Introduction
Hypoxic-ischemic brain damage (HIBD) is
irreversible; it releases cytokines and oxygen free radicals that
exacerbate ischemic brain damage through inflammation and apoptosis
(1). In the necrotic area and
ischemic brain tissues following cerebral infarction, excessive
inflammatory and immune responses are the pathophysiological basis
of tissue damage (2). Toll-like
receptors (TLRs) are indispensable in the inflammatory response and
may recognize endogenous damage-associated molecular patterns
(DAMPs) and exogenous pathogen-associated molecular patterns
(PAMPs) when affiliated ligands are activated. In addition, TLRs
induce nuclear factor-κB (NF-κB)-associated pro-inflammatory
cytokines through a myeloid differentiation factor 88
(MyD88)-dependent pathway and produce interferon regulatory factor
(IRF) through a MyD88-independent pathway to promote downstream
pathways (3).
TLRs are activated when bacteria or virus-associated
molecular patterns are identified, and then promote an inflammatory
response mediated by macrophages, neutrophils and complement
(4). It has been suggested that a
small dose of a bacterial endotoxin (PAMP) may induce endotoxin
tolerance through the modulation of TLRs and reduce the damage
mediated by an excessive inflammatory response (5–7).
Previous studies have shown that small doses of endotoxin may
result in tolerance to ischemic brain injury and protective effects
(8,9), suggesting that pretreatment with
exogenous PAMP may induce endogenous DAMP tolerance. However, the
mechanism by which brain tissues become ischemia tolerant with
endogenous DAMP pretreatment remains unknown.
Transient ischemic preconditioning is commonly used
for studying the role of endogenous TLR ligands and has been shown
to have protective effects against ischemia-reperfusion injury in
the brain (10). A previous study
identified that neuroprotective functions were weaker following
transient cerebral ischemia in TLR4 gene-deficient mice, suggesting
that the release of endogenous TLR ligands induces cerebral
protective mechanisms (11). The
exact mechanisms of action of the TLRs and the downstream
MyD88-dependent and -independent pathways require further study. In
the present study, an ischemic preconditioning model with a line
embolism blocking the middle cerebral artery was established in
rats and the expression levels of MyD88, NF-κB, Toll/interleukin-1
receptor-domain-containing adaptor-inducing interferon-β (TRIF) and
IRF-3 were detected at three time-points following reperfusion.
Materials and methods
Animal grouping
Twenty-seven male Sprague Dawley rats were purchased
from the Vital River Laboratory Animal Technology Co., Ltd.
(Beijing, China) and were randomly divided into three groups: The
sham group (Group A) received sham treatment twice (n=3); the
ischemia-reperfusion group (Group B) received one sham surgery and
one ischemia-reperfusion treatment (n=12); and the ischemic
preconditioning group (Group C) received one ischemic
preconditioning and one ischemia-reperfusion surgery (n=12).
According to different time-points of animal sacrifice, the groups
were each divided into three subgroups. The subgroups were A12,
A24, A48, B12, B24, B48, C12, C24 and C48, with the letter
corresponding to the group name, and the number to the time
following reperfusion; for example, A12 included animals from Group
A sacrificed 12 h following reperfusion. The present study was
approved by the Ethics Committee of Shengjing Hospital (Shenyang,
China).
Establishment of the ischemic
preconditioned rat model
The rats in Group C were anaesthetized with 10%
chloral hydrate (3.5 ml/kg) and the common carotid artery, external
carotid artery and internal carotid artery were blunt separated.
The proximal and distal ends of the common carotid artery were
ligated with surgical sutures (size, 3–0), bifurcation of the
internal and external carotid artery was closed with artery
occlusion; an additional artery occlusion was used to close the
external carotid artery. A small cut was made between the common
carotid artery ligation and the vascular clamp. The line embolism
was inserted into the middle cerebral artery and reperfusion was
induced following 10 min of blocking. The subcutaneous tissues and
skin tissues were sutured at each layer. In Groups A and B, only
the common, external and internal carotid arteries were blunt
separated, and the subcutaneous tissues and skin tissues were
sutured at each layer. All animals were conventionally fed for two
days. After these two days, the separated common, external and
internal carotid arteries were identified. In Group C, the distal
end and the bifurcation of the internal and external carotid
arteries were closed with artery occlusion and a new surgical line
replaced the old one that had been inserted two days previously. An
additional artery occlusion was performed to close the external
carotid artery and prevent line embolism in the external carotid
artery. A small cut was made between the common carotid artery
ligation and vascular clamp, a line embolism was inserted in the
middle cerebral artery and reperfusion was induced for 12, 24 and
48 h following 60 min of blocking. In Group B, the
ischemia-reperfusion method was the same as that in Group C. In
Group A, only the common, external and internal carotid arteries
were blunt separated, and the subcutaneous and skin tissues were
sutured at each layer.
Specimen grouping at 12, 24 and 48 h
In Group A, one rat was sacrificed at 12 h, one at
24 h and one at 48 h after ischemia-reperfusion, to constitute the
A12, A24 and A48 subgroups, respectively. In Groups B and C, four
rats were sacrificed at each of 12, 24 and 48 h after
ischemia-reperfusion to constitute the B12, B24 and B48 and C12,
C24 and C48 subgroups, respectively. In addition, brain tissue from
each rat was collected for further experiments.
Western blot analysis of MyD88, NF-κB,
TRIF and IRF-3
Protein extracts were prepared using
radioimmunoprecipitation assay (RIPA) lysis buffer and the protein
concentrations were measured by the Bradford method using a BCA
Protein assay kit (Pierce Biotechnology, Inc., Rockford, IL, USA).
Equal quantities of protein extracts (30 μg) were separated by 12%
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE) and then transferred onto 0.45 μm polyvinylidene
difluoride (PVDF) membranes (Millipore, Billerica, MA, USA) in wet
conditions. The membranes were blocked in 1X Tris-buffered Saline
and Tween 20 (TBST) containing 5% non-fat dry milk for 1 h and then
incubated at 4°C overnight with rabbit anti-MyD88, -NF-κB, -TRIF
and -IRF-3 antibodies (sc-179; Santa Cruz Biotechnology, Inc.,
Santa Cruz, CA, USA) or rabbit anti-β-actin antibody (AP0060;
Bioworld Technology, Inc., St. Louis Park, MN, USA), both diluted
1:1,000 in 1X TBST containing 5% non-fat dry milk. This was
followed by incubation with a secondary horseradish
peroxidase-conjugated anti-rabbit antibody (BS13278; Bioworld
Technology, Inc.) diluted 1:5,000 in 1X TBST containing 5% non-fat
dry milk for 1 h at room temperature. Proteins were detected using
an enhanced chemiluminescence system (Pierce Biotechnology, Inc.)
in a dark room. Western band densities were quantified using ImageJ
software, version 1.44p (National Institutes of Health; http://rsbweb.nih.gov/ij/. Accessed September 15,
2013).
Immunofluorescence staining of NF-κB and
IRF-3
Tissues were fixed in 4% paraformaldehyde for 24 h
and then embedded in paraffin. Sections were prepared for
hematoxylin and eosin staining. Immunohistochemical staining of
formalin-fixed paraffin-embedded sections was performed. Tissue
sections (4 μm thick) were dewaxed in toluene, rehydrated,
permeabilized in citrate buffer (pH 6.0) and quenched with 3%
H2O2 for 15 min to eliminate endogenous
peroxidase activity. The sections were then incubated overnight at
4°C with a primary antibody for rabbit anti-NF-κB, anti-NF-κb-Cy3,
anti-IRF-3-FITC (diluted 1:100 in PBS). After washing in
phosphate-buffered saline, tissues were incubated with biotinylated
goat anti-rabbit immunoglobulin G (Dako, Glostrup, Denmark)
followed by treatment with peroxidase-conjugated streptavidin and
staining with diaminobenzidine. Counterstaining was performed with
hematoxylin. Immunostaining was evaluated by two independent
observers unaware of the origin of the tissue.
Statistical analyses
All data are expressed as the mean ± standard
deviation. SPSS software, version 18.0 (SPSS, Inc., St. Louis, MO,
USA) was used for statistical analysis of the data. Data between
two groups was analyzed by Student’s t-test and data from several
groups was evaluated with variance analysis. P<0.05 was
considered to indicate a statistically significant difference.
Results
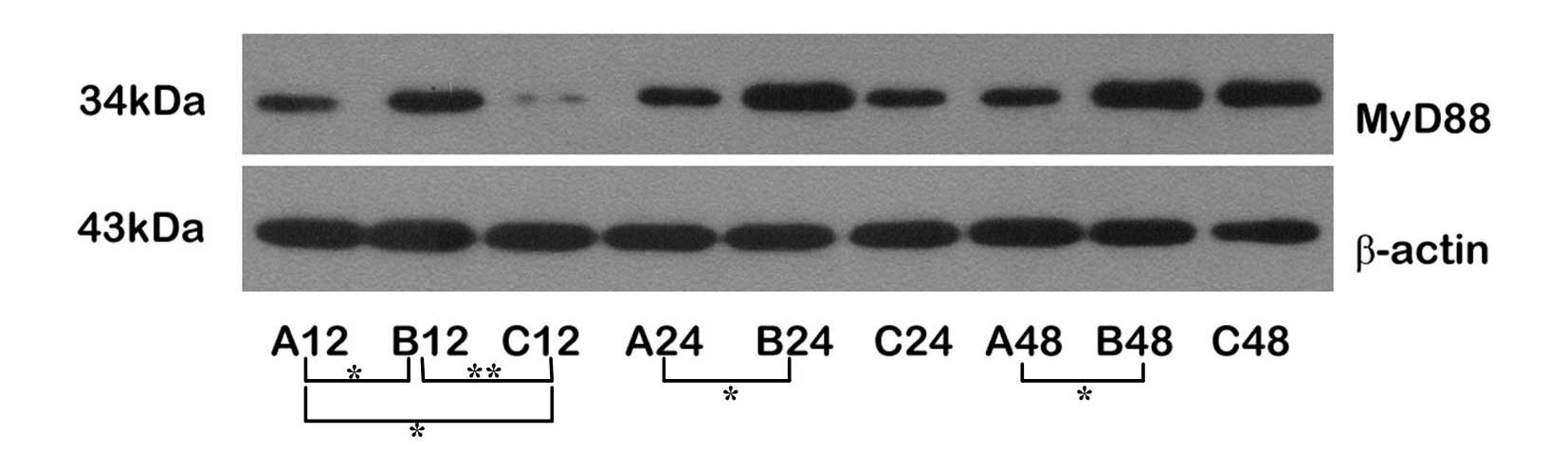
Downregulation of MyD88 and NF-κB protein
expression by ischemic preconditioning
Following transient ischemic preconditioning (Group
C), the level of MyD88 protein expression was downregulated at all
time-points. In the ischemia-reperfusion group without
preconditioning (Group B), MyD88 protein expression was upregulated
at all time-points. The MyD88 expression level of the C12 subgroup
was significantly lower than that of the B12 subgroup (P<0.01)
and was also significantly lower than that of A12. The MyD88
expression level of B12 was significantly higher than that of A12.
These results showed that the expression level of MyD88 protein was
significantly increased following 60 min of ischemia followed by
reperfusion for 12 h and confirmed that transient ischemic
preconditioning inhibited the expression of MyD88. We observed that
MyD88 expression was upregulated in Group B, and the expression
levels were significantly higher than those of Group A. In
addition, MyD88 protein expression was upregulated in Group C, but
at a low level. The MyD88 expression level of the C24 subgroup was
lower than that in normal brain tissue (Group A), indicating that
ischemic preconditioning is able to inhibit the overexpression of
MyD88 (Fig. 1).
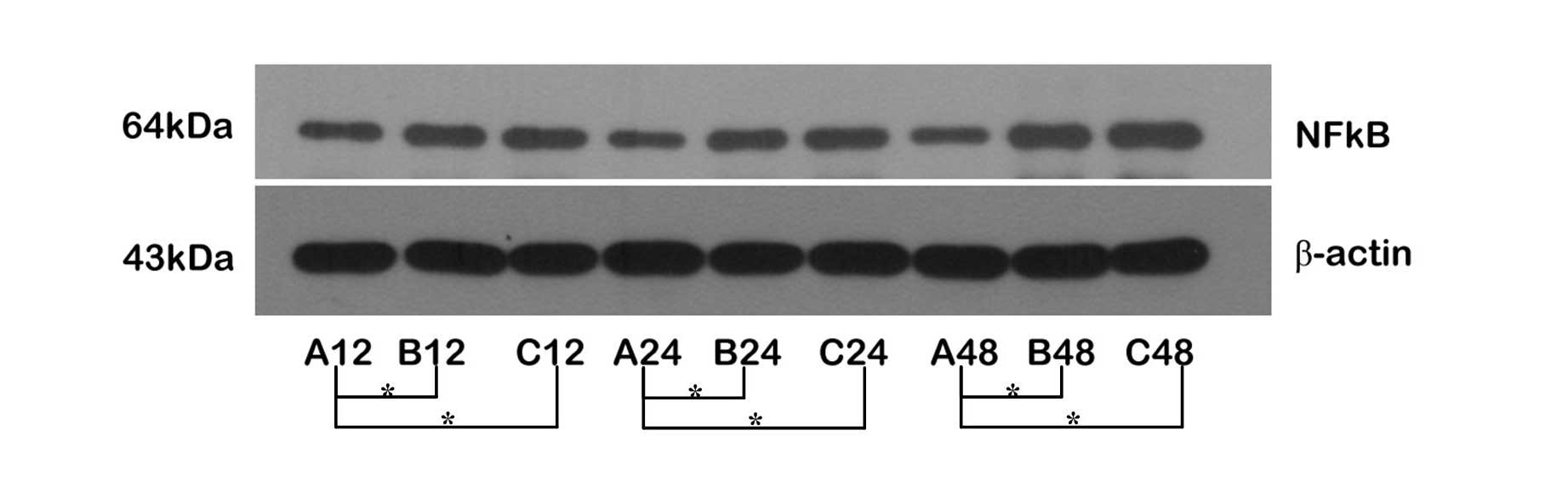
The expression of NF-κB was upregulated at all
time-points in Groups B and C (Fig.
2); the expression of NF-κB in the C48 subgroup was marginally
higher than that in the B48 subgroup, but there were no significant
differences between Groups B and C. These results indicate that the
expression of NF-κB was inconsistent with the expression of MyD88,
suggesting the upregulation of NF-κB was regulated through
additional pathways.
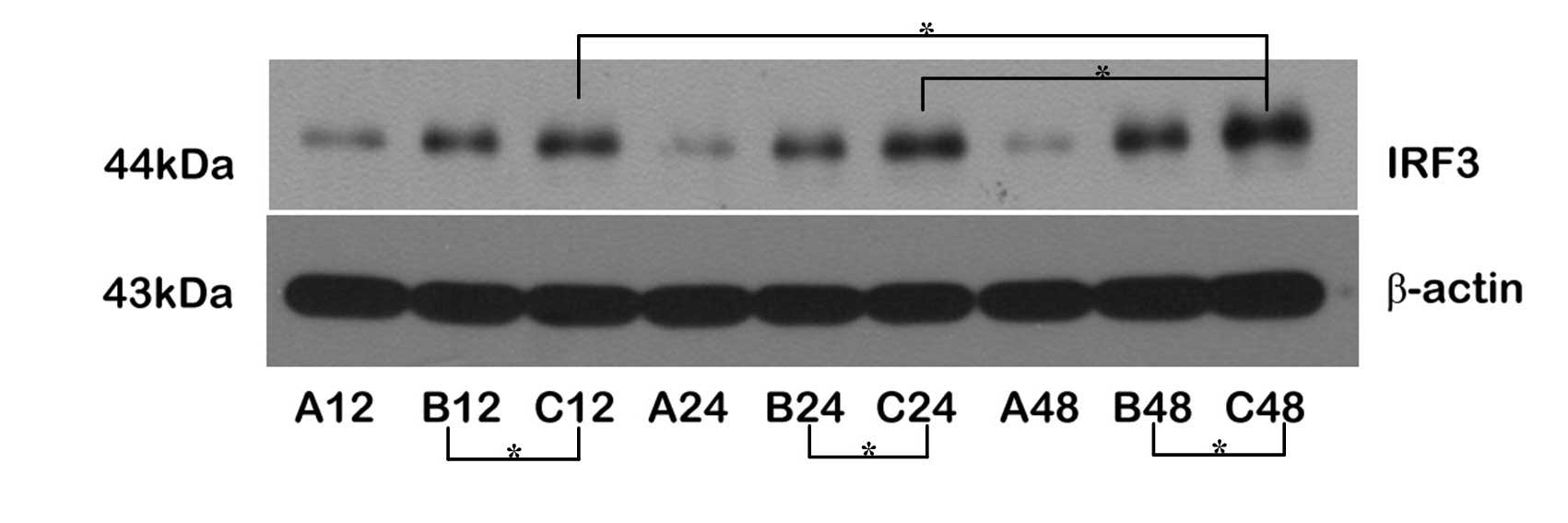
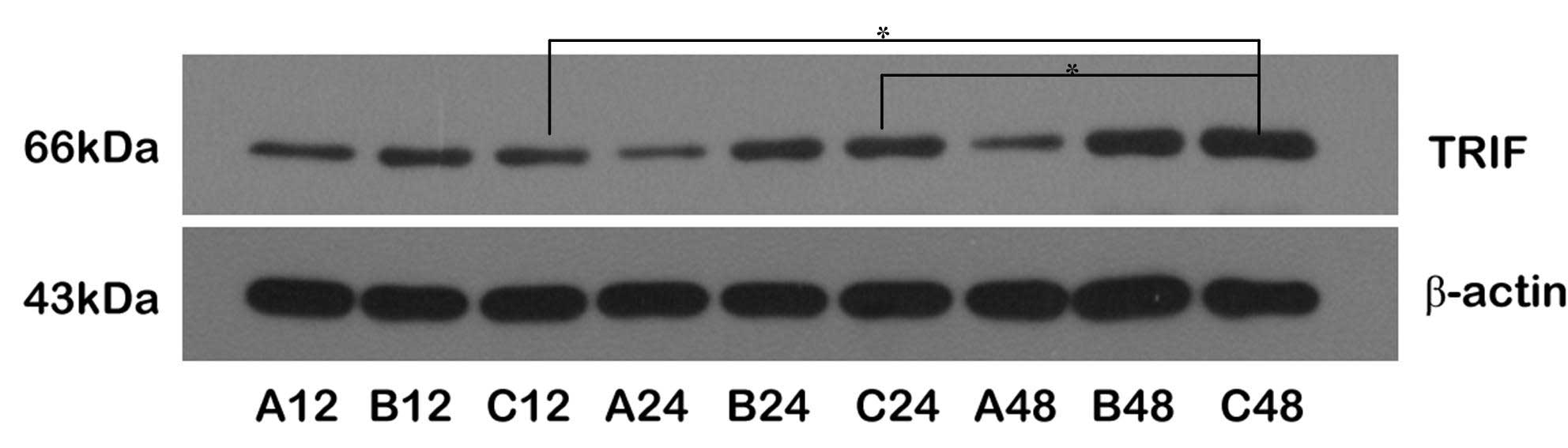
Upregulation of TRIF/IRF-3 protein
expression through the MyD88-independent pathway
The expression levels of the TRIF and IRF-3 proteins
were upregulated following reperfusion. The expression levels of
IRF-3 protein were higher in Group C than in Group B at all
time-points; the expression levels of TRIF and IRF-3 were
significantly increased in the C48 subgroup, but only the
expression of IRF-3 was marginally increased at C12, suggesting
that the expression levels of TRIF and IRF-3 were upregulated
through transient ischemic preconditioning. Comparing the
expression levels of TRIF and IRF-3 proteins between Groups B and
C, the changes in the expression levels of IRF-3 were larger than
those of TRIF, indicating that the expression of IRF-3 was
regulated by TRIF during the cerebral protective process, and
ischemic pretreatment may have facilitated this process, resulting
in brain ischemic tolerance (Figs.
3 and 4).
Coordination of TRIF/IRF-3 and NF-κB
proteins in ischemic preconditioning
The upregulation of NF-κB and TRIF/IRF-3 protein
expression showed coordination at certain time-points; in
particular, NF-κB and TRIF proteins were upregulated at 12 and 24 h
following reperfusion in Groups B and C. The expression levels of
NF-κB, TRIF and IRF-3 were increased 48 h following reperfusion and
the protein expression levels were higher in Group C than in Group
B (P>0.05).
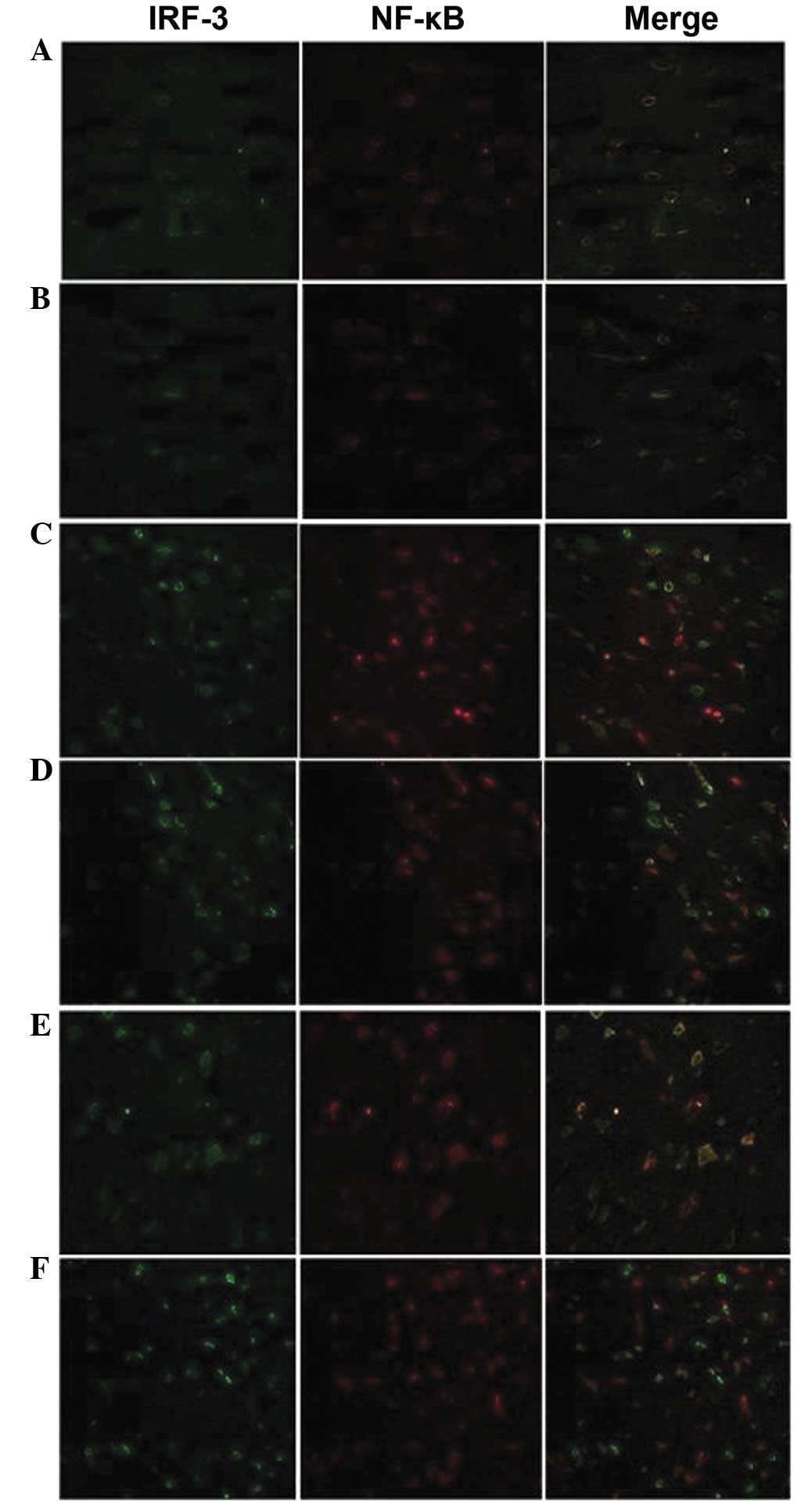
Location of NF-κB and IRF-3 proteins
Without ischemic preconditioning and
ischemia-reperfusion, the NF-κB and IRF-3 proteins were located in
the cytoplasm and the fluorescent brightness was consistent with
the results of the semi-quantitative western blot analysis
(Fig. 5A and B).
The fluorescent brightness of NF-κB and IRF-3
proteins was elevated in the B12 and B24 subgroups compared with
those in Group A, and the proteins, particularly NF-κB, were
transferred into the nucleus. However, the distribution of NF-κB
and IRF-3 proteins showed no significant changes in the B48
subgroup from those in B12 and B24, and the IRF-3 protein was
mainly distributed in the nucleus (Fig. 5C and D).
Notably, the distribution and fluorescent brightness
of NF-κB and IRF-3 proteins in the C12 subgroup was similar to that
of the B48 subgroup and higher than that of Group A. The proteins,
particularly IRF-3, were transferred into the nucleus. The NF-κB
protein was transferred from the nucleus to cytoplasm in the C24
and C48 subgroups, and IRF-3 protein was mainly expressed in the
nucleus. The fluorescence brightness of the IRF-3 protein was
higher in the C48 subgroup than in the C12 and C24 subgroups
(Fig. 5E and F).
Discussion
A previous study showed that the ischemic tolerance
of endotoxins through TLR4 was regulated by NF-κB of the
MyD88-dependent pathway and IRF-3 of the MyD88-independent pathway.
The study focused on the endogenous DAMPs recognized by TLR4 and
the ischemic tolerance mechanisms (12).
The results of the present study identified that
MyD88 protein expression was significantly inhibited following
transient cerebral ischemia and the expression of MyD88 remained
low even following reperfusion for 12 h. We speculate that the
inhibitory effects are transient and the delayed expression of
MyD88 protein may subsequently prevent the inflammatory response,
which is caused by activation of the inflammatory pathway. The
expression level of MyD88 protein was increased at 24 and 48 h
following reperfusion in the ischemic-preconditioned group and the
level of expression at 48 h was higher than that in the control
group; however, the MyD88 expression level was lower than that of
the ischemia-reperfusion group. It is known that the
MyD88-dependent pathway induces the inflammatory response, and is
associated with the proliferation and differentiation of nerve
cells following injury (13). The
inflammatory cytokines and anti-inflammatory cytokines may achieve
a steady balance so as to repair nerve injury (14). It has been demonstrated that
ischemic tolerance is induced by inflammatory cytokines, such as
tumor necrosis factor-α, but inhibited by glucocorticoids (15). Therefore, the MyD88 protein was
marginally reduced, but not completely inhibited following
transient ischemic preconditioning; with a suitable time of
ischemia-reperfusion, the appropriate inflammatory cytokines may
induce ischemic tolerance.
NF-κB is the downstream cytokine of the
MyD88-dependent pathway and its expression levels were inconsistent
with those of MyD88. In the present study, we observed that the
expression levels of NF-κB were not affected by ischemic
preconditioning after 12 and 24 h of reperfusion and the expression
levels were higher than those of the normal control group. The
expression of the TRIF protein was upregulated following ischemic
preconditioning, but there were no significant differences in TRIF
expression between the ischemia-reperfusion and ischemic
preconditioning groups at 12 and 24 h. At 48 h, the expression
levels of TRIF, IRF-3 and NF-κB were significantly increased, and
significant differences were identified between the
ischemia-reperfusion and ischemic preconditioning groups. The
TRIF/IRF-3 pathways is the main signaling pathway for
neuroprotection with endotoxin or ischemia and hypoxia
preconditioning (16,17). In the present study, we observed
that the TRIF/IRF-3 expression levels were upregulated following
transient ischemic preconditioning. In addition, the changes in the
expression of NF-κB protein were induced by the action of the MyD88
and TRIF pathways, which suggests that the expression changes of
NF-κB protein were mainly mediated by the late NF-κB activation of
the TRIF pathway. The sudden elevation of the NF-κB expression
levels at 48 h may have been caused by the activation of the
TBK1/IKKɛ pathway (18). We also
observed that NF-κB migrated from the nucleus to the cytoplasm and
IRF-3 gradually transferred to the nucleus with reperfusion.
Notably, in Group B, NF-κB was mainly expressed in the nucleus at
12 and 24 h, but IRF-3 was mainly expressed in the nucleus at 48 h.
NF-κB is the cytokine necessary for regulating the inflammatory
response, growth and differentiation. In lymphocytes, NF-κB
interacts with the inhibitory protein, IκB, to maintain an inactive
state; however, when IκB protein is degraded, NF-κB translocates to
the nucleus to combine with genes (19). NF-κB activity was significantly
inhibited by endotoxins following ischemia-reperfusion for 24 h and
inhibitory proteins, such as Ship1 and Tollip, were found in the
cytoplasm, which would not affect the expression of
pro-inflammatory genes (17).
Therefore, in our study, although the expression of NF-κB was
increased, it was maintained in an inactive state in the cytoplasm
and IRF-3 protein was upregulated in the nucleus to achieve
cerebral protection. However, the inflammatory cytokines and
pro-inflammatory cytokines were not investigated.
In conclusion, to the best of our knowledge, we
conducted transient ischemic preconditioning in rats for the first
time. We investigated the associated patterns of endogenous injury
recognized by TLRs, and observed the expression of proteins
associated with the MyD88-dependent and -independent pathways at
12, 24 and 48 h following ischemia-reperfusion. In addition, we
outlined the cerebral protective mechanisms of ischemic
preconditioning and provided a reference for further study. Our
findings indicate a potential therapeutic approach for the
treatment of hypoxic-ischemic brain damage.
References
|
1
|
Kuroda S and Siesjö BK: Reperfusion damage
following focal ischemia: pathophysiology and therapeutic windows.
Clin Neurosci. 4:199–212. 1997.PubMed/NCBI
|
|
2
|
Zhang X, Li H, Hu S, Zhang L, Liu C, Zhu
C, Liu R and Li C: Brain edema after intracerebral hemorrhage in
rats: the role of inflammation. Neurol India. 54:402–407. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tang SC, Arumugam TV, Xu X, Cheng A,
Mughal MR, Jo DG, Lathia JD, Siler DA, Chigurupati S, Ouyang X, et
al: Pivotal role for neuronal Toll-like receptors in ischemic brain
injury and functional deficits. Proc Natl Acad Sci USA.
104:13798–13803. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Takeda K, Kaisho T and Akira S: Toll-like
receptors. Annu Rev Immunol. 21:335–376. 2003. View Article : Google Scholar
|
|
5
|
Granowitz EV, Porat R, Mier JW, Orencole
SF, Kaplanski G, Lynch EA, Ye K, Vannier E, Wolff SM and Dinarello
CA: Intravenous endotoxin suppresses the cytokine response of
peripheral blood mononuclear cells of healthy humans. J Immunol.
151:1637–1645. 1993.
|
|
6
|
Erroi A, Fantuzzi G, Mengozzi M, Sironi M,
Orencole SF, Clark BD, Dinarello CA, Isetta A, Gnocchi P,
Giovarelli M, et al: Differential regulation of cytokine production
in lipopolysaccharide tolerance in mice. Infect Immun.
61:4356–4359. 1993.PubMed/NCBI
|
|
7
|
Marsh B, Stevens SL, Packard AE, Gopalan
B, Hunter B, Leung PY, Harrington CA and Stenzel-Poore MP: Systemic
lipopolysaccharide protects the brain from ischemic injury by
reprogramming the response of the brain to stroke: a critical role
for IRF3. J Neurosci. 29:9839–9849. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tasaki K, Ruetzler CA, Ohtsuki T, Martin
D, Nawashiro H and Hallenbeck JM: Lipopolysaccharide pre-treatment
induces resistance against subsequent focal cerebral ischemic
damage in spontaneously hypertensive rats. Brain Res. 748:267–270.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rosenzweig HL, Lessov NS, Henshall DC,
Minami M, Simon RP and Stenzel-Poore MP: Endotoxin preconditioning
prevents cellular inflammatory response during ischemic
neuroprotection in mice. Stroke. 35:2576–2581. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Stenzel-Poore MP, Stevens SL, Xiong Z,
Lessov NS, Harrington CA, Mori M, Meller R, Rosenzweig HL, Tobar E,
Shaw TE, et al: Effect of ischaemic preconditioning on genomic
response to cerebral ischaemia: similarity to neuroprotective
strategies in hibernation and hypoxia-tolerant states. Lancet.
362:1028–1037. 2003. View Article : Google Scholar
|
|
11
|
Pradillo JM, Fernández-López D,
García-Yébenes I, Sobrado M, Hurtado O, Moro MA and Lizasoain I:
Toll-like receptor 4 is involved in neuroprotection afforded by
ischemic preconditioning. J Neurochem. 109:287–294. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Häcker H, Redecke V, Blagoev B,
Kratchmarova I, Hsu LC, Wang GG, Kamps MP, Raz E, Wagner H, Häcker
G, et al: Specificity in Toll-like receptor signalling through
distinct effector functions of TRAF3 and TRAF6. Nature.
439:204–207. 2006.PubMed/NCBI
|
|
13
|
Rolls A, Shechter R, London A, Ziv Y,
Ronen A, Levy R and Schwartz M: Toll-like receptors modulate adult
hippocampal neurogenesis. Nat Cell Biol. 9:1081–1088. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ekdahl CT, Kokaia Z and Lindvall O: Brain
inflammation and adult neurogenesis: the dual role of microglia.
Neuroscience. 158:1021–1029. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rosenzweig HL, Minami M, Lessov NS, Coste
SC, Stevens SL, Henshall DC, Meller R, Simon RP and Stenzel-Poore
MP: Endotoxin preconditioning protects against the cytotoxic
effects of TNFalpha after stroke: a novel role for TNFalpha in
LPS-ischemic tolerance. J Cereb Blood Flow Metab. 27:1663–1674.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Stevens SL, Leung PY, Vartanian KB,
Gopalan B, Yang T, Simon RP and Stenzel-Poore MP: Multiple
preconditioning paradigms converge on interferon regulatory
factor-dependent signaling to promote tolerance to ischemic brain
injury. J Neurosci. 31:8456–8463. 2011. View Article : Google Scholar
|
|
17
|
Vartanian KB, Stevens SL, Marsh BJ,
Williams-Karnesky R, Lessov NS and Stenzel-Poore MP: LPS
preconditioning redirects TLR signaling following stroke: TRIF-IRF3
plays a seminal role in mediating tolerance to ischemic injury. J
Neuroinflammation. 8:1402011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guo B and Cheng G: Modulation of the
interferon antiviral response by the TBK1/IKKi adaptor protein
TANK. J Biol Chem. 282:11817–11826. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Piccioli P, Porcile C, Stanzione S,
Bisaglia M, Bajetto A, Bonavia R, Florio T and Schettini G:
Inhibition of nuclear factor-kappaB activation induces apoptosis in
cerebellar granule cells. J Neurosci Res. 66:1064–1073. 2001.
View Article : Google Scholar : PubMed/NCBI
|