenon in that increased myocardial collagen content
demonstrated by histology and reduced cardiac function shown by
transthoracic echocardiography at 4 weeks may be the sequential
changes induced by microembolism. Briefly, coronary microembolism/
coronarymicrothrombosis may result in endothelial damage/
dysfunction as well as arteriolar spasm, leading to coronary
vascular resistance increase, no reflow, microinfarctlets
inflammatory reaction, myocardial remodeling and cardiac
dysfunction.
The aim of this study was to explore the feasibility
of using the present rat CME model to reflect the therapeutic
effects of clinically effective medication on CME injury. The
effects of intravenous diltiazem were therefore evaluated in this
animal model. Diltiazem is a calcium channel antagonist that
inhibits myocardial calcium entry by blockade of voltage-dependent
membrane calcium channels. Although calcium channel antagonists all
inhibit calcium channel conductance, they exhibit considerable
selectivity of action in terms of vasodilation, negative inotropic
and chronotropic effects (37,39).
Previous clinical studies have shown that calcium antagonists may
attenuate microvascular spasm by relaxing small vascular smooth
muscle (7,8,40),
and intravascular application of diltiazem may attenuate coronary
artery spasm in patients with microvascular angina (19,20).
In the current study, it was demonstrated that intravenous
diltiazem (1 mg/ml, 50 μg/min/kg) administered at 5 min
post-automicrothrombotic particulate injection for 175 min improved
cardiac function, attenuated the reduction in the number of
arterioles (diameter 10–50 μm) and reduced the NF in the
present CME model, possibly through attenuating microvascular
spasm. Moreover, diltiazem also reduced myocardial ischemia,
endothelia dysfunction and inflammatory responses, as indicated by
changes in c-TnI, plasma vWF and ET-1 levels, as well as the
myocardial protein expression levels of TNF-α and IL-6. It was also
demonstrated that the number of red thrombi reduced immediately
after diltiazem application in H&E-stained myocardial tissue,
the ischemic area in HBFP-stained myocardial samples was reduced 3
h postinjection and the CVF in Masson-stained myocardial samples
was reduced 28 days postinjection. These results are thus in line
with previous findings showing that treatment of the ischemic
myocardium with calcium channel blockers attenuates ultrastructural
myocardial injury (41,42), decreases calcium influx (42,43)
and improves postischemic left ventricular segmental function
(43–48). The NF area was reduced but did not
disappear completely in the CME+DIL group. This may indicate that
part of the capillary network was already undergoing necrotic
changes and was unable to be recovered following diltiazem
treatment. However, diltiazem did improve automicrothrombotic
particulate-induced functional no-reflow by reducing myocardial
spasm (47).
Large animal CME models have been widely used in
microembolism research (49,50).
The rat CME model used in the present study differs from the large
animal CME models in the following respects: i) This model mimicked
in vivo arteriole blockade by various sizes of microthrombi
following atherosclerotic plaque rupture; ii) the components of
auto-microthrombotic particulates are similar to thrombi in
vivo, including fibrin, platelets and blood corpuscle; iii)
automicrothrombotic particulates are easy to obtain and do not
require elaborate equipment in the laboratory; iv) rat models are
more economical compared with large animal models; and v) rat
hearts are smaller and the entire heart may be easily sampled with
few histological sections.
Notably, it is difficult to define the exact
mechanism of action for diltiazem and to differentiate the
anti-vasospasm and -vasoconstriction effects of diltiazem with the
data available for this model. Therefore, further studies are
required to explore these points.
In conclusion, this animal model mimicked certain
pathological changes induced by coronary embolization that have
been observed in clinical patients with acute coronary syndromes
and in patients who have undergone revascularization procedures
(fibrinolytics or transcatheter recanalization during surgical or
percutaneous procedures, or prior embolization before procedures).
Intravenous diatiazem reduced automicrothrombotic particulate
injection-induced myocardial injury. Thus, this model may be used
to test the effects of drugs that have the potential to attenuate
CME and arteriolar thrombosis-induced myocardial injury.
Acknowledgements
This study was supported by the
National Natural Science Foundation of China (81270266).
References
|
1.
|
Abdelmeguid AE, Topol EJ, Whitlow PL, Sapp
SK and Ellis SG: Significance of mild transient release of creatine
kinase-MB fraction after percutaneous coronary interventions.
Circulation. 94:1528–1536. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Califf RM, Abdelmeguid AE, Kuntz RE, et
al: Myonecrosis after revascularization procedures. J Am Coll
Cardiol. 31:241–251. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Herrmann J, Haude M, Lerman A, et al:
Abnormal coronary flow velocity reserve after coronary intervention
is associated with cardiac marker elevation. Circulation.
103:2339–2345. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Mehran R, Dangas G, Mintz GS, et al:
Atherosclerotic plaque burden and CK-MB enzyme elevation after
coronary interventions: intravascular ultrasound study of 2256
patients. Circulation. 101:604–610. 2000. View Article : Google Scholar
|
|
5.
|
Erbel R and Heusch G: Coronary
microembolization. J Am Coll Cardiol. 36:22–24. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Golino P, Piscione F, Benedict CR, et al:
Local effect of serotonin released during coronary angioplasty. N
Engl J Med. 330:523–528. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Wilson RF, Laxson DD, Lesser JR and White
CW: Intense microvascular constriction after angioplasty of acute
thrombotic coronary arterial lesions. Lancet. 1:807–811. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Piana RN, Paik GY, Moscucci M, et al:
Incidence and treatment of ‘no-reflow’ after percutaneous coronary
intervention. Circulation. 89:2514–2518. 1994.
|
|
9.
|
Gu Y, Bai Y, Wu J, Hu L and Gao B:
Establishment and characterization of an experimental model of
coronary thrombotic microembolism in rats. Am J Pathol.
177:1122–1130. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Heusch G, Kleinbongard P, Böse D, et al:
Coronary micro-embolization: from bedside to bench and back to
bedside. Circulation. 120:1822–1836. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Herrmann J: Peri-procedural myocardial
injury: 2005 update. Eur Heart J. 26:2493–2519. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Heusch G, Schulz R, Haude M and Erbel R:
Coronary microembolization. J Mol Cell Cardiol. 37:23–31. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Lee KW and Norell MS: Management of
‘no-reflow’ complicating reperfusion therapy. Acute Card Care.
10:5–14. 2008.
|
|
14.
|
Pasceri V, Patti G and Di Sciascio G:
Prevention of myocardial damage during coronary intervention.
Cardiovasc Hematol Disord Drug Targets. 6:77–83. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Valero SJ, Moreno R, Reyes RM, et al:
Pharmacological approach of no-reflow phenomenon related with
percutaneous coronary interventions. Cardiovasc Hematol Agents Med
Chem. 6:125–129. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Kleinbongard P, Konorza T, Böse D, et al:
Lessons from human coronary aspirate. J Mol Cell Cardiol.
52:890–896. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Werner GS, Lang K, Kuehnert H and Figulla
HR: Intracoronary verapamil for reversal of no-reflow during
coronary angioplasty for acute myocardial infarction. Catheter
Cardiovasc Interv. 57:444–451. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
McIvor ME, Undemir C, Lawson J and
Reddinger J: Clinical effects and utility of intracoronary
diltiazem. Cathet Cardiovasc Diagn. 35:287–293. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Sütsch G, Oechslin E, Mayer I and Hess OM:
Effect of diltiazem on coronary flow reserve in patients with
microvascular angina. Int J Cardiol. 52:135–143. 1995.PubMed/NCBI
|
|
20.
|
Zheng ZF, Pu XQ, Yang TL, et al: Effects
of intracoronary diltiazem on no-reflow phenomenon after emergent
percutaneous coronary intervention in patients with acute
myocardial infarction. Zhong Nan Da Xue Xue Bao Yi Xue Ban.
31:917–920. 2006.(In Chinese).
|
|
21.
|
Brown C: Blood collection from the tail of
a rat. Lab Anim (NY). 35:24–25. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Kudo M, Aoyama A, Ichimori S and Fukunaga
N: An animal model of cerebral infarction. Homologous blood clot
emboli in rats. Stroke. 13:505–508. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Grossman W: Pressure measurement.
Grossman’s Cardiac Catheterition, Angiography, and Intervention.
Baim DS: 7th edition. Lippincott Williams & Wilkins;
Philadelphia: pp. 139–141. 2006
|
|
24.
|
Genda S, Miura T, Miki T, Ichikawa Y and
Shimamoto K: K(ATP) channel opening is an endogenous mechanism of
protection against the no-reflow phenomenon but its function is
compromised by hypercholesterolemia. J Am Coll Cardiol.
40:1339–1346. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Eitzman DT, Bodary PF, Shen Y, et al:
Fabry disease in mice is associated with age-dependent
susceptibility to vascular thrombosis. J Am Soc Nephrol.
14:298–302. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Fujita M, Fujioka Y and Ommura Y:
Histopathological diagnosis of early stages of myocardial
infarction - applications of the improved hematoxylin basic fuchsin
picric acid (HBFP) staining method to human autopsy hearts.
Hokkaido Igaku Zasshi. 60:313–320. 1985.(In Japanese).
|
|
27.
|
Goldner J: A modification of the Masson
trichrome technique for routine laboratory purposes. Am J Pathol.
14:237–243. 1938.PubMed/NCBI
|
|
28.
|
Blann AD: Plasma von Willebrand factor,
thrombosis, and the endothelium: the first 30 years. Thromb
Haemost. 95:49–55. 2006.PubMed/NCBI
|
|
29.
|
With Notø AT, Bøgeberg Mathiesen E, Amiral
J, Vissac AM and Hansen JB: Endothelial dysfunction and systemic
inflammation in persons with echolucent carotid plaques. Thromb
Haemost. 96:53–59. 2006.PubMed/NCBI
|
|
30.
|
Stewart DJ, Kubac G, Costello KB and
Cernacek P: Increased plasma endothelin-1 in the early hours of
acute myocardial infarction. J Am Coll Cardiol. 18:38–43. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Tønnessen T, Giaid A, Saleh D, Naess PA,
Yanagisawa M and Christensen G: Increased in vivo expression and
production of endothelin-1 by porcine cardiomyocytes subjected to
ischemia. Circ Res. 76:767–772. 1995.PubMed/NCBI
|
|
32.
|
Ruggeri ZM: Von Willebrand factor,
platelets and endothelial cell interactions. J Thromb Haemost.
1:1335–1342. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Bonvini RF, Hendiri T and Camenzind E:
Inflammatory response post-myocardial infarction and reperfusion: a
new therapeutic target? Eur Heart J Suppl. 7(Suppl I): I27–I36.
2005. View Article : Google Scholar
|
|
34.
|
Skyschally A, Schulz R, Haude M, Erbel R
and Heusch G: Coronary microembolization: perfusion-contraction
mismatch secondary to myocardial inflammation. Herz. 29:777–781.
2004.(In German).
|
|
35.
|
Dörge H, Neumann T, Behrends M, et al:
Perfusion-contraction mismatch with coronary microvascular
obstruction: role of inflammation. Am J Physiol Heart Circ Physiol.
279:H2587–H2592. 2000.PubMed/NCBI
|
|
36.
|
Dörge H, Schulz R, Belosjorow S, et al:
Coronary microembolization: the role of TNF-alpha in contractile
dysfunction. J Mol Cell Cardiol. 34:51–62. 2002.PubMed/NCBI
|
|
37.
|
Triggle DJ and Swamy VC: Calcium
antagonists. Some chemical-pharmacologic aspects. Circ Res.
52:I17–I28. 1983.PubMed/NCBI
|
|
38.
|
Kleinbongard P, Heusch G and Schulz R:
TNFalpha in atherosclerosis, myocardial ischemia/reperfusion and
heart failure. Pharmacol Ther. 127:295–314. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39.
|
Fleckenstein A: History of calcium
antagonists. Circ Res. 52:I3–I16. 1983.PubMed/NCBI
|
|
40.
|
Beltrame JF, Turner SP, Leslie SL, Solomon
P, Freedman SB and Horowitz JD: The angiographic and clinical
benefits of mibefradil in the coronary slow flow phenomenon. J Am
Coll Cardiol. 44:57–62. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41.
|
Hamm CW and Opie LH: Protection of
infarcting myocardium by slow channel inhibitors. Comparative
effects of verapamil, nifedipine, and diltiazem in the
coronary-ligated, isolated working rat heart. Circ Res.
52:I129–I138. 1983.
|
|
42.
|
Nayler WG, Ferrari R and Williams A:
Protective effect of pretreatment with verapamil, nifedipine and
propranolol on mitochondrial function in the ischemic and
reperfused myocardium. Am J Cardiol. 46:242–248. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
43.
|
Bourdillon PD and Poole-Wilson PA: The
effects of verapamil, quiescence, and cardioplegia on calcium
exchange and mechanical function in ischemic rabbit myocardium.
Circ Res. 50:360–368. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
44.
|
Przyklenk K and Kloner RA: Effect of
verapamil on postischemic ‘stunned’ myocardium: importance of the
timing of treatment. J Am Coll Cardiol. 11:614–623. 1988.
|
|
45.
|
Bush LR, Buja LM, Tilton G, et al: Effects
of propranolol and diltiazem alone and in combination on the
recovery of left ventricular segmental function after temporary
coronary occlusion and long-term reperfusion in conscious dogs.
Circulation. 72:413–430. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
46.
|
Higgins AJ and Blackburn KJ: Prevention of
reperfusion damage in working rat hearts by calcium antagonists and
calmodulin antagonists. J Mol Cell Cardiol. 16:427–438. 1984.
View Article : Google Scholar : PubMed/NCBI
|
|
47.
|
Galiuto L: Optimal therapeutic strategies
in the setting of post-infarct no reflow: the need for a
pathogenetic classification. Heart. 90:123–125. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48.
|
Kleinbongard P, Baars T and Heusch G:
Calcium antagonists in myocardial ischemia/reperfusion - update
2012. Wien Med Wochenschr. 162:302–310. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49.
|
Huang Y, Hunyor S, Jiang L, et al:
Remodeling of the chronic severely failing ischemic sheep heart
after coronary microembolization: functional, energetic,
structural, and cellular responses. Am J Physiol Heart Circ
Physiol. 286:2141–2150. 2004. View Article : Google Scholar
|
|
50.
|
Gill RM, Jones BD, Corbly AK, et al:
Exhaustion of the Frank-Starling mechanism in conscious dogs with
heart failure induced by chronic coronary microembolization. Life
Sciences. 79:536–544. 2006. View Article : Google Scholar : PubMed/NCBI
|