Introduction
It has previously been shown that extracellular
signal-regulated kinase 1/2 (ERK1/2) is a member of the
mitogen-activated protein kinase (MAPK) family, which is abundantly
expressed in the neuron body and dendrites (1) Moreover, ERK1/2 acts as part of a
signaling pathway activated by multiple upstream factors, such as
growth factors, inflammatory factors, neurotransmitters and calcium
influx, via the MAPK and ERK activator kinase (MEK). Furthermore,
these two activator kinases are capable of phosphorylating ERK1/2
at threonine and tyrosine residues with the latter resulting in the
dissociation of ERK1/2 from MEK1/2. Phosphorylated ERK1/2
(p-ERK1/2) enters the nucleus by passive diffusion of the monomer,
active transport of the dimer or by a direct interaction of ERK1/2
with the nuclear pore complex, where it activates nuclear factors
(2–5). Epilepsy is characterized by the
excessively synchronized discharge of cerebral neurons and
comprises a diverse group of syndromes with different etiologies.
In epileptic seizures, normal brain tissue undergoes damage that
produces permanent plasticity changes and results in
epileptogenesis. Studies have revealed that in addition to ERK1/2
affecting neuronal excitability, mossy fiber sprouting and synaptic
remodeling mechanisms are also involved in epileptogenesis
(6–8).
Valproate sodium (VPAS) is a classical and
broad-spectrum antiepileptic drug that is widely used in different
types of epilepsy. VPAS predominantly exerts its antiepileptic
effect through the γ-aminobutyric acid (GABA) system (9). In the present study, concentration-
and time-response experiments were performed to measure the effect
of VPAS on p-ERK1/2 expression, in order to provide an enhanced
understanding of the cellular and molecular mechanisms underlying
the antiepileptic effect of VPAS.
Materials and methods
Neuron culture and patch-clamp
recording
Neonatal Sprague-Dawley (SD) rats, aged <24 h,
were purchased from the Experimental Animal Center of Chongqing
Medical University of China (Chongqing, China). Following
anesthesia, the brain tissue was exposed and the bilateral
hippocampi were excised using a dissecting microscope (Nikon Corp.,
Tokyo, Japan), prior to the meninges and superficial blood vessels
being removed. The hippocampus was then minced in ice-cold D-
Hank’s balanced salt solution (Sigma, St. Louis, MO, USA) and
incubated in five-fold volumes of 0.125% parenzyme (Sigma) at 37ºC
and in 5% CO2 for 20 min. The digestion was terminated
by the addition of an equal volume of growth medium composed of
Neurobasal® Medium, 2% B-27, 0.5 mM L-glutamine and 0.5%
fetal bovine serum (FBS; all from Gibco-BRL, Grand Island, NY,
USA). Following centrifugation at 212 × g for 5 min, the
supernatant was discarded and fresh growth medium was added. The
tissue was then dissociated and the cell suspension was filtered
through a 200-mesh cell sieve (Nanjing jing yu sheng instrument
Co., Ltd., Nangjing, China). The cells were diluted to a
concentration of 5×105 cells/ml, plated on coverslips
coated with polylysine (0.1%; Gibco-BRL) and maintained at 37ºC in
5% CO2. Twenty-four hours subsequent to plating, the
medium was changed to a maintenance medium (Neurobasal Medium, 2%
B-27, and 0.5 mM L-glutamine). Half the volume of the maintenance
medium was changed every three days. Following nine days in
vitro, cell excitability was measured using previously
established procedures (6).
Briefly, the cells were placed in magnesium-free artificial
cerebrospinal fluid (ACSF; Chongqing chemical reagent factory,
Chongqing, China) containing 124 mM NaCl, 3 mM KCl, 2 mM
CaCl2, 2 mM MgCl2, 1.23 mM
NaH2PO4, 26 mM NaHCO3, 10 mM
glucose and 0.002 mM glycine (pH 7.3, 325 mOsm) for 3 h. To measure
cell excitability, the whole-cell current-clamp technique was used
to record the epileptiform activity. For whole-cell recording, the
cultures were placed on the stage of an inverted microscope (Nikon
Corp.) and continuously perfused with ACSF. Patch electrodes (2–4
MV resistance) were filled with the following internal solution: 60
mM K2SO4, 60 mM N-methyl-D-glucamine (NMG),
40 mM HEPES, 4 mM MgCl2, 0.5 mM
1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (BAPTA),
12 mM phosphocreatine, 2 mM Na2ATP, 0.2 mM
Na3GTP and 0.1 mM leupeptin (pH 7.2–7.3, 265–270 mOsm/l)
(Sigma). At the zero holding current in the whole-cell model, the
membrane potential was measured immediately. Neuronal recording was
then performed using an Axon Axopatch 200B Capacitor Feedback Patch
Clamp Amplifier (Axon Instruments, Inc., Molecular Devices Corp.,
Palo Alto, CA, USA) that was controlled and monitored using pCLAMP™
9 Electrophysiology Data Acquisition and Analysis Software with an
Axon DigiData 1320 Series Interface unit (Axon Instruments,
Inc.).
Immunofluorescence measurement of
p-ERK1/2 in the concentration-response experiment
The wells of cultured neurons were randomly divided
into control and VPAS groups. The control neurons displaying
epileptiform activity were incubated in magnesium-free medium for 3
h and were examined at 0 min. The VPAS-treated neurons were
incubated in magnesium-free ACSF to which VPAS (50, 75 and 100
mg/l, respectively) was added 30 min prior to the epileptiform
discharge, and then were examined at time-points corresponding with
those of the control group. p-ERK1/2 levels were detected
absolutely using immunofluorescence. Briefly, the cultured neurons
were washed with phosphate-buffered saline (PBS) for 3–5 min, fixed
in 4% paraformaldehyde for 30 min and washed with PBS for 3–5 min.
The neurons were then treated with 0.5% Triton X-100 (Gibco-BRL)
for 20 min at room temperature, washed with PBS for a further 3–5
min and blocked with 10% goat serum for 20 min at room temperature.
The neurons were subsequently incubated with mouse anti-p-ERK1/2
(1:100; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA)
overnight at 4ºC. Secondary antibodies [goat anti-mouse-fluorescein
isothiocyanate (FITC), 1:50; Santa Cruz Biotechnology, Inc.] were
applied for 2 h at room temperature, prior to the neurons being
washed with PBS for 3–5 min. Images of each sample were captured
using a laser scanning confocal microscope (Leica Microsystems,
Wetzlar, Germany).
Western blot analysis of p-ERK1/2 in the
time-response experiment
The neurons were divided into two groups, identical
to those in the concentration-response experiment. The levels of
p-ERK1/2 in the control and VPAS (50 mg/l) groups were measured
using western blotting at different time-points (30 min prior to
epileptiform discharge and 0 min, 30 min, 2 h and 6 h subsequent to
epileptiform discharge). The cultured neurons were washed with cold
PBS, collected using centrifugation (11,190 × g for 5 min) and
lysed in cell lysis buffer (100 ml) containing Tris-HCl (50 mM; pH
8.0), NaCl (150 mM), EDTA (1 mM), ethylene
glycol-O,O′-bis(2-aminoethyl)-N,N,N′,N′-tetraacetic acid (EGTA; 1
mM), Triton X-100 (1%), phenylmethylsulfonyl fluoride (1 mM) and a
freshly added protease inhibitor cocktail (Calbiochem, La Jolla,
CA, USA) on ice for 30 min. The protein (50 mg) was then resolved
on a 10% polyacrylamide gel, transferred to a polyvinylidene
difluoride (PVDF) membrane and blocked for 1 h at room temperature
with 5% nonfat dried milk in PBS. The membranes were subsequently
incubated with primary antibodies (mouse anti-p-ERK1/2 at 1:1,000
or rabbit anti-β-actin at 1:2,000; Santa Cruz Biotechnology, Inc.)
in blocking buffer. The blots were washed for 3–10 min each with
PBS plus Tween-20 (0.1%) and then incubated with the appropriate
diluted horseradish peroxidase (HRP)-tagged secondary antibody
(1:1,000; Santa Cruz Biotechnology, Inc.) for 1 h at room
temperature. The blots were developed in accordance with the
manufacturer’s instructions using SuperSignal™ West Pico
Chemiluminescent HRP substrate (Pierce Protein Biology Products,
Thermo Fisher Scientific, Inc., Rockford, IL, USA), and visualized
following exposure to X-ray film. The band intensities were
calculated using the GelWorks 4.1 image analysis system (UVP
products, Upland, CA, USA).
Statistical analysis
Data are presented as the mean ± standard deviation
(SD), and SPSS version 18.0 statistical software (SPSS, Inc.,
Chicago, IL, USA) was used for the statistical analysis.
Significant differences between the groups were assessed using
one-way analysis of variance (ANOVA). P<0.05 was considered to
indicate a statistically significant difference.
Results
Effects of different concentrations of
VPAS on the phosphorylation of ERK1/2 following hippocampal
neuronal epileptiform discharge
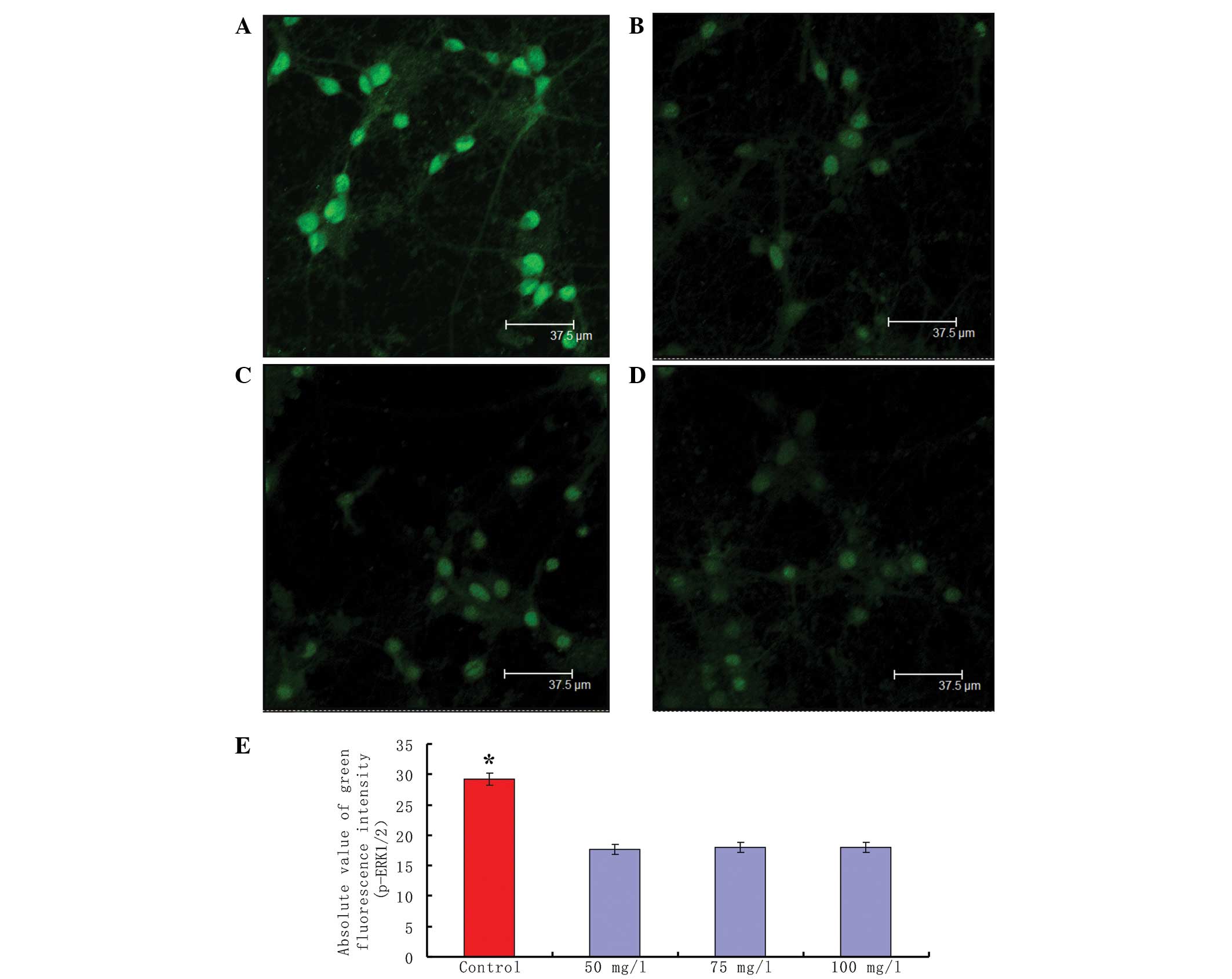
Significant differences were observed in the
expression of p-ERK1/2 between untreated and VPAS-treated
hippocampal neurons displaying epileptiform discharge, as assessed
using immunofluorescence labeling. In the control neurons, a high
intensity of green fluorescence, reflecting a high level of
p-ERK1/2, was observed in the nucleus and cytoplasm (Fig. 1A). However, decreased p-ERK1/2
green fluorescence intensity was apparent in the VPAS-treated
neurons. VPAS treatment 30 min prior to discharge at different
concentrations (50, 75 and 100 mg/l) significantly inhibited the
green fluorescence intensity of p-ERK1/2 in the hippocampal neurons
compared with that in the control group, (Fig. 1B–D). Moreover, the phosphorylation
level of ERK1/2 did not vary with the change in VPAS concentration.
A total of 20 neurons were randomly selected from each group and a
comparison of the absolute fluorescence intensity of p-ERK1/2
staining between the control and VPAS groups was performed.
Statistical analysis using the Student’s t-test revealed
significant differences in the fluorescence intensity of p-ERK1/2
staining between the untreated and VPAS-treated hippocampal neurons
(P<0.01; Fig. 1E).
Effects of VPAS on the phosphorylation of
ERK1/2 at different time-points following hippocampal neuronal
epileptiform discharge
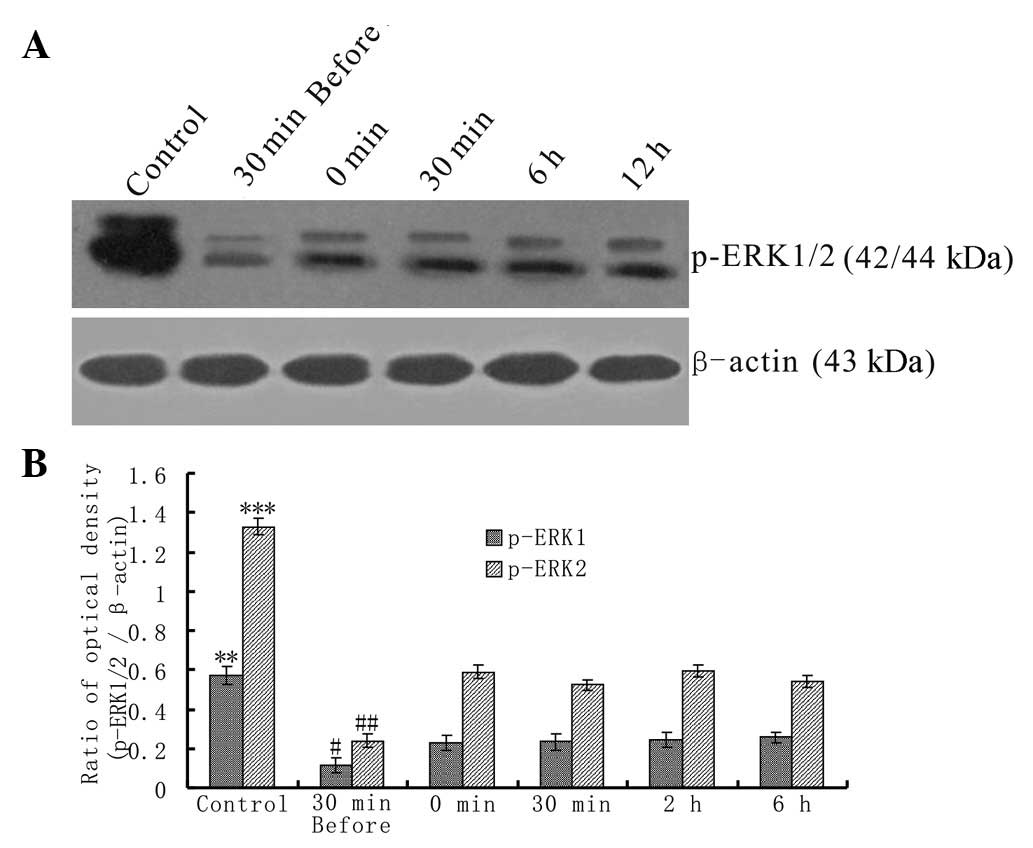
The protein levels of p-ERK1/2 in the untreated and
VPAS-treated neurons were examined using western blotting.
Measurements of p-ERK1/2 levels in untreated neurons were made
following the culture of the neurons with magnesium-free ACSF for 3
h (Fig. 2A, Control). The
expression level of p-ERK1/2 was significantly reduced at all
time-points in the VPAS-treated neurons (Fig. 2A, 30 min before, 0 min, 30 min, 6 h
and 12 h) compared with the expression level in the control group.
The lowest expression of p-ERK1/2 was observed at the ‘30 min
before’ time-point. The relative expression levels of p-ERK1/2 in
the two groups were divergent at all the examined time-points
(P<0.01).
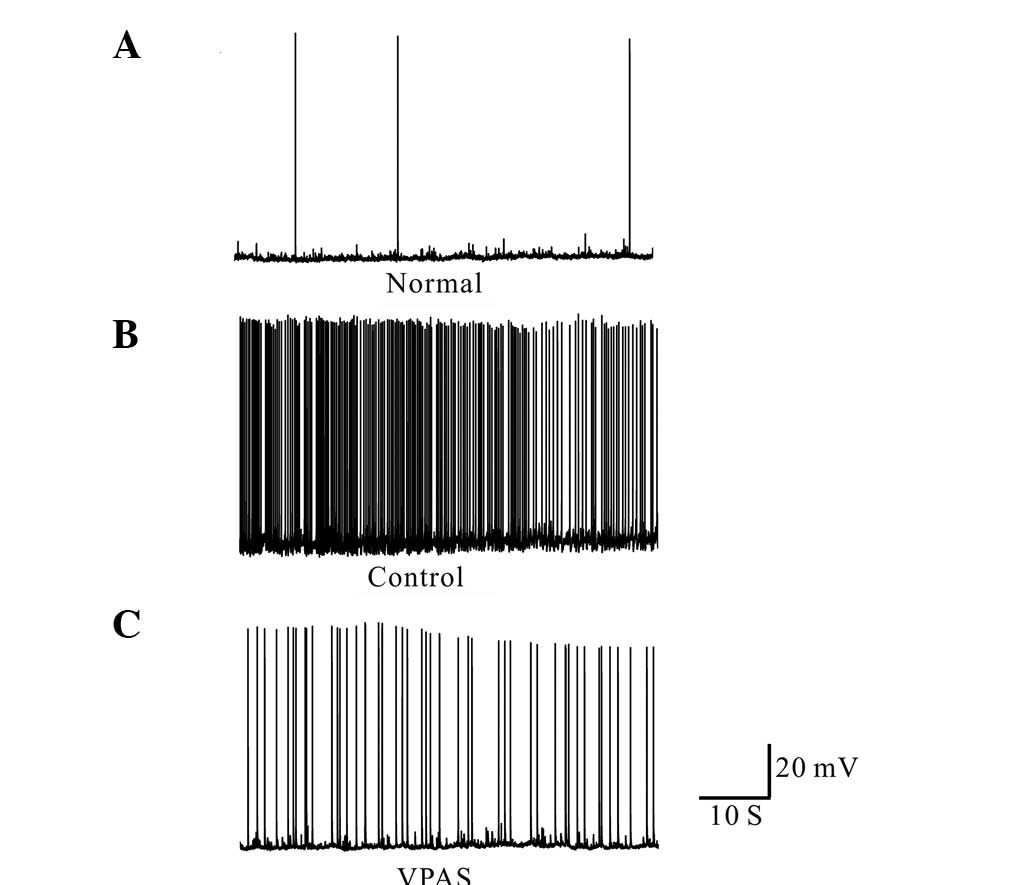
Effects of VPAS on action potential
frequency following hippocampal neuronal epileptiform
discharge
Current recordings from the normal neurons displayed
occasional action potentials, as shown in Fig. 3A (Normal). By contrast, culturing
the neurons in magnesium-free medium for 3 h resulted in the
development of continuous, high-frequency burst discharges that
evolved into recurrent epileptiform discharges. The spike frequency
of these neurons was >3 Hz. This epileptiform discharge was
continuous throughout the 60 min of recording (Fig. 3B). At 15 min following 50 mg/l VPAS
perfusion, the spike frequency was significantly decreased
(Fig. 3C).
Discussion
Treatment of rat neurons with three different
concentrations of VPAS 30 min prior to epileptiform discharge in
magnesium-free ACSF blocked the expression of p-ERK1/2. No
significant differences were identified among the phosphorylation
levels of the neurons treated with 50, 75 and 100 mg/l VPAS. This
result suggested that the effect of VPAS was not
concentration-dependent. Furthermore, following the incubation of
the neurons with 50 mg/l VPAS and the analysis of p-ERK1/2 levels
at five different time-points, it was observed that the lowest
expression of p-ERK1/2 was at ‘30 min before’ and the downward
trend was continued at 0 min, 30 min, 2 h and 6 h subsequent to
epileptiform discharge. It was observed that 50 mg/l VPAS was
capable of significantly decreasing the epileptiform activity. The
study therefore demonstrated that ERK1/2 signaling acted as a
downstream target of VPAS in the regulation of epilepsy.
MAPKs belong to a large family of proline-directed
serine-threonine protein kinases that are fundamental in cellular
functions. MAPKs include ERK1/2, ERK3/4, ERK5, ERK7/8, c-Jun
N-terminal kinase (JNK)-1/2/3 and p38 (α/β/γ/δ) MAPK (10–12).
The activation of MAPK proceeds through a cascade of upstream
molecules in an orderly fashion, with activation occurring at a low
level in certain MAPKs and particularly in ERK1/2, which is most
likely due to basal signaling and metabolic needs (13). The nature of the upstream molecules
depends on the stimulatory trigger, cell type and the subcellular
location of activation. The activation of MEK1/2 leads to the
phosphorylation of threonine and tyrosine residues of ERK1/2, with
recognition sites of Thr-Glu-Tyr (TEY). ERK1 and ERK2 are
homologous isoforms that share the same substrate specificities
in vitro. These two proteins, which phosphorylate a
multitude of protein substrates, have ~85% amino acid identity and
are expressed in almost all tissues with much greater identity in
the core regions (14). Studies
focusing on the extracellular signal transduction pathway are
likely to enhance the understanding of epilepsy with new theories
and technologies. Using in vitro and in vivo epilepsy
models, it has been demonstrated that ERK1/2 is involved in the
occurrence and development of epilepsy. The phosphorylation of
ERK1/2 was shown to be significantly enhanced in a kainic acid
mouse model (15). Furthermore, in
a pilocarpine-evoked model, ERK1/2 was rapidly activated,
particularly in the hippocampal dentate gyrus granule cells, and
the activation was completed prior to the seizure (8). In our previous study, it was observed
that there was marked expression of p-ERK1/2 in epileptic neurons,
and the level of expression was demonstrated to be greater than
that in normal neurons. Moreover, the phosphorylation level of
ERK1/2 peaked at 30 min following epileptiform discharge (6). The study showed that the expression
of p-ERK1/2 was marked in the neurons at the end of the 3-h
treatment with magnesium-free ACSF. In an epileptic model evoked by
the potassium channel inhibitor 4-aminopyridine, the peak
phosphorylation level of ERK1/2 was observed at 20 min.
Pretreatment with an inhibitor of ERK1/2 inhibited ERK1/2
phosphorylation and also blocked the epileptiform discharge
completely during the ictal period (16). In the fragile X syndrome mouse
model simulated using gene knockout technology, ERK1/2 was observed
to participate in group I metabotropic glutamate receptor (I
mGluR)-mediated epileptiform discharge (17). In addition, the expression of
p-ERK1/2 was shown to be notable in the temporal lobe and
hippocampus of patients with drug-resistant epilepsy (18). It has been shown that the
phosphorylation of EKR1/2 is one of the early cell responses in
seizures, and may therefore be regarded as a potential therapeutic
target to prevent chronic epilepsy (19).
VPAS, a broad-spectrum and first-line antiepileptic
drug, is capable of controlling most types of seizures, including
absence, myoclonic and generalized tonic-clonic seizures, as well
as status epilepticus. The primary antiepileptic pharmacological
effect of VPAS is to inhibit the GABA enzyme and succinic
semialdehyde dehydrogenase. In order to increase the concentration
of GABA in the brain, VPAS is also able to inhibit
N-methyl-D-aspartate (N-methyl-D-aspartic acid, NMDA)
receptor-mediated neuron depolarization, and to inhibit the
Ca2+ influx leading to K+ conduction
(20). The results of the present
study suggest that VPAS decreases the action potential frequency
induced by magnesium-free ACSF. However, its potential and multiple
mechanisms have not yet been fully elucidated. An investigation
into VPAS and the signal transduction pathway reported that VPAS
was capable of significantly reducing protein kinase C (PKC) and G
protein activity, with specificity for α and ɛ moieties (9). Tang et al(21) demonstrated that PKC small
interfering RNA (siRNA) completely inhibited acetylcholine-induced
mesenchymal stem cell migration by blocking ERK1/2 phosphorylation
(21). Furthermore, the PKC
pathway has been shown to protect LNCaP prostate cancer cells from
phorbol ester-induced apoptosis by promoting ERK1/2 (22). The results of the present study
indicate that the enhancement of ERK1/2 phosphorylation following
epileptiform discharge is significantly decreased by VPAS in
primary cultured hippocampal neurons. The results showed notable
timeliness, with a low-dose effective concentration of VPAS
inhibiting the phosphorylation of ERK1/2 at an earlier period of
neuronal epileptiform discharge. The negative regulatory mechanism
of VPAS on the signal transduction pathway has yet to be
elucidated. Studies have shown that brain-derived neurotrophic
factor (BDNF) is an activator of ERK1/2, and that its upregulation
may evoke excessive neuronal excitability and trigger mossy fiber
sprouting (23,24). Future studies of the specific
mechanisms are required.
In conclusion, the association between VPAS and the
cell signal transduction pathways is complex and diverse, and the
effect of VPAS on the level of ERK1/2 phosphorylation is only one
of numerous factors. The results of the present study have provided
a new experimental basis for further investigation into the
mechanism underlying the VPAS-induced suppression of seizure-onset.
This may facilitate the identification of a novel target for the
development of future anticonvulsant therapies.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81260201) and the Master of
Scientific Research Foundation of the Affiliated Hospital of Zunyi
Medical College (grant no. 209.001.097.14).
References
|
1
|
Thomas KL and Hunt SP: The regional
distribution of extracelluarly regulated kinase-1 and -2 messenger
RNA in the adult rat central nervous system. Neuroscience.
56:741–757. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li YQ, Xue T, Xu J, Xu ZC, Liu H and Chen
YM: ERK1/2 activation in reactive astrocytes of mice with
pilocarpine-induced status epilepticus. Neurol Res. 31:1108–1114.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Matsubayashi Y, Fukuda M and Nishida E:
Evidence for existence of a nuclear pore complex-mediated,
cytosol-independent pathway of nuclear translocation of ERK MAP
kinase in permeabilized cells. J Biol Chem. 276:41755–41760. 2001.
View Article : Google Scholar
|
|
4
|
Whitehurst AW, Wilsbacher JL, You Y,
Luby-Phelps K, Moore MS and Cobb MH: ERK2 enters the nucleus by a
carrier-independent mechanism. Proc Natl Acad Sci USA.
99:7496–7501. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kondoh K, Torii S and Nishida E: Control
of MAP kinase signaling to the nucleus. Chromosoma. 114:86–91.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu ZC, Chen YM, Xu P, Liu H, Xie YL and
Zeng KB: Epileptiform discharge upregulates p-ERK1/2,
growth-associated protein 43 and synaptophysin in cultured rat
hippocampal neurons. Seizure. 18:680–685. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hu B, Liu C, Bramlett H, Sick TJ, Alonso
OF, Chen S and Dietrich WD: Changes in trkB-ERK1/2-CREB/Elk-1
pathways in hippocampal mossy fiber organization after traumatic
brain injury. J Cereb Blood Flow Metab. 24:934–943. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Berkeley JL, Decker MJ and Levey AL: The
role of muscarinic acetylcholine receptor-mediated activation of
extracelluar signal-regulated kinase1/2 in pilocarpine-induced
seizures. J Neurochem. 82:192–201. 2002. View Article : Google Scholar
|
|
9
|
Bowden CL: New concepts in mood
stabilization: evidence for the effectiveness of valproate and
lamotrigine. Neuropsychopharmacology. 19:194–199. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Roux PP and Blenis J: ERK and p38
MAPK-activated protein kinases: a family of protein kinases with
diverse biological functions. Microbiol Mol Biol Rev. 68:320–344.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rincón M and Davis RJ: Regulation of the
immune response by stress-activated protein kinases. Immunol Rev.
228:212–224. 2009.PubMed/NCBI
|
|
12
|
Brown MD and Sacks DB: Protein scaffolds
in MAP kinase signalling. Cell Signal. 21:462–469. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Alam R and Gorska MM: Mitogen-activated
protein kinase signalling and ERK1/2 bistability in asthma. Clin
Exp Allergy. 41:149–159. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mebratu Y and Tesfaigzi Y: How ERK1/2
activation controls cell proliferation and cell death: Is
subcellular localization the answer? Cell Cycle. 8:1168–1175. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
de Lemos L, Junyent F, Verdaguer E, et al:
Differences in activation of ERK1/2 and p38 kinase in Jnk3 null
mice following KA treatment. J Neurochem. 114:1315–1322.
2010.PubMed/NCBI
|
|
16
|
Merlo D, Cifelli P, Cicconi S, Tancredi V
and Avoli M: 4-Aminopyridine-induced epileptogenesis depends on
activation of mitogen-activated protein kinase ERK. J Neurochem.
89:654–659. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chuang SC, Zhao W, Bauchwitz R, Yan Q,
Bianchi R and Wong RK: Prolonged epileptic form discharges induced
by altered group I metabotroptic glutamate receptor-mediated
synaptic responses in hippocampal slices of a fragile X mouse
model. J Neurosci. 25:8048–8055. 2005. View Article : Google Scholar
|
|
18
|
Xi ZQ, Wang XF, He RQ, et al:
Extracellular signal-regulated protein kinase in human intractable
epilepsy. Eur J Neurol. 14:865–872. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Di Maio R, Mastroberardino PG, Hu X,
Montero L and Greenamyre JT: Pilocapine alters NMDA receptor
expression and function in hippocampal neurons: NADPH oxidase and
ERK1/2 mechanisms. Neurobiol Dis. 42:482–495. 2011.PubMed/NCBI
|
|
20
|
Johannessen CU and Johannessen SI:
Valproate: Past, present, and future. CNS Drug Rev. 9:199–216.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tang JM, Yuan J, Li Q, et al:
Acetylcholine induces mesenchymal stem cell migration via
Ca2+/PKC/ERK1/2 signal pathway. J Cell Biochem.
113:2704–2713. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen J, Giridhar KV, Zhang L, Xu S and
Wang QJ: A protein kinase C/protein kinase D pathway protects LNCaP
prostate cancer cells from phorbol ester-induced apoptosis by
promoting ERK1/2 and NF-{kappa}B activities. Carcinogenesis.
32:1198–1206. 2011.PubMed/NCBI
|
|
23
|
Bittigau P, Sifringer M and Ikonomidou C:
Antiepileptic drugs and apoptosis in the developing brain. Ann NY
Acad Sci. 993:103–114. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Koyama R, Yamada MK, Fujisawa S,
Katoh-Semba R, Matsuki N and Ikegaya Y: Brain-derived neurotrophic
factor induces hyperexcitable reentrant circuits in the dentate
gyrus. J Neurosci. 24:7215–7224. 2004. View Article : Google Scholar : PubMed/NCBI
|