Introduction
Pulmonary fibrosis is a chronic, progressive and
lethal diffuse interstitial lung disease. It has re-emerged as a
focus of scientific study, due to its increasing incidence
(1). Transforming growth factor β1
(TGF-β1) is a profibrotic cytokine that has an important function
in pulmonary fibrosis. The c-Jun N-terminal kinase (JNK) signaling
pathway is a significant downstream kinase pathway in the TGF-β
signaling pathway (2,3,4). The
excessive activation of JNK is related to pulmonary fibrosis.
Thalidomide is an effective bidirectional immunomodulatory agent
that is able to suppress the generation of tumor necrosis factor-α
(TNF-α) and inhibit collagen synthesis, in addition to having
inhibitory effects on liver fibrosis and cirrhosis (5). However, whether thalidomide inhibits
pulmonary fibrosis through the TGF-β1/JNK signaling pathway has yet
to be elucidated. Furthermore, the mechanisms underlying the
effects of thalidomide in pulmonary fibrosis remain unclear. In the
present study, a bleomycin (BLM)-induced model of pulmonary
fibrosis was used to determine whether thalidomide acted to reduce
pulmonary fibrosis through the TGF-β1/JNK signaling pathway. The
effects of thalidomide on the model of pulmonary fibrosis were
observed using immunohistochemistry and western blotting.
Simultaneously, the mechanisms underlying the effects of
thalidomide were explored, in order to provide the basis for new
clinical treatments for pulmonary fibrosis.
Materials and methods
Animals
Ninety male Wistar rats were used in this study. The
study was carried out in strict accordance with the recommendations
in the Guide for the Care and Use of Laboratory Animals (Institute
of Laboratory Animal Resources Commission on Life Sciences,
National Academy Press, Washington DC, 1996). The animal-use
protocol was reviewed and approved by the Institutional Animal Care
and Use Committee (IACUC) of the First Hospital of Shanxi Medical
University (Taiyuan, China).
Model of pulmonary fibrosis
Male Wistar rats were injected intraperitoneally
with 3% chloral hydrate (1 ml/100 g). Following anesthetization,
BLM (0.3 ml, 5 mg/kg) was intratracheally administered to the rats.
The rats were placed in a vertical position and rotated for several
times in order to distribute the drug in the lung tissues
uniformly, and 0.3 ml dimethyl sulfoxide (Wuhan Boster Biological
Technology, Ltd., Wuhan, China) solution was injected
intraperitoneally on the same day.
Treatment groups
Ninety healthy male Wistar rats (200±20 g) were
randomly assigned into control (N), model (M), SP600125 (SP; a JNK
inhibitor), thalidomide (T) and SP600125 plus thalidomide (SP + T)
groups (n=18 per group). Pulmonary fibrosis models were established
in groups M, SP, T and SP + T by the intratracheal injection of 5
mg/kg bleomycin (BLM) on the first day, as described above, whereas
group N was injected with normal saline. The rats of groups T and
SP + T were treated with a gavage of thalidomide (100 mg/kg) in
saline (3 ml, 0.9%) once daily, whereas the rats in the other
groups were administered a gavage of the same volume of saline
without thalidomide. The rats of groups SP and SP + T were injected
intraperitoneally with SP600125 (15 mg/kg, dissolved in DMSO)
following BLM administration, whereas the rats in the other groups
received DMSO alone. Rats were randomly sacrificed by abdominal
aortic phlebotomy on days 7, 14 and 28 and their lung tissues were
collected for analysis. Pathological changes were examined under a
light microscope by hematoxylin and eosin (H&E) staining;
hydroxyproline (HYP) was detected in the lung tissues by alkaline
hydrolysis; and the expression levels of phosphorylated JNK (p-JNK)
protein and α-SMA were measured by immunohistochemical staining and
western blot analysis, as described below.
H&E staining
Lung tissues were fixed in 10% (w/v)
neutral-buffered formalin for 24 h, dehydrated in a graded ethanol
series, and embedded in paraffin. Sequential 6-μm sections of the
lungs were placed on slides and stained with H&E for
morphological analysis using a standard protocol. The slides were
then investigated under a light microscope.
HYP assay
The collagen content in the lung homogenates was
examined by HYP assay using a HYP detection kit from Nanjing
Jiancheng Bioengineering Institute (Nanjing, China). All steps of
the HYP assay were performed according to the manufacturer’s
instructions. The absorbance of each sample at 550 nm was read with
a microplate reader (Thermo Fisher Scientific, Waltham, MA,
USA).
Immunohistochemical assay
The lung tissues from the rats were fixed in 4%
paraformaldehyde, dehydrated, embedded in paraffin and sectioned.
The sections were incubated overnight at 4°C with a 1:200 dilution
of mouse anti-mouse α-smooth muscle actin (α-SMA) monoclonal
antibody (Wuhan Boster Biological Technology, Ltd.) or a 1:1,000
dilution of rabbit anti-mouse p-JNK monoclonal antibody (Cell
Signaling Technology, Inc., Danvers, MA, USA). This procedure was
followed by incubation with goat anti-mouse/goat anti-rabbit
secondary antibody (Wuhan Boster Biological Technology, Ltd.) for
20 min at 37°C, and with avidin-biotin-conjugated horseradish
peroxidase following the manufacturer’s instructions (Wuhan Boster
Biological Technology, Ltd.). Samples were stained with hematoxylin
for 30 sec subsequent to being fixed by dehydration. The
immunohistochemical staining results were studied by computer image
analysis (Image-Pro Plus 6.0; Photometrics, Tucson, AZ, USA). The
positively stained gray value was determined and this was used as
the mean value for statistical analysis. A low average gray value
and a deep color indicated a high protein content.
Western blot analysis
Lung tissues were homogenized in ice-cold
radioimmunoprecipitation lysis buffer (Wuhan Boster Biological
Technology, Ltd.). After centrifugation at 12,000 × g for 10 min at
4°C, the supernatant was collected, and the protein concentration
was determined using a bicinchoninic acid protein assay kit (Boster
Company, Wuhan, China). Proteins (30 μg) were separated by sodium
dodecyl sulfate polyacrylamide gel electrophoresis, and transferred
to polyvinylidene fluoride membranes. The blotted membranes were
blocked with 5% bovine serum albumin in Tris-buffered saline with
0.1% Tween-20 (TBS-T), and incubated at 4°C overnight with a 1:200
dilution of mouse anti-mouse α-SMA monoclonal antibody (Wuhan
Boster Biological Technology, Ltd.) or 1:1,000 dilution of rabbit
anti-mouse p-JNK monoclonal antibody (Cell Signaling Technology,
Inc.), or anti-β-actin antibodies (Wuhan Boster Biological
Technology, Ltd.). After rinsing five times with TBS-T at 5-min
intervals, the membranes were incubated for 2 h at 4°C a 1:5,000
dilution of horseradish peroxidase-labeled goat anti-mouse/goat
anti-rabbit secondary antibody (BOSTER Company, Wuhan, China).
Immunodetection was performed with enhanced chemiluminescence
detection reagents (Applygen Technologies Inc., Beijing, China) and
a chemiluminescence gel imaging system (FluorChem HD2;
ProteinSimple, Santa Clara, CA, USA), with β-actin as the internal
control.
Statistical analysis
Results are presented as the mean ± standard error
the mean. The groups were compared using one-way analysis of
variance (ANOVA). P<0.05 was considered to indicate a
statistically significant difference.
Results
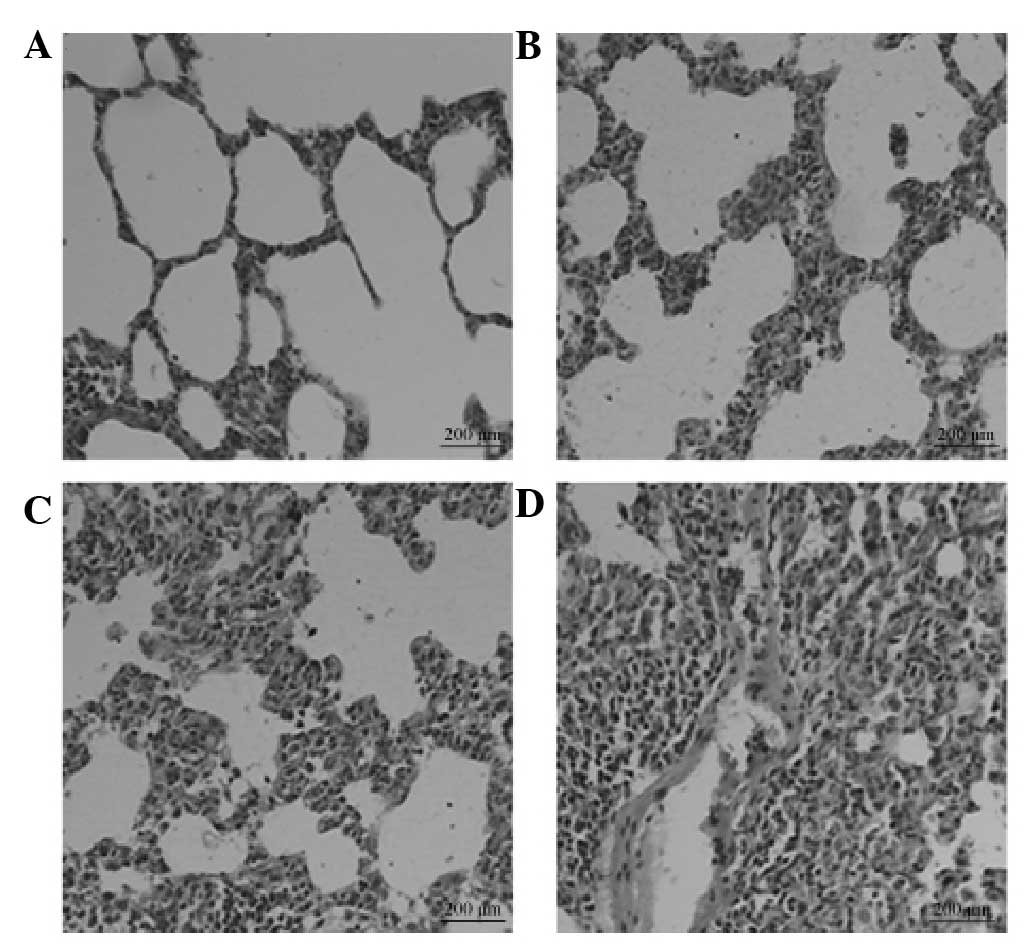
Pathological changes
In group M, alveolitis was most severe on day 7 and
then eased on day 14. However, marked pulmonary fibrosis was
observed in this group on day 28 (Fig.
1). The degree of fibrosis in groups SP, T and SP + T was
attenuated compared with that in group M, with the SP + T group
exhibiting the most marked improvement.
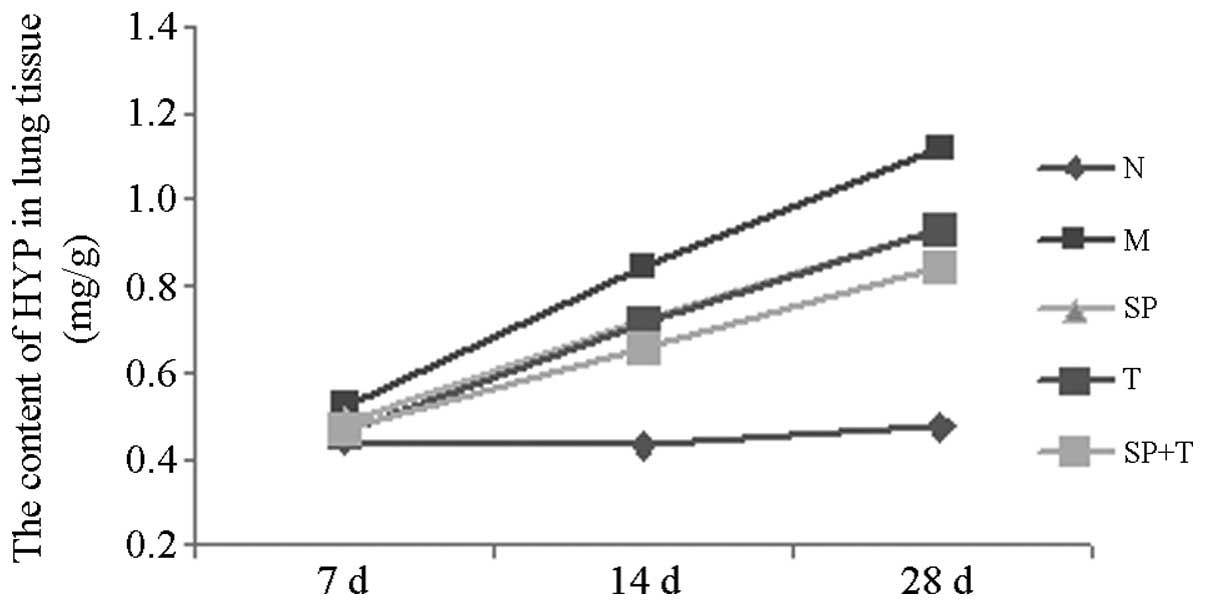
Hydroxyproline (HYP) levels
No significant differences were observed among the
HYP levels in group N on days 7, 14 and 28. The HYP level increased
gradually with time and peaked on day 28 in groups M, SP, T and SP
+ T. On days 14 and 28, the HYP levels in groups M, SP, T and SP +
T were significantly higher than that in group N (P<0.05);
however, the levels in groups SP, T and SP + T were significantly
lower than that in group M (P<0.05). On day 28, the HYP levels
were significantly lower in the group SP + T than those in the
other BLM-treated groups (P<0.05; Table I, Fig.
2).
 | Table IHydroxyproline (HYP) levels. |
Table I
Hydroxyproline (HYP) levels.
| | HYP (mg/g) |
|---|
| |
|
|---|
| Group | No. | 7 days (n=6) | 14 days (n=6) | 28 days (n=6) |
|---|
| N | 18 | 0.442±0.036 | 0.427±0.049 | 0.476±0.030 |
| M | 18 | 0.523±0.045a | 0.847±0.086a | 1.120±0.081a |
| SP | 18 | 0.485±0.062 | 0.725±0.071a,b | 0.931±0.070a,b |
| T | 18 | 0.463±0.059 | 0.713±0.079a,b | 0.934±0.080a,b |
| SP+T | 18 | 0.467±0.050 | 0.659±0.074a,b | 0.846±0.066a–d |
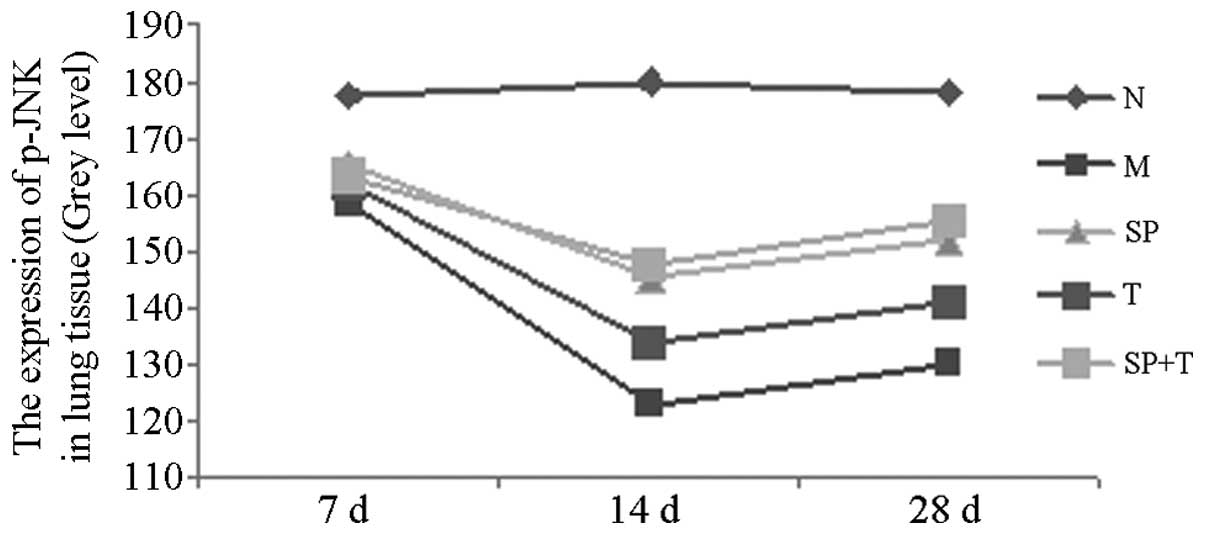
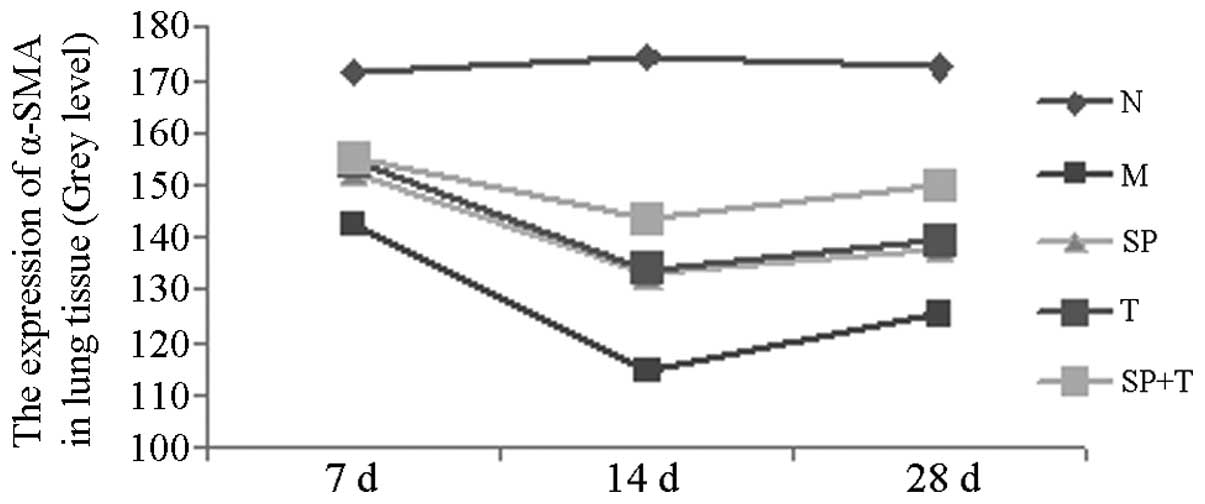
Immunohistochemical staining
The protein expression levels of phosphorylated JNK
(p-JNK) and α-smooth muscle actin (α-SMA) in group M were
significantly higher than those in group N (indicated by a low
average gray value), with the most marked difference on day 14
(P<0.05). The protein expression levels of p-JNK and α-SMA in
groups SP, T and SP + T were significantly lower than those in
group M, with the most notable differences on day 14 (P<0.05).
The expression level of α-SMA in the SP + T group was lower than
those in groups SP and T on days 14 and 28 (P<0.05). In
addition, the expression of p-JNK protein in group T was
significantly higher than those in groups SP and SP + T on days 14
and 28 (P<0.05; Tables II and
III, Figs. 3 and 4).
| Table IIExpression of phosphorylated c-Jun
N-terminal kinase (p-JNK). |
Table II
Expression of phosphorylated c-Jun
N-terminal kinase (p-JNK).
| | p-JNK (gray
level) |
|---|
| |
|
|---|
| Group | No. | 7 days (n=6) | 14 days (n=6) | 28 days (n=6) |
|---|
| N | 18 | 177.69±7.06 | 180.05±8.12 | 177.89±9.43 |
| M | 18 | 158.34±4.33a | 123.03±8.29a | 130.09±8.15a |
| SP | 18 | 165.26±6.45a | 145.44±9.44a,b | 152.04±7.85a,b |
| T | 18 | 161.91±7.18a | 133.91±7.96a–d | 141.09±10.26a–d |
| SP+T | 18 | 163.52±6.73a | 147.81±9.28a,b | 155.40±7.62a,b |
| Table IIIExpression of α-smooth muscle actin
(α-SMA). |
Table III
Expression of α-smooth muscle actin
(α-SMA).
| | α-SMA (gray
level) |
|---|
| |
|
|---|
| Group | No. | 7 days (n=6) | 14 days (n=6) | 28 days (n=6) |
|---|
| N | 18 | 171.59±7.06 | 174.28±7.93 | 172.46±10.03 |
| M | 18 | 142.74±6.78a | 114.27±8.06a | 125.65±6.69a |
| SP | 18 | 152.31±7.31a,b | 132.81±7.29a,b | 138.11±8.31a,b |
| T | 18 | 154.42±9.35a,b | 134.00±7.53a,b | 139.30±7.46a,b |
| SP+T | 18 | 155.16±7.29a,b | 143.54±7.35a–d | 149.94±9.56a–d |
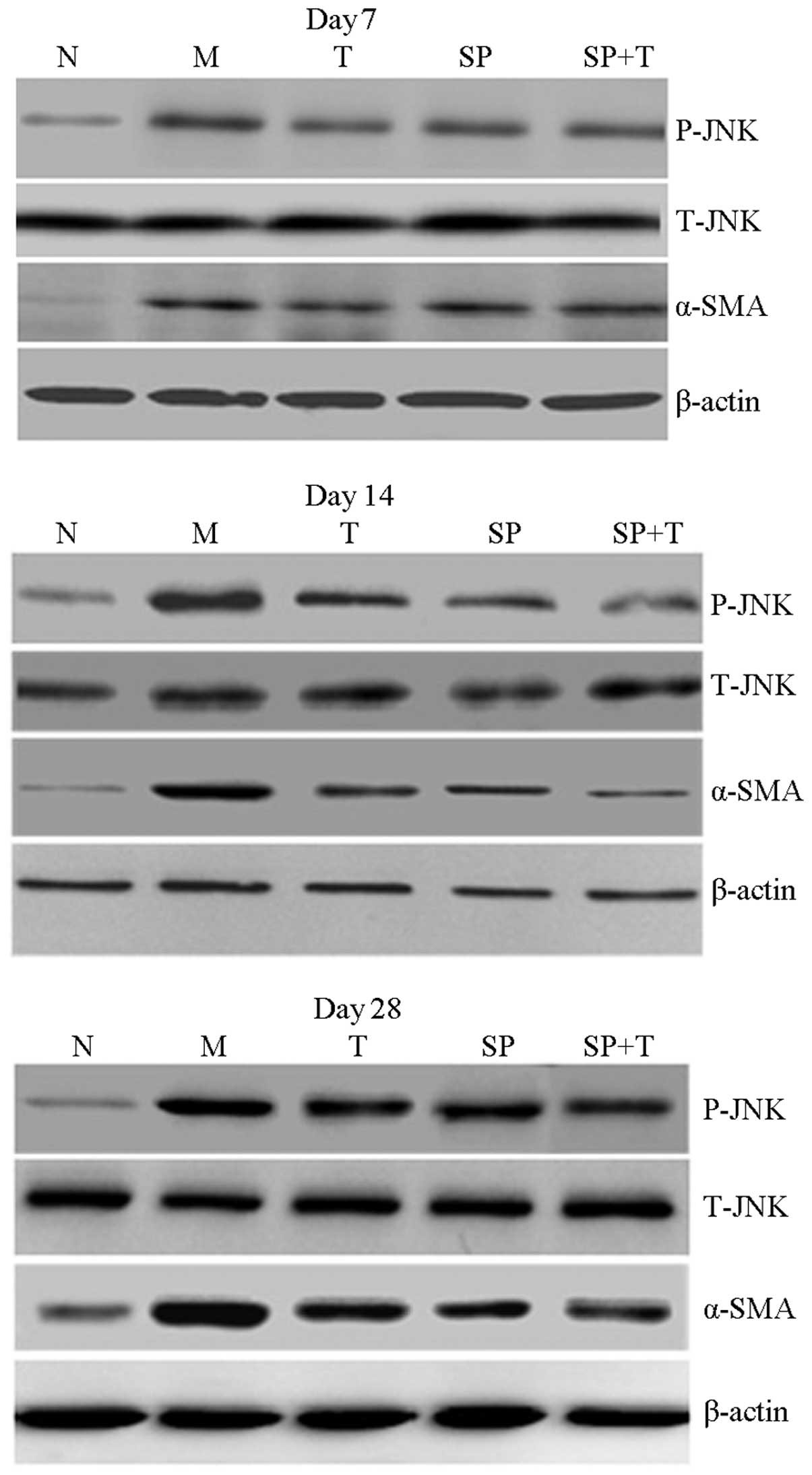
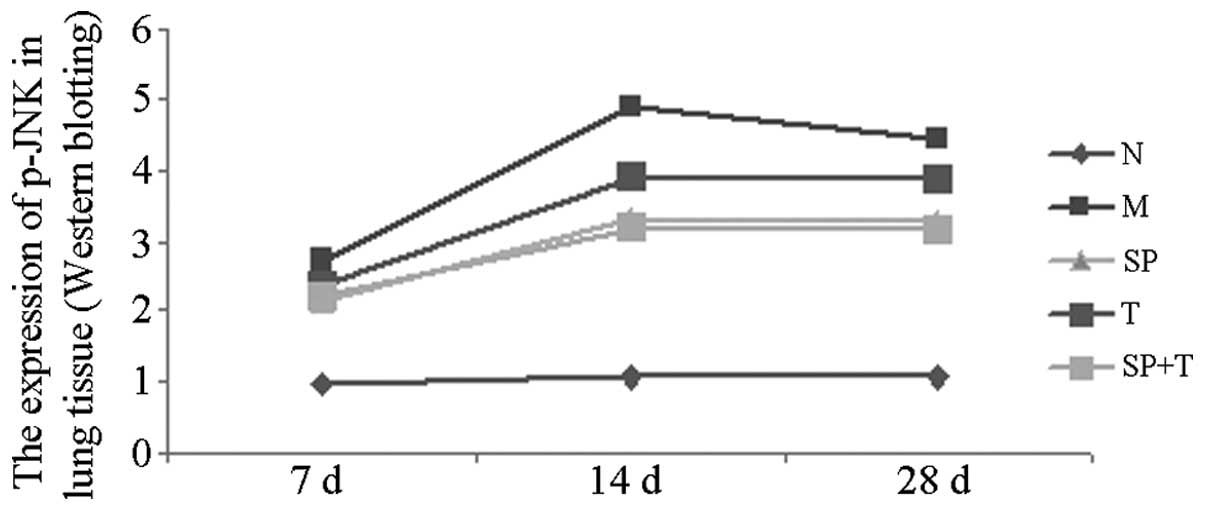
Western blot analysis
The protein expression levels of p-JNK and α-SMA in
group M were significantly higher than those in group N, with the
most marked difference on day 14 (P<0.05). In addition, the
protein expression levels of p-JNK and α-SMA in groups SP, T and SP
+ T were significantly lower than those in group M and higher than
those in group N (P<0.05). The expression of α-SMA in the SP + T
group was lower than that in groups SP and T on days 14 and 28
(P<0.05), whereas the expression level of p-JNK protein in group
T was significantly higher than that in groups SP and SP + T on
days 14 and 28 (P<0.05). A significant positive correlation was
observed between p-JNK protein and α-SMA levels in group M
(r=0.858, P<0.05; Figs.
5–7).
Discussion
Pulmonary fibrosis is a chronic, progressive and
lethal diffuse interstitial lung disease that involves pulmonary
interstitial substances, pulmonary alveoli and/or bronchioles.
Pulmonary fibrosis is characterized by fibroblast proliferation and
deposition of extracellular matrix materials. Effective therapeutic
methods for the disease exist. The disease has high morbidity and
mortality rates, with a five-year mortality rate of >50%. To
improve the quality of life among patients with pulmonary fibrosis,
the pathogenesis and treatment of the disease were investigated in
the present study. Numerous studies have shown that thalidomide
exhibits anti-inflammatory and immunomodulatory properties and
exerts beneficial effects in myelofibrosis (6–8),
hepatic fibrosis, renal fibrosis and pulmonary fibrosis.
Furthermore, it has been demonstrated that thalidomide inhibits the
production of inflammatory cytokines, including TNF-α, TGF-β1 and
nuclear factor-κB (NF-κB), thereby reducing the degree of pulmonary
fibrosis. However, the mechanisms underlying the effects of
thalidomide in pulmonary fibrosis remain unclear. TGF-β1 is a
profibrotic cytokine that has an important function in pulmonary
fibrosis. The JNK signaling pathway is an important downstream
signaling pathway of TGF-β1 (4).
It has been indicated that the excessive activation of JNK is
associated with pulmonary fibrosis (9,10).
However, whether thalidomide acts through the TGF-β1/JNK signal
transduction pathway remains unclear.
In recent years, the number of studies investigating
signaling pathways has increased. It has been shown that TGF-β1 is
an important fibrosis factor that is able to promote the
transformation of epithelial cells to mesenchymal cells, induce
lung fibroblasts to differentiate into muscle cells, upregulate the
expression of α-SMA and collagen fibers and inhibit myofibroblast
apoptosis (11). JNK is involved
in an important signaling pathway downstream of TGF-β1 (10). Three kinds of JNK proteins exist,
JNK1, JNK2 and JNK3; JNK1 and JNK2 are widely distributed in
tissues, whereas JNK3 is distributed in the heart, brain and testes
(13). In general, JNK
predominantly exists in the cytoplasm. JNK is phosphorylated and
activated when stimulated by upstream signals. The translocation of
JNK into the nucleus and the activation of the nuclear
transcription factor c-Jun enhance transcriptional activity. JNK is
also able to activate the transcription factors activator protein-1
and Elk-1, further increasing the transcription of specific genes,
proliferation, differentiation and apoptosis, which are involved in
the regulation of many cellular activities. Hashimoto et al
(9) demonstrated that the JNK
signaling pathway was important in the differentiation of
fibroblasts to myofibroblasts in the human lungs. Furthermore, the
absence of the JNK1 gene may prevent pulmonary fibrosis in rats
(10). SP600125 is a benzothiazole
derivative and a specific inhibitor of JNK, with mechanisms
involving reversing the ATP-competitive inhibitor and blocking JNK
(14). In our previous
experiments, administering SP600125 to Wistar rats with pulmonary
fibrosis inhibited JNK (15). The
reduced expression levels of α-SMA and collagen deposition in the
lung tissues of rats with pulmonary fibrosis inhibited the
differentiation of fibroblasts to myofibroblasts and decreased the
degree of pulmonary fibrosis. Thus, the TGF-1/JNK signaling pathway
has an important function in pulmonary fibrosis.
The results of the present study showed that only a
low level of p-JNK protein expression was apparent in group N at
each time point. However, in group M, p-JNK protein expression
increased on day 7, reached its peak on day 14 and then decreased
on day 28. The expression of a large quantity of α-SMA resulted in
the substantial phosphorylation of JNK protein and activation of
pulmonary fibrotic activity. Thus, the p-JNK protein may have an
important function in the development of pulmonary fibrosis.
Thalidomide is a derivative of glutamic acid that
has been used as a two-way immune regulator with anti-inflammatory,
immunoregulatory and anti-angiogenic effects (16). Arai et al (17) demonstrated that thalidomide was
able to reduce the levels of the vascular endothelial growth
factor, TGF-β1 and α-SMA to prevent the occurrence of peritoneal
fibrosis in rats. Thalidomide has also been shown to inhibit the
TGF-β1-induced differentiation of lung fibroblasts into
myofibroblasts and the synthesis of α-SMA and collagen.
Furthermore, Chong et al (18) revealed that thalidomide
downregulated the expression of inflammatory factors in animal
experiments and reduced the degree of liver fibrosis, while Ye
et al (19) demonstrated
that thalidomide reduced levels of interleukin-8 and TNF. Thus, it
has been indicated that thalidomide has therapeutic potential in
the treatment of pulmonary fibrosis. Our previous study (20) showed that thalidomide inhibits the
over-expression of type I collagen in pulmonary fibrosis rats via
inhibition the JNK signaling pathway. Choe et al (5) demonstrated that thalidomide acted
against liver fibrosis by inhibiting the TGF-β1/extracellular
signal-regulated kinase 1/2 signal pathway. Our previous study
(21) further indicated that
thalidomide was able to downregulate the levels of TNF-α and TGF-β
in the lungs of rats, and exhibit an attenuating effect on
pulmonary fibrosis. In addition, Horton and Hallowell (22) proposed that thalidomide had
potential as a drug for the treatment of idiopathic pulmonary
fibrosis, due to its immune regulation and anti-angiogenic effects.
A randomized trial conducted by the Johns Hopkins University showed
that thalidomide was able to improve coughing in patients with
idiopathic pulmonary fibrosis and enhance their quality of life
(23). However, whether
thalidomide has an important function in the occurrence and
development of pulmonary fibrosis by the TGF-β1/JNK signaling
pathway has yet to be elucidated. In the present study, using
thalidomide and SP600125, a blocker of the JNK signaling pathway,
to target pulmonary fibrosis in rats, the protein expression levels
of p-JNK and α-SMA were observed in different groups to determine
the mechanism by which thalidomide acted in pulmonary fibrosis.
The results of hematoxylin and eosin staining showed
that the degree of fibrosis in the lung tissues of each rat was
reduced in the model of pulmonary fibrosis following thalidomide
injection. The HYP content in the lung tissues in group M was low
on days 14 and 28, although higher than that in group N
(P<0.05). Immunohistochemistry and western blot analysis showed
that although the expression of α-SMA in group N lung tissues was
high, it was lower than that in group M on days 14 and 28
(P<0.05). Thus, thalidomide reduced pulmonary fibrosis in
rats.
The results of the immunohistochemistry and western
blot analysis showed that p-JNK and α-SMA protein expression levels
increased on day 7, peaked on day 14 and then declined on day 28 in
the BLM-treated rats. The protein expression levels of p-JNK and
α-SMA in group T were lower on days 14 and 28 than those in group
M; however, they were higher than those of group N (P<0.05).
Levels in groups SP and SP + T increased significantly compared
with group T (P<0.05). Thalidomide reduced the activation of
p-JNK and α-SMA protein expression; however, this effect was
attenuated by the specific JNK inhibitor SP600125. Thus,
thalidomide is indicated to act via the JNK signaling pathway. JNK
has a key function in pulmonary fibrosis; however, it is not the
only mechanism.
In conclusion, thalidomide is able to inhibit the
JNK signaling pathway in vivo. This pathway has a key
function in the treatment of pulmonary fibrosis. Given that the
pathogenesis of pulmonary fibrosis is complicated, further studies
are required to investigate the pathogenesis and treatment of
pulmonary fibrosis.
Acknowledgements
The authors would like to thank Liu Xuejun, Qian Li
and Hao Xiaoyan for their technological guidance in this study.
References
|
1
|
Collard HR and Pantilat SZ: Dyspnea in
interstitial lung disease. Curr Opin Support Palliat Care.
2:100–104. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hou N, Torii S, Saito N, Hosaka M and
Takeuchi T: Reactive oxygen species-mediated pancreatic beta-cell
death is regulated by interactions between stress-activated protein
kinases, p38 and c-Jun N-terminal kinase, and mitogen-activated
protein kinase phosphatases. Endocrinology. 149:1654–1665. 2008.
View Article : Google Scholar
|
|
3
|
Horton MR, Santopietro V, Mathew L, et al:
Thalidomide for the treatment of cough in idiopathic pulmonary
fibrosis: a randomized trial. Ann Intern Med. 157:398–406. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Smith JL, Schaffner AE, Hofmeister JK, et
al: ets-2 is a target for an akt (Protein kinase B)/jun N-terminal
kinase signaling pathway in macrophages of motheaten-viable mutant
mice. Mol Cell Biol. 20:8026–8034. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Choe JY, Jung HJ, Park KY, et al:
Anti-fibrotic effect of thalidomide through inhibiting
TGF-beta-induced ERK1/2 pathways in bleomycin-induced lung fibrosis
in mice. Inflamm Res. 59:177–188. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
D’Amato RJ, Loughnan MS, Flynn E and
Folkman J: Thalidomide is an inhibitor of angiogenesis. Proc Natl
Acad Sci USA. 91:4082–4085. 1994.
|
|
7
|
Moreira AL, Sampaio EP, Zmuidzinas A,
Frindt P, Smith KA and Kaplan G: Thalidomide exerts its inhibitory
action on tumor necrosis factor alpha by enhancing mRNA
degradation. J Exp Med. 177:1675–1680. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Koch HP: Thalidomide and congeners as
anti-inflammatory agents. Prog Med Chem. 22:165–242. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hashimoto S, Gon Y, Takeshita I, Matsumoto
K, Maruoka S and Horie T: Transforming growth Factor-betal induces
phenotypic modulation of human lung fibroblasts to myofibroblast
through a c-Jun-NH2- terminal kinase-dependent pathway. Am J Respir
Crit Care Med. 163:152–157. 2001. View Article : Google Scholar
|
|
10
|
Alcorn JF, Guala AS, van der Velden J, et
al: Jun N-terminal kinase 1 regulates epithelial-to-mesenchymal
transition induced by TGF-beta1. J Cell Sci. 121:1036–1045. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kapoun AM, Gaspar NJ, Wang Y, et al:
Transforming growth factor-beta receptor type 1 (TGFbetaRI) kinase
activity but not p38 activation is required for TGF betaRI-induced
myofibroblast differentiation and profibrotic gene expression. Mol
Pharmacol. 70:518–531. 2006. View Article : Google Scholar
|
|
12
|
Bogoyevitch MA, Boehm I, Oakley A,
Ketterman AJ and Barr RK: Targeting the JNK MAPK cascade for
inhibition: basic science and therapeutic potential. Biochim
Biophys Acta. 1697:89–101. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu J and Lin A: Role of JNK activation
apoptosis: a double-edged sword. Cell Res. 15:36–42. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bennett BL, Sasaki DT, Murray BW, et al:
SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase.
Proc Natl Acad Sci USA. 98:13681–13686. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang C-X and Liu X-J: The expression and
significance of p-JNK protein and α-SMA in rats with pulmonary
fibrosis. Chinese Remedies & Clinics. 12:36–38. 2012.(In
Chinese).
|
|
16
|
Joglekar S and Levin M: The promise of
thalidomide: evolving indications. Drugs Today (Barc). 40:197–204.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Arai H, Furusu A, Nishino T, et al:
Thalidomide prevents the progression of peritoneal fibrosis in
mice. Acta Histochem Cytochem. 44:51–60. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chong LW, Hsu YC, Chiu YT, Yang KC and
Huang YT: Anti-fibrotic effects of thalidomide on hepatic stellate
cells and dimethylnitrosamine-intoxicated rats. J Biomed Sci.
13:403–418. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ye Q, Chen B, Tong Z, et al: Thalidomide
reduces IL-18, IL-8 and TNF-alpha release from alveolar macrophages
in interstitial lung disease. Eur Respir J. 28:824–831. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu X, Qian L, Nan H, et al: Thalidomide
inhibits the over-expression of type I collagen in pulmonary
fibrosis rats via inhibition the JNK signaling pathway. Chinese
Journal of Geriatrics. (In Press).
|
|
21
|
Luo X and Liu X-J: Thalidomide suppresses
bleomycin-induced pulmonary fibrosis by down-regulating expressions
of TGF-β1 and TNF -α in rats. Chinese Remedies & Clinics.
10:1346–1349. 2010.(In Chinese).
|
|
22
|
Horton MR and Hallowell RW: Revisiting
thalidomide: fighting with caution against idiopathic pulmonary
fibrosis. Drugs Today (Barc). 48:661–671. 2012.PubMed/NCBI
|
|
23
|
Horton MR, Santopietro V, Mathew L, et al:
Thalidomide for the treatment of cough in idiopathic pulmonary
fibrosis: a randomized trial. Ann Intern Med. 157:398–406. 2012.
View Article : Google Scholar : PubMed/NCBI
|