Introduction
Hearing loss is the most common human birth defect,
with an incidence of approximately one case among 1,000 newborns.
The American newborn hearing screening project began in 1964 and
has gradually spread globally. Newborn hearing screening has been
gradually implemented in large and middle-sized cities across China
and an increasing number of children with hearing loss are
diagnosed shortly after birth. The program has a profound effect on
the language development, communication, cognition, mental health
and career planning of the children if early intervention is
achieved (1).
With the launch of newborn hearing screening,
assessment of its effectiveness reveals its limitations. First, the
major screening target is permanent hearing loss of >35 dB,
which is not detected if the newborns have low-grade hearing loss.
Secondly, late-onset or progressive hearing loss is not detected
since newborn hearing is normal at birth. Cytomegalovirus
infection, Pendred syndrome, autosomal dominant nonsyndromic
hearing loss, recessive vestibular aqueduct expansion and
mitochondrial DNA (mtDNA) mutations (such as 12S rRNA gene
1555A>G and 1494C>T) also lead to undetected hearing loss at
birth, whereas late-onset hearing loss occurs later (2).
The development of molecular genetics has
demonstrated that 50% of nonsyndromic deafness has genetic factors,
which makes genetic testing a powerful weapon for screening
children with hearing loss (3). A
preliminary survey of Chinese domestic genetic epidemiology for
deafness showed that the GJB2, SLC26A4 and mitochondrial 12S rRNA
(MTRNR1) genes are common mutation hot spots in Chinese
nonsyndromic hearing loss (4).
Association detection of these three genes indicated that 26.65% of
the Northern Chinese population are prelingually deaf (5). Therefore, association detection of
GJB2, SLC26A4 and mitochondrial 12S rRNA was combined with hearing
screening to determine the common sites and frequencies of newborn
deafness gene mutation, which may be used to develop a more
effective and earlier intervention for hearing disorders.
Materials and methods
Subjects
One thousand newborns in the Handan Center Hospital
(Handan, China) between November 2010 and October 2011 were
studied. The GJB2, SLC26A4 and MTRNR1 genes were tested
simultaneously in 532 males and 468 females. This study was
conducted in accordance with the Declaration of Helsinki and with
approval from the Research Ethics Committee of the Central Hospital
of Handan City (Handan, China). Written informed consent was
obtained from all guardians/parents of the participants.
Hearing screening
Hearing screening was performed in the maternity
ward and the newborns were tested under quiet natural sleeping
conditions using ambient noise <30 dB. An AccuScreen Screening
Instrument (Madsen, Copenhagen, Denmark) was used for the hearing
test. At the third day after birth, every newborn was tested at
different frequencies and volumes using otoacoustic emissions
(OAEs). If the newborn failed the test, the OAE test was repeated
at 42 days after birth in a hearing diagnostic laboratory.
Genetic information and blood
samples
Approximately 2 ml of cord blood was collected from
each newborn and then stored in an EDTA-anticoagulated vacutainer
for gene screening (6). The
parents were asked to provide information, including names, age,
hospital number, home address, telephone number, pregnancy
information, family genetic history, neonatal gender, weight and
birth information.
Deafness gene screening
DNA was extracted from the cord blood to screen for
the GJB2, SLC26A4 and MTRNR1 genes. A MassARRAY system (Sequenom
Inc., San Diego, CA, USA) was used to screen for the deafness
mutation sites of GJB2 and SLC26A4 genes. The MTRNR1 gene was
screened for the 1449C>T and 1555A>G mutation sites through
direct sequencing.
Genomic DNA was extracted using a kit (Axygen
Biotechnology Co., Ltd., Silicon Valley, CA, USA) according to the
manufacturer’s instructions. A NanoDrop 1000 spectrophotometer
(Thermo Fisher Scientific Inc., Wilmington, DE, USA) was used to
detect the concentration and purity of the extracted DNA. Following
completion of the extraction process, a final concentration of 10
ng/μl genomic DNA was achieved from all samples in the 384-well
plates and preserved at −20°C.
Genetic typing of the GJB2 and SLC26A4
genes
The Assay Designer package (Sequenom Inc., San
Diego, CA, USA) was used to design the polymerase chain reaction
(PCR) primers and single-base extension primers for the deafness
loci on the GJB2 and SLC26A4 genes. The primer probe design was
strictly in accordance with the requirements of the MassARRAY
system and the probes were synthesized by Shanghai Invitrogen
Biotechnology Co., Ltd. (Shanghai, China). A 14-plex PCR
amplification reaction was performed in one well, and the detected
point mutations are shown in Table
I.
 | Table IFourteen SNPs used for genotyping. |
Table I
Fourteen SNPs used for genotyping.
| Gene name | Mutation site | rs |
|---|
| GJB2 | c.101T>C | rs35887622 |
| c.235delC | rs80338943 |
| c.592G>A | |
| c.427C>T | rs80338948 |
| SLC26A4 | IVS7-2A>G | rs111033313 |
| c.916-c.917insG | |
| c.754T>C | |
| c.281C>T | |
| IVS15+5G>A | |
| c.2027T>A | rs111033318 |
| c.2168A>G | rs121908362 |
| c.439A>G | |
| c.1226G>A | rs111033305 |
| c.589G>A | rs111033380 |
Genomic DNA (1 μl; 10 ng/μl) and 4 μl of PCR mixture
(Sequenom Inc.) were added into 384-well plates. The reaction
conditions were as follows: 95°C for 2 min; 95°C for 30 sec, 56°C
for 30 sec and 72°C for 1 min, for 45 cycles; then 72°C for 5
min.
Shrimp alkaline phosphatase (SAP; New England
Biolabs Inc., Beverly, MA, USA) reaction mixture (2 μl) was added
into 384-well plates for the SAP reaction. The reaction conditions
were as follows: 37°C for 40 min and 85°C for 5 min. After
completing the reaction, the mixture was cooled to room temperature
and stored at 4°C.
The extension reaction conditions were as follows:
94°C for 30 sec; 94°C for 5 sec, 52°C for 5 sec, 80°C for 5 sec,
for 40 cycles; 72°C for 3 min. The products were then preserved at
4°C.
Approximately 16 μl of deionized water and 6 mg of
resin were added to the 384-well plates for desalination and then
were analyzed with matrix-assisted laser desorption/ionization-time
of flight mass spectrometry (MALDI-TOF-MS). Final results were read
by the MassARRAY RT real-time sotware sustems. Genotype analyses
were completed by the MassARRAY Typer software.
Sanger sequencing of the MTRNR1 gene
The two mutation points of the MTRNR1 gene,
1494C>T and 1555A>G, were combined to design the primers. The
online software Primer3 (Whitehead Institute for Biomedical
Research, Cambridge, MA, USA) was used for designing the
mitochondrial amplification primers (http://bioinfo.ut.ee/primer3-0.4.0/primer3/. Accessed:
October 23, 2013). The following parameters were set to improve the
effectiveness and specificity of PCR amplification: the GC content
in the primers was set between 40 and 60%, the annealing
temperature was set between 55 and 60°C, the 3′-end of the primers
were generally set to end with G or C to improve the integration of
the primers into the template strand, and the amplified fragment
was generally set between 200 and 500 bp to facilitate sequencing.
The sequences of the mitochondrial primers were as follows: forward
primer, 5′-CAACCTCACCACCTCTTGCT-3′ and reverse,
5′-GTAAGGTGGAGTGGGTTTGG-3′. The fragment length was 497 bp. The
primers were synthesized by Shanghai Invitrogen Biotechnology Co.,
Ltd.
The PCR conditions were as follows: predenaturation
at 94°C of 5 min; 35 cycles of 94°C for 30 sec, annealing at 60°C
for 30 sec and extension at 72°C of 35 sec; and a final extension
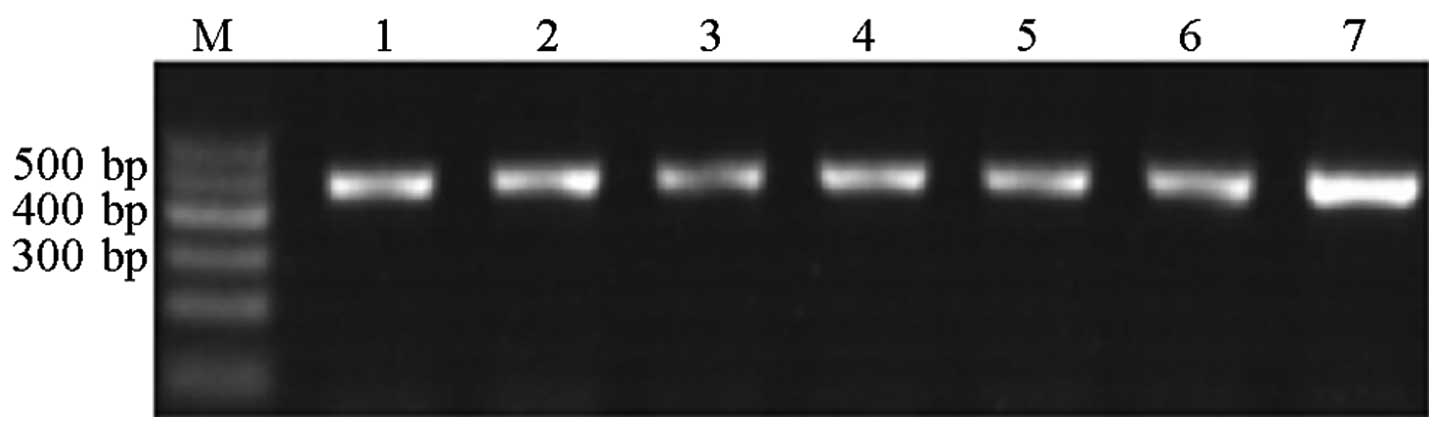
at 72°C for 5 min. Agarose gel electrophoresis (1.5% gel) was used
to verify the results of the PCR amplification. The bands obtained
were clearly visible and did not overlap (Fig. 1).
SAP mixture (1.5 μl) and the PCR product (1 μl) were
added to 384-well plates. The reaction conditions were as follows:
37°C for 60 min and 80°C for 20 min. After the completion of the
reaction, the reaction mixture was cooled to room temperature and
stored at 4°C.
BigDye Terminator (BDT; 0.5 μl) and a DNA sequencing
of the PCR-amplified product was performed bidirectionally on an
ABI3730XL automated sequencer (Applied Biosystems, Foaster City,
CA, USA) using the same primers. Sequence data were analyzed by
evaluating samples for alignment with the National Center for
Biotechnology Information reference (NCBI) sequence of MTRNR1
(NC_012920.1) using Sequencher Demo 3.0 and Mutation Surveyor Demo
V4.0.
Results
Summary of results
Among the 1,000 newborns tested, 996 cases were
rescreened. Only one of the 61 cases who failed the initial hearing
screening test also failed the single-ear rescreening test. All 61
cases were screened for the three genes, and only the infant who
failed the rescreening exhibited homozygous 427C>T mutation of
the GJB2 gene. All other subjects passed the genetic screening,
which indicated that no disease-causing mutation was present.
The two cases with the 1555A>G mutation and the
two cases with the 1494C>T mutation of MTRNR1 and the other 16
cases that carried pathogenic mutations in the GJB2 and SLC26A4
genes passed the newborn hearing screening test.
Hearing screening
In the initial screening of the hearing of 1,000
newborns, 939 (93.9%) of the newborns passed the initial OAE
screening, whereas 25 cases failed the left-ear hearing test and 21
cases failed the right-ear hearing test, for total of 46 (4.6%)
cases. Fifteen (1.5%) cases failed both the right- and the left-ear
hearing tests. A total of 61 (6.1%) newborns failed the initial
screening. At 42 days after birth, only one of the 61 cases who
failed the initial hearing screening also failed the single-ear
rescreening. Thus, one (0.1%) case did not pass OAE
rescreening.
Genetic screening
A total of 1,000 newborns were screened for
mutations in the GJB2, SLC26A4 and MTRNR1 genes. Ten cases
exhibited a heterozygous 235delC mutation of the GJB2 gene, and two
cases exhibited a homozygous 427C>T mutation. Four cases
exhibited heterozygous IVS7-2A>G mutation of the SLC26A4 gene
and one case exhibited heterozygous 1226G>A mutation. Two cases
exhibited homogeneous 1494C>T mutation of the MTRNR1 gene and
two cases exhibited homogeneous 1555A>G mutation. The overall
carrier rate was 2.1% (21/1,000). The specific carrier rates of the
three genes among the 1,000 cases were then analyzed.
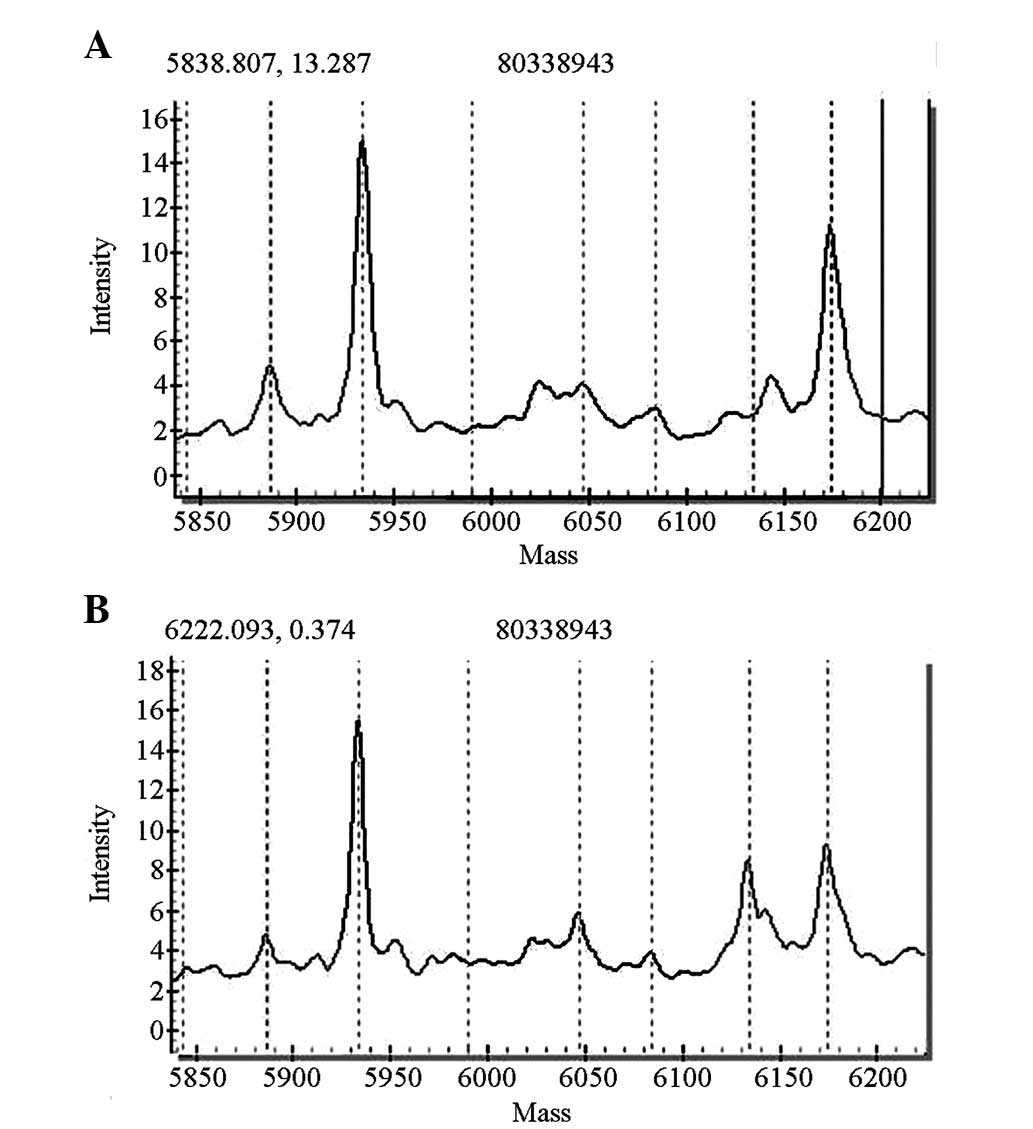
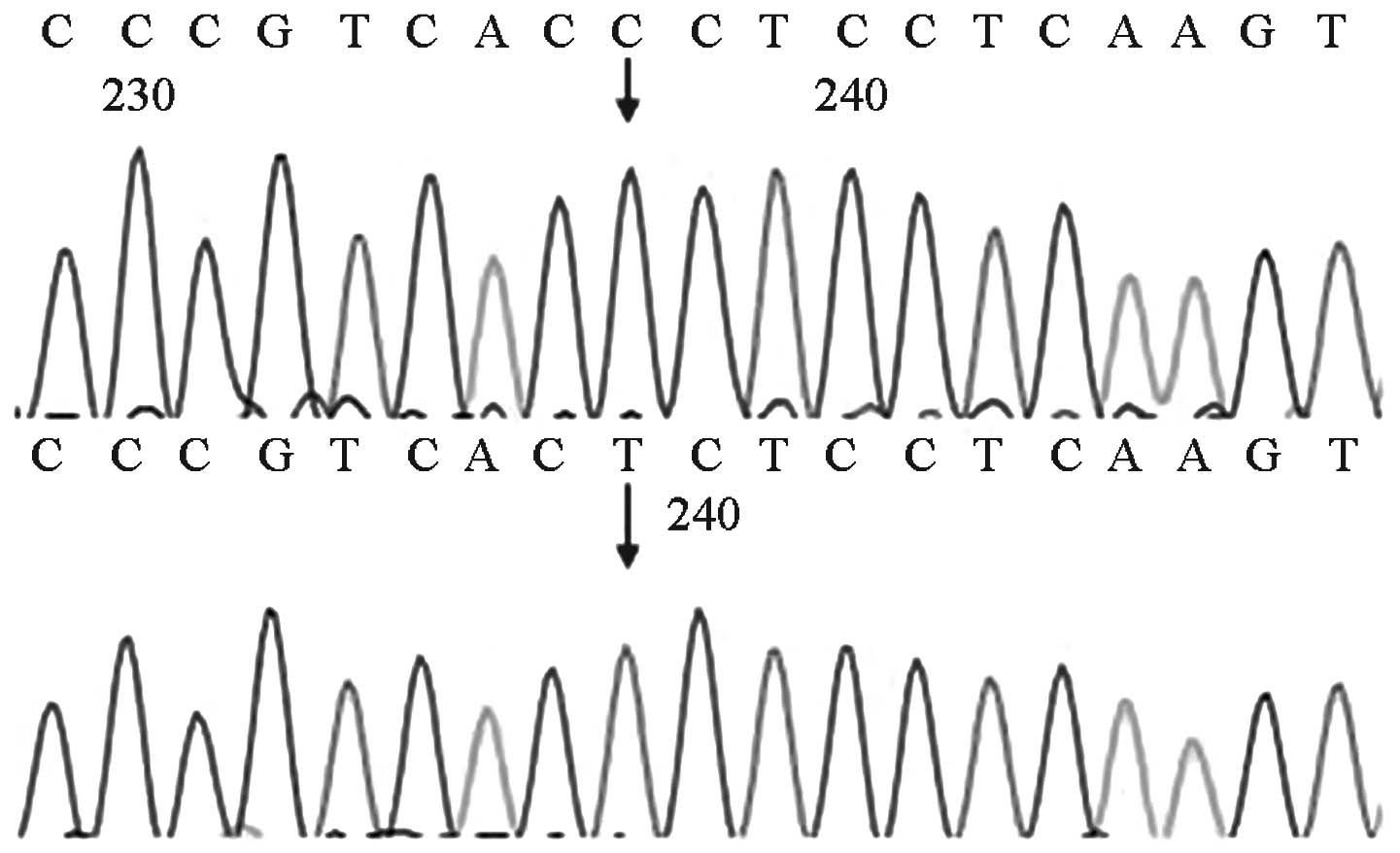
GJB2 gene screening indicated that 10 cases had
miscellaneous 235delC mutations and two cases had homozygous
427C>T mutations. The pathogenic carrier rate was 1.2%
(12/1,000), among whom 11 cases passed the initial hearing
screening test. One case did not pass the primary screening and the
rescreening, and was identified to carry a homozygous 427C>T
mutation (Figs. 2 and 3).
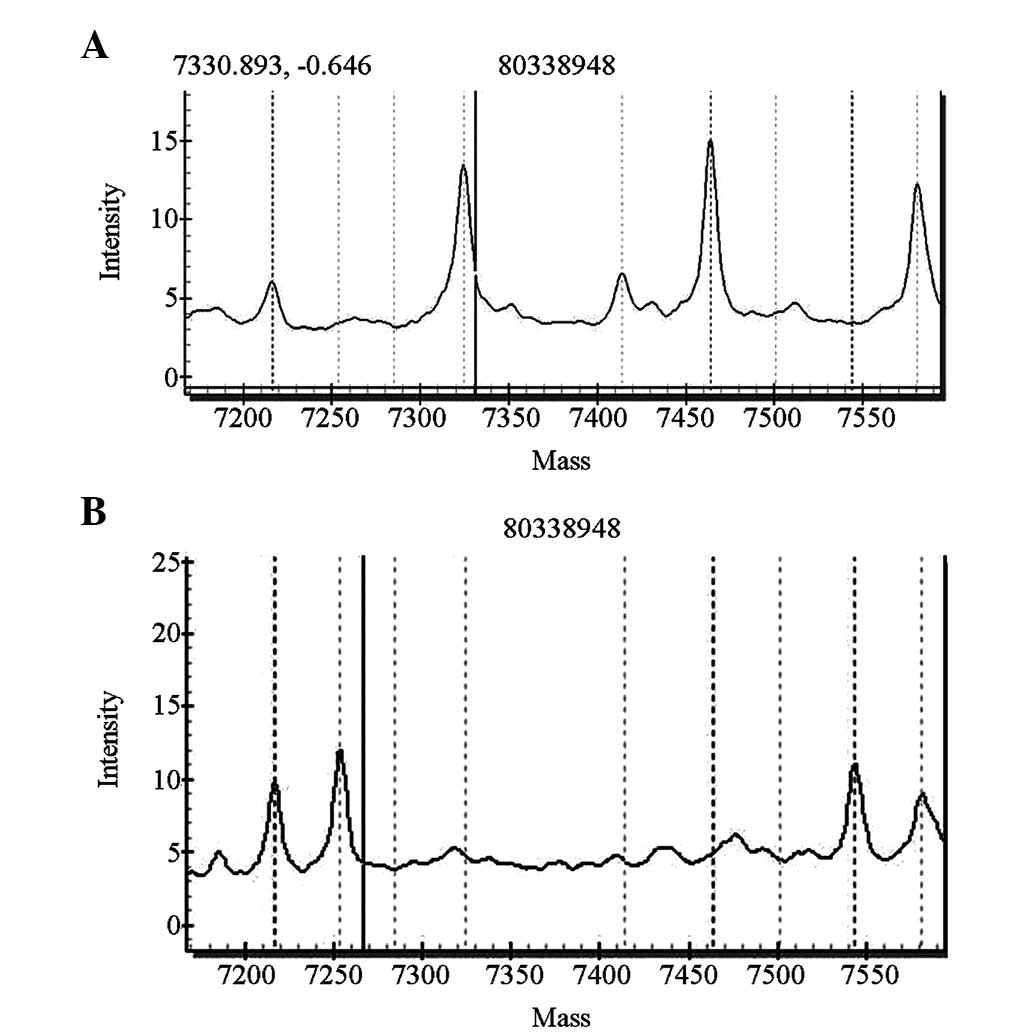
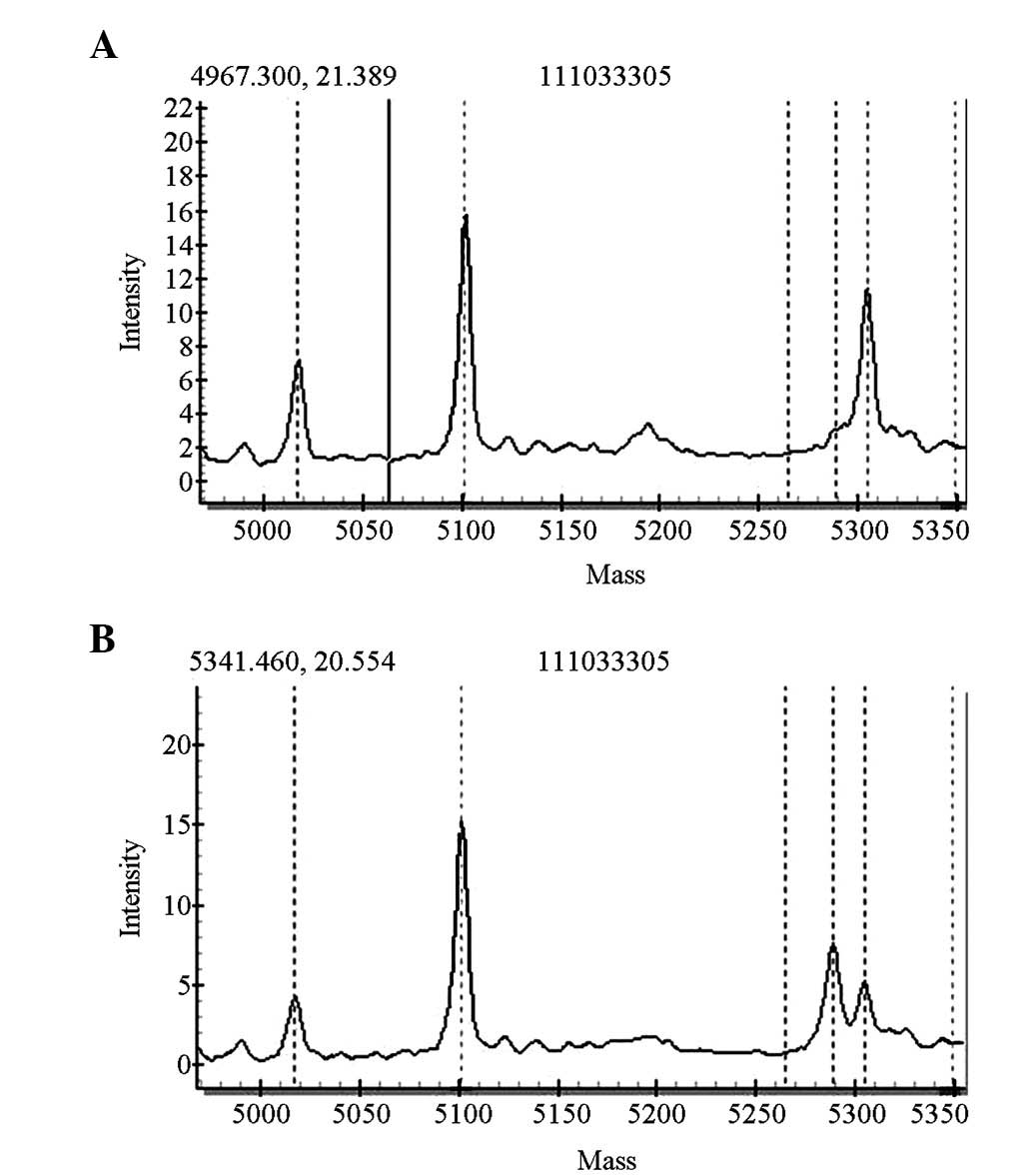
For the SLC26A4 gene screening, four cases carried
heterozygous IVS7-2A>G mutations of the SLC26A4 gene and one
case carried a heterozygous 1226G>A mutation. The pathogenic
carrier rate was 0.5% (5/1,000). The five newborns with these
mutations passed the initial hearing screening test. The
distributions of the mutations are shown in Figs. 4 and 5.
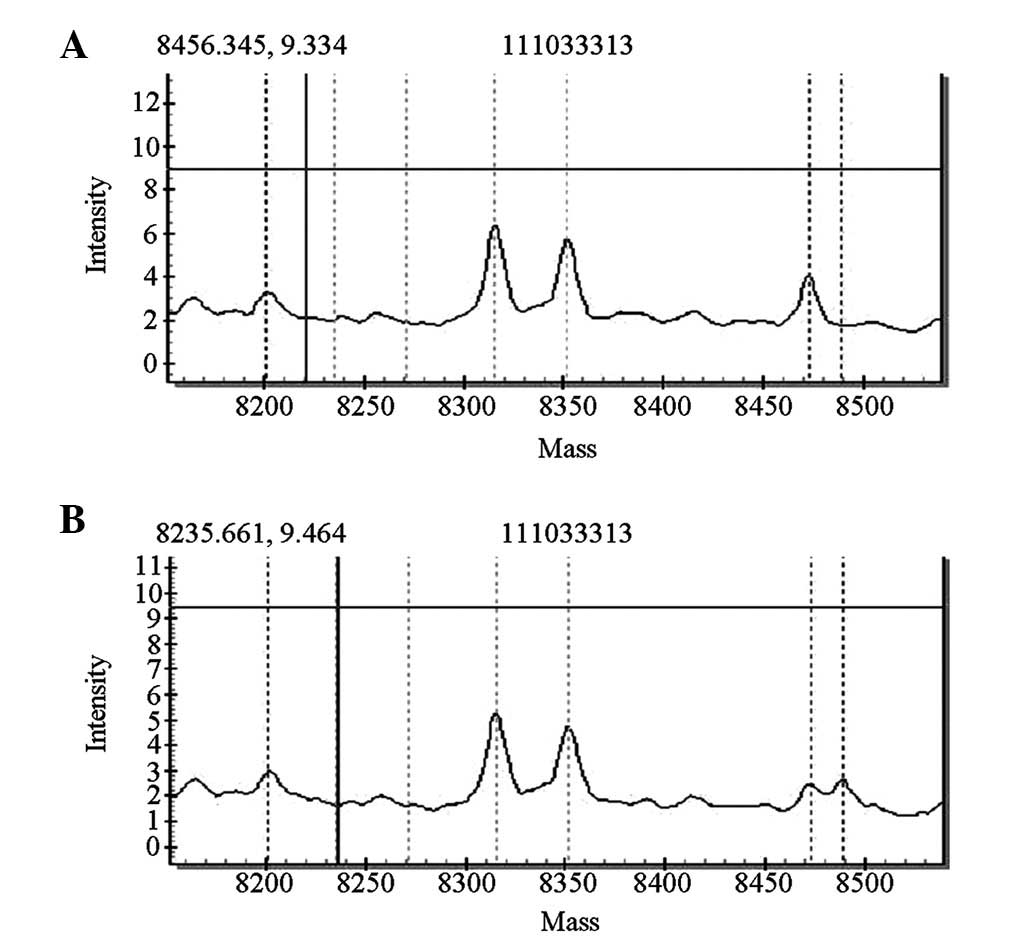
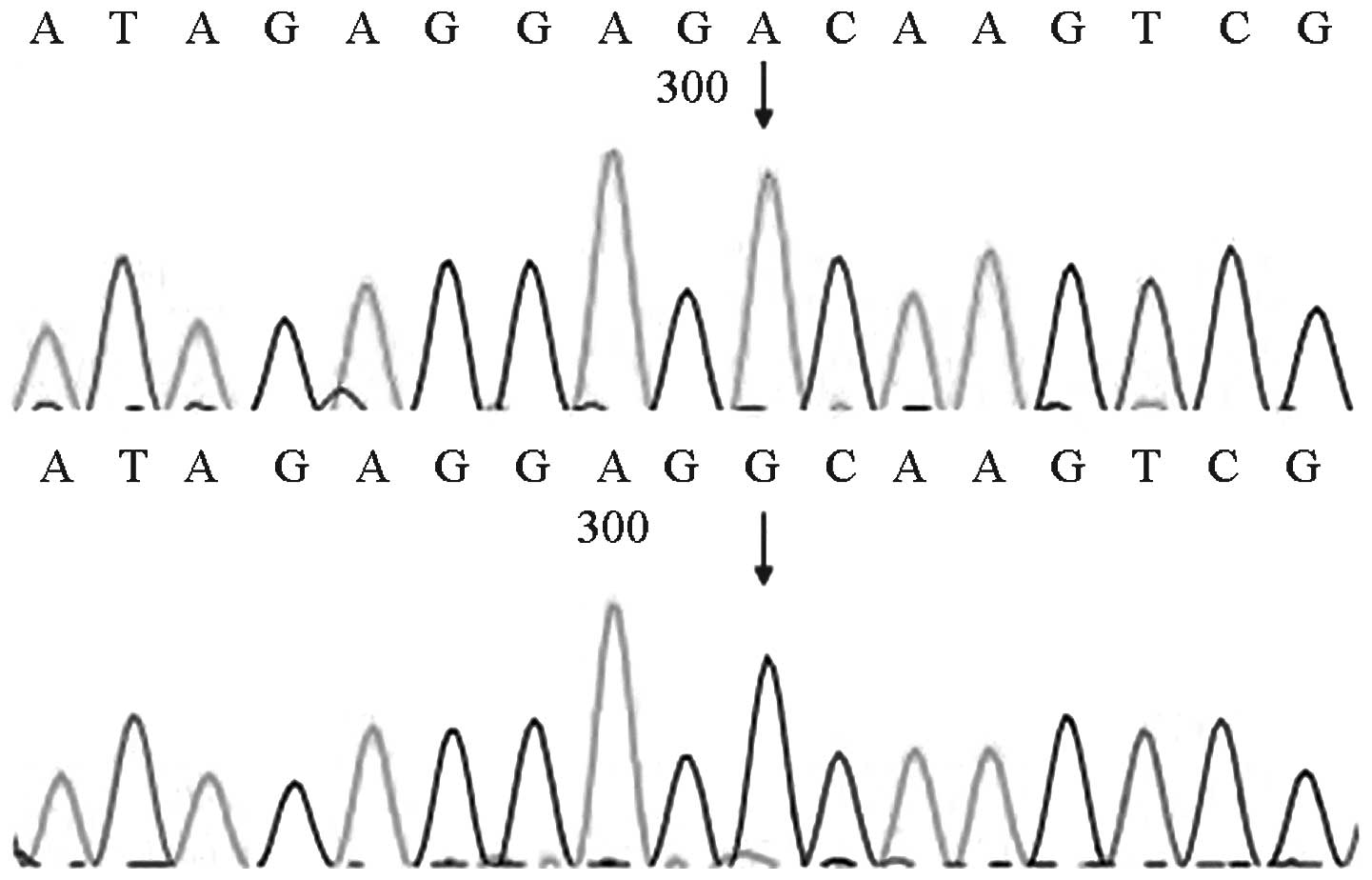
MTRNR1 genetic screening revealed that four of the
1,000 cases carried gene mutations [pathogenic carrier rate of 0.4%
(4/1,000)]. Two of the four cases exhibited homozygous 1494C>T
mutations, whereas the other two cases exhibited homozygous
1555A>G mutations. The mutations detected by the forward
sequencing were identified via reverse sequencing. The four cases
passed the initial hearing screening test. The forward sequencing
results are shown in Figs. 6 and
7.
Discussion
In the present study, a program for the simultaneous
screening of newborn hearing and genes was implemented, that is,
the hearing of the newborns was tested and GJB2, SLC26A4 and MTRNR1
genes were screened simultaneously. Of the 999 newborns that passed
the newborn hearing screening, 20 exhibited mutations in
deafness-associated genes. The carrier rate of disease-causing
mutations reached 2.1%, which is 20 times higher than the incidence
of congenital deafness (0.1%) from hearing screening. Notably,
these cases were not identified through hearing screening. Ten
cases exhibited a heterozygous mutation of GJB2 235delC (1%;
10/1,000) and all passed the hearing screening test. The
possibility of late-onset type hearing loss among these 10 cases is
significantly increased during development compared with that of
newborns with the normal gene. Four cases of IVS7-2A>G
heterozygous mutation were identified during SLC26A4 gene
screening. Individuals with this mutation should avoid strenuous
exercise, trauma and collision, and follow-up examinations should
be conducted at regular intervals. The aforementioned 14 newborns
should also avoid marriage with carriers of the same genotype since
their offspring would have a 25% chance of deafness. Two cases
exhibited homozygous 427C>T mutation of GJB2 and one of these
cases failed the hearing screening test in 1998. The 427C>T
mutation was first reported in the GJB2 gene, with the mutation
causing the amino acid at position 143 to change from arginine to
tryptophan, thereby causing autosomal recessive deafness (7). One case of heterozygous 1226G>A
mutation of the SLC26A4 gene was observed; this mutation was first
reported in 1998 (8). The arginine
at position 409 is changed into histidine in the vestibular
aqueduct expansion deafness phenotype, which is commonly associated
with autosomal recessive deafness (8). Temporal bone computed tomography (CT)
is recommended for individuals that carry the heterozygous mutation
in the SLC26A4 gene to detect whether vestibular aqueduct
enlargement has occurred, and regular follow-up examinations should
be performed. In the present study, four cases of MTRNR1 gene
mutations were also identified. Among them two cases had a
1555A>G mutation and two cases had a 1494C>T mutation. The
MTRNR1 pathogenic mutation carrier rate was 0.4% (4/1,000). These
four babies are extremely sensitive to aminoglycosides (including
streptomycin, neomycin, kanamycin and gentamicin) and any exposure
to these drugs is likely to lead to irreversible deafness. If the
babies are not exposed to these drugs, head trauma, exposure to
noisy environments and various infections that may cause hearing
loss, they may have normal hearing during their lifetime. Due to
the maternal transmission of mtDNA, these genetic test results may
be treated as an early warning for maternal family members of these
four cases. The carrier rate of pathogenic mutations in
mitochondrial genes in this region is much higher than in other
regions (9), which indicates the
requirement for gene screening among the newborns from this
region.
In the current newborn deafness gene screening
program, a high-flux method was used for detecting gene mutations,
namely, the MassARRAY system. The basic principle of the system is
based on MALDI-TOF-MS technology, combined with a highly specific
and sensitive chip technology. The system facilitates the research
and application of single-nucleotide polymorphism (SNP) genotyping,
gene expression, copy number variation, gene methylation analysis,
pathogen typing and prenatal diagnosis in one platform. The system
integrates the high sensitivity of PCR and high accuracy of mass
spectrometry (10). The
advantageous feature of the system is its ability to perform rapid
genotype identification with high accuracy and directly measure
target DNA with SNPs or mutations (11). The MassARRAY system is non-hybrid
dependent, free from potential hybrid mismatch interference, does
not require various biomarkers, and completes a large number of
loci detection and fully automatic analysis within a short time
through its high-density Spectro CHIP lattice chip analysis system.
We designed 14 reactions/well by selecting the mutation hot spots
of the Chinese deafness genes. The selection of the MassARRAY
system greatly reduced the screening cost, established a high-flux
genetic mutation detection method, and provided a new application
for clinical MassARRAY detection of deafness-associated
mutations.
The present study initially explored the
distribution of deafness-susceptibility genes in newborn hearing
screening and analyzed the conditions of newborn deafness gene
carriers. The results demonstrate the necessity and feasibility of
genetic screening for deafness in newborns. Newborn hearing
screening combined with deafness-susceptibility gene screening may
be a promising strategy for the early diagnosis of prelingual
hearing loss, for individuals with a high risk of delayed-type
deafness or deafness gene carriers (7). In the present study, the total gene
mutation carrier rate and the individual carrier rate of the three
genes were extremely high. Therefore, deafness-susceptibility gene
screening is important for identifying hereditary hearing loss.
However, the genetic screening results do not provide accurate
information regarding the hearing situation and prognosis of the
newborns (12) due to numerous
uncertainties and factors in the molecular detection results,
including gene polymorphisms and single-locus heterozygous
mutations. Therefore, hearing screening and genetic screening
should be considered together.
Acknowledgements
The authors would like to thank the patients who
participated in this study and the research teams of the Birth
Defect Research Center & Pathology Research Center, Fudan
University, Shanghai, China, who assisted with this research. This
work was supported by the Key Program of Handan (1113108017) and
the Science and Technology Support Program of Hebei
(11276102D).
References
|
1
|
Johnson JL, White KR, Widen JE, et al: A
multicenter evaluation of how many infants with permanent hearing
loss pass a two-stage otoacoustic emissions/automated auditory
brainstem response newborn hearing screening protocol. Pediatrics.
116:663–672. 2005. View Article : Google Scholar
|
|
2
|
Wang QJ: Newborn hearing loss genetic
screening. Journal of Audiology and Speech Pathology. 16:83–88.
2008.(In Chinese).
|
|
3
|
Smith RJ, Bale JF Jr and White KR:
Sensorineural hearing loss in children. Lancet. 365:879–890. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ouyang XM, Yan D, Yuan HJ, et al: The
genetic bases for non-syndromic hearing loss among Chinese. J Hum
Genet. 54:131–140. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Guo YF, Liu XW, Guan J, et al: GJB2,
SLC26A4 and mitochondrial DNA A1555G mutations in prelingual
deafness in Northern Chinese subjects. Acta Otolaryngologica.
128:297–303. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hwa HL, Ko TM, Hsu CJ, et al: Mutation
spectrum of the connexin 26 (GJB2) gene in Taiwanese patients with
prelingual deafness. Genet Med. 5:161–165. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yoshinaga-Itano C, Sedey AL, Coulter DK
and Mehl AL: Language of early- and later-identified children with
hearing loss. Pediatrics. 102:1161–1171. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Van Hauwe P, Everett LA, Coucke P, et al:
Two frequent missense mutations in Pendred syndrome. Hum Mol Genet.
7:1099–1104. 1998.PubMed/NCBI
|
|
9
|
Wu CC, Hung CC, Lin SY, et al: Newborn
genetic screening for hearing impairment: a preliminary study at a
tertiary center. PLoS One. 6:e223142011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Oeth P, del Mistro G, Marnellos G, et al:
Qualitative and quantitative genotying using single base primer
axtension coupled with matrix-assisted laser desorption/ionization
time-of-flight mass spectrometry (MassARRAY). Methods Mol Biol.
578:307–343. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Troxell ML, Levine J, Beadling C, et al:
High prevalence of PIK3CA/AKT pathway mutations in papillary
neoplasms of the breast. Mod Pathol. 23:27–37. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bae JW, Lee KY, Choi SY, Lee SH, Park HJ
and Kim UK: Molecular analysis of mitochondrial gene mutations in
Korean patients with nonsyndromic hearing loss. Int J Mol Med.
22:175–180. 2008.PubMed/NCBI
|