Introduction
S-1 is widely administered to gastric cancer
patients and consists of tegafur (FT), gimeracil (CDHP) and
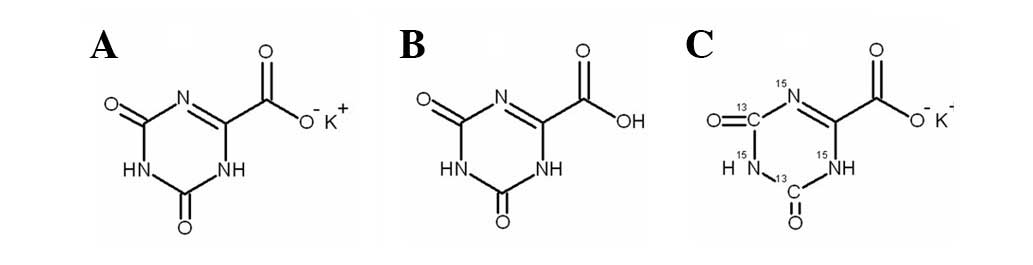
potassium oxonate (Oxo;
1,2,3,4-etetrahydro-2,4-dioxo-1,3,5-triazin-6-carboxylate) in a
molar ratio of 1:0.4:1 (1–7). FT is converted to 5-fluorouracil
(5-FU), which has an anticancer role in vivo. However, 5-FU
has some disadvantages, including a short half-life in vivo,
which causes gastrointestinal tract and bone marrow toxicity. CDHP
and Oxo act as modulators, which have no anticancer activity when
used singly. CDHP competitively inhibits dihydropyrimidine
dehydrogenase to maintain a high plasma concentration of 5-FU
(8). Oxo relieves the
gastrointestinal tract toxicity induced by 5-FU (9). CDHP and Oxo facilitate the function
of 5-FU, which leads to improved therapeutic effects (10). Oxo is converted into oxonic acid
(Fig. 1) in vivo.
To date, there are several methods reported in the
literature to determine Oxo, including enzyme immunoassay (11), GC-NICI-MS (gas
chromatography-negative ion chemical ionization mass spectrometry)
(12,13) and LC-MS/MS (liquid
chromatography-tandem mass spectrometry) (14–17).
The enzyme immunoassay method is not suitable for analysis in large
batches. The GC-NICI-MS method has been shown to be unstable when
used clinically. The LC-MS/MS method is reported to require long
and complex pre-processing for derivatization. Therefore, a simple
and novel method is required. In this study, a novel HPLC-MS/MS
method was developed and successfully applied to a pharmacokinetic
study, after single oral administration of a combination of FT,
CDHP and Oxo to 12 tumor patients. This method was determined to be
simple, rapid and stable for use in the quantitation of Oxo in
human plasma.
Materials and methods
Chemicals and reagents
Potassium oxonate (purity, 99.60%) was supplied by
the National Institute for the Control of Pharmaceutical and
Biological Products (Beijing, China).
[13C2,15N3]-Oxo
(purity, 99.8%) was purchased from Toronto Research Chemicals Inc.
(Toronto, ON, Canada). Methanol, acetonitrile and formic acid (LC
grade) were purchased from Dima Technology Inc. (Richmond Hill, ON,
Canada). The other chemicals (analytical grade) were supplied by
Beijing Chemical Co. (Beijing, China). Deionized H2O was
prepared using a Milli-Q H2O purifying system, purchased
from Millipore Corporation (Bedford, MA, USA). Blank (drug-free)
human plasma was obtained from healthy subjects.
Preparation of stock and working
solutions
The stock solutions were prepared separately in
methanol-H2O [1:1, v/v; 400.0 μg/ml for Oxo and
40.0 μg/ml for the internal standard (IS)]. Routine daily
calibration curves were prepared in drug-free plasma. Appropriate
volumes of stock solutions and drug-free human plasma were added to
each test tube to prepare working solutions at concentrations of
2.0, 5.0, 10.0, 25.0, 50.0, 100.0 and 200.0 ng/ml. Contemporary
quality control samples, which were run in each assay, were also
prepared at concentrations of 5.0, 25.0 and 160.0 ng/ml. Working
solutions of IS (250.0 ng/ml) were obtained by diluting the stock
solution with methano-H2O (1:1, v/v). The standard
solutions and IS were stored at −20°C.
HPLC-MS/MS analysis
HPLC was performed on a Shimadzu HPLC system
consisting of a LC-20AD binary pump, a DGU-20A3 degasser, a SIL-20A
autosampler and a CTO-10ASVP column oven
(Shimadzu Corporation, Kyoto, Japan). Chromatographic separation
was achieved on a Waters:Atlantis dC18 column (150x4.6
mm, 5.0 μm, Waters, Milford, MA, USA) maintained at 20°C in
the column oven. The signal acquisition and peak integration were
performed by Analyst 1.4.2 software (Applied Biosystems, Foster
City, CA, USA). The mobile phase was H2O containing 0.1%
formic acid-acetonitrile (90:10, v/v), with a flow rate of 1.2
ml/min (70% shunting). Detection was carried out on an API 3200
MS/MS System (Applied Biosystems) equipped with an electrospray
ionization (ESI) source in the negative ionization mode. Multiple
reaction monitoring (MRM) at unit resolution was employed to
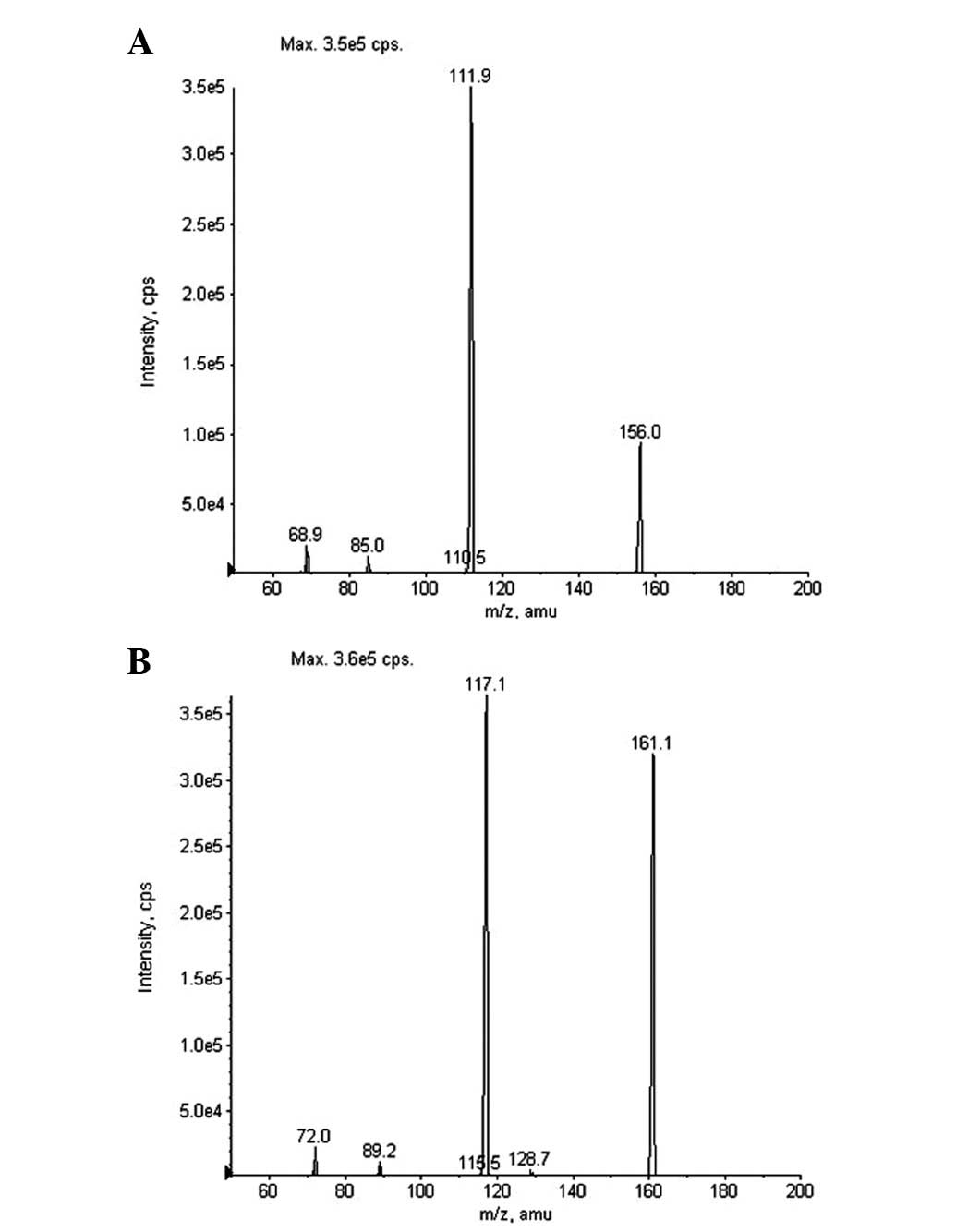
monitor the transitions of Oxo at m/z 156.0→111.9 and IS at m/z
161.1→117.1. The operating parameters of ESI-mass spectrometry
(ESI-MS) were as follows: curtain gas, 15 psi; gas 1, 40 psi; gas 2
(nitrogen), 80 psi; dwell time, 150 msec; ion spray voltage, −4.5
kV; ion source temperature, 550°C; declustering potential (DP), −20
V; collision energy (CE), −11 V.
Sample preparation
Briefly, 50 μl IS working solution and 200
μl deionized H2O were added to a 50-μl
aliquot of human plasma in a 1.5-ml Eppendorf tube. The mixture was
vortex-mixed for 30 sec at 20°C. The SPE column was activated by
the elution of 350 μl methanol and 350 μl
H2O. Plasma samples were loaded to the SPE column and
the column was eluted by 350 μl H2O and 350
μl methanol. Subsequently, 25 μl of the mixture of
methanol and formic acid (50:50, v/v) was applied to extract Oxo
twice. The eluted solution was vortex-mixed with 50 μl
H2O. Finally, 5 μl of the previously mentioned
solutions was injected and analyzed using HPLC-MS/MS.
Pharmacokinetic study
Plasma concentrations of Oxo in 12 tumor patients
were determined up to 48 h following single oral administration of
a 40-mg S-1 capsule. Blood samples were drawn at various
time-points (0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 24 and 48 h)
after ingestion of S-1. This clinical pharmacokinetic study was
approved by the Ethics Committee of the Affiliated Hospital of the
Academy of Military Medical Sciences. All tumor patients provided
written informed consent to participate in the study, in accordance
with the principles of the Declaration of Helsinki.
Results and Discussion
Selection of IS
It is necessary to use an IS to obtain high accuracy
when a mass spectrometer is used as the HPLC detector.
[13C2,15N3]-Oxo was
adopted as the IS due to the similarities in its retention and
ionization characteristics with those of Oxo and the minimal
endogenous interferences of
[13C2,15N3]-Oxo in the
MRM channels.
Chromatography
The stock solutions of Oxo and IS were diluted to a
concentration of 1 μg/ml with methanol. The diluted
solutions (5 μl) were injected and analyzed by the
HPLC-MS/MS method. The mass spectra fragmentation of the ions from
Oxo and IS are shown in Fig. 2.
Molecular ions of Oxo and IS exhibited m/z 156.0 and 161.1
([M–H]−), respectively. The product ion scan spectra
revealed high abundance fragment ions at m/z 111.9 and 117.1
([M–H]−) for Oxo and IS, respectively. Therefore, MRM
transitions of m/z 156.0→111.9 and 161.1→117.1 were adopted for
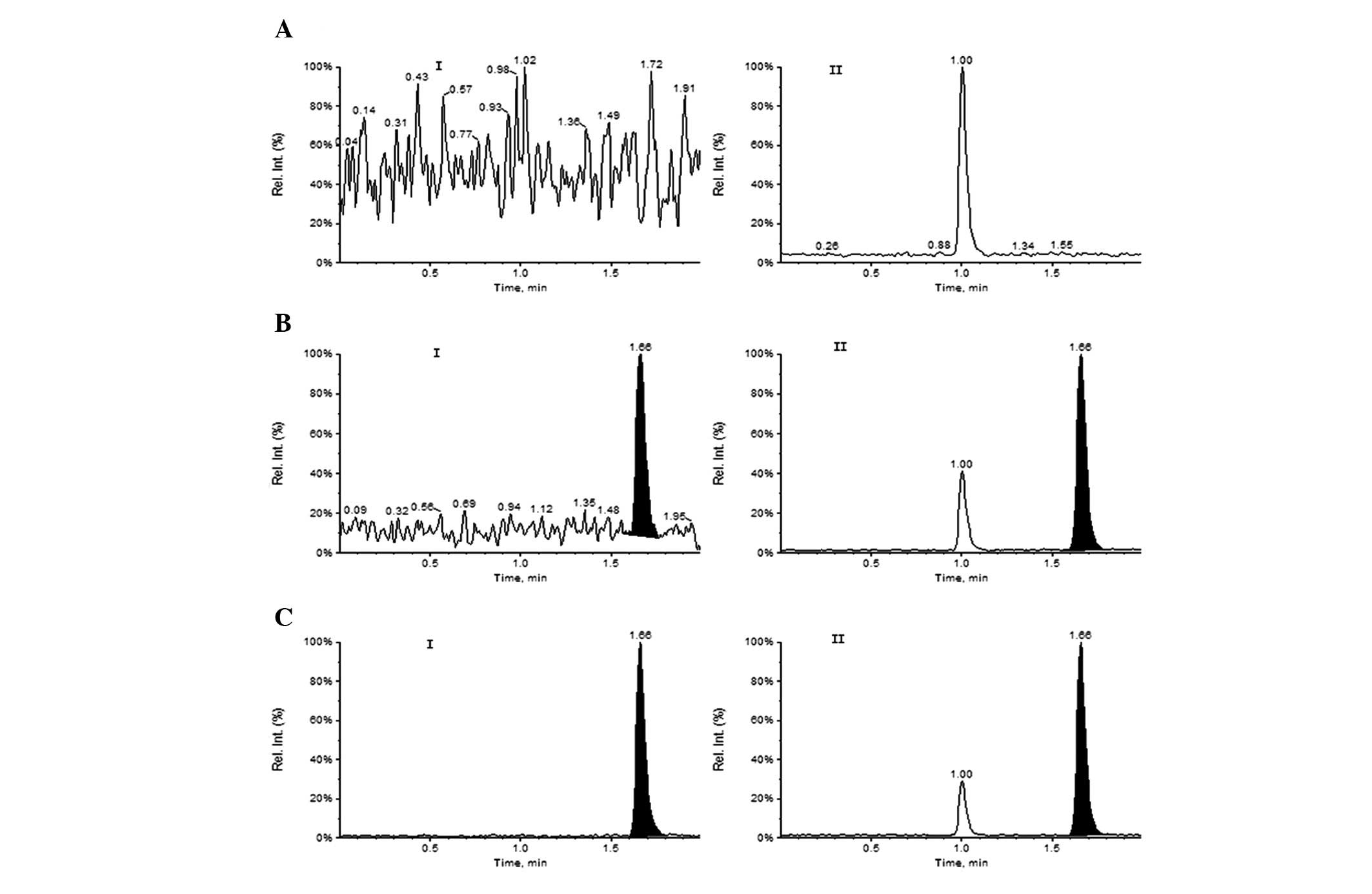
quantification of Oxo and IS, respectively. The typical HPLC-MS/MS
MRM chromatograms of a blank human plasma sample and a human plasma
sample spiked with Oxo and IS at the lower limit of quantification
(LOQ; 2.0 ng/ml) are shown in Fig.
3. There was almost no interference peak at the retention time
of Oxo (1.66 min) and IS (1.66 min). This result demonstrated that
this method was sensitive and specific, allowing for the analysis
of samples in batches and exhibiting suitability for
pharmacokinetic studies.
Linearity and LOQ
The calibration standards of seven Oxo concentration
levels at 2.0, 5.0, 10.0, 25.0, 50.0, 100.0 and 200.0 ng/ml were
injected and assayed. The calibration curve was constructed by
plotting the peak area ratios (Y) of Oxo to the IS versus the
concentrations (X) of Oxo, using weighted least squares linear
regression (the weighting factor was 1/X2). In our
study, the mean standard curve for Oxo was Y=0.0057X+0.000353
(r=0.9991). The concentrations of Oxo in unknown samples were
obtained from the regression line. The LOQ was defined as the
lowest concentration on the calibration curve, where precision was
within ±20% and accuracy was within ±20%. This was established
using six independent samples of standards.
Intra-day and inter-day precision and
accuracy
Intra-assay precision and accuracy were assessed by
measuring the concentration of Oxo in six aliquots of three
different quality control samples, which were extracted and
analyzed on the same day. Inter-assay precision and accuracy were
determined from the results of three different quality control
samples, which were extracted and analyzed six-fold on three
consecutive days. The results are presented in Table I.
 | Table I.Extraction recoveries, intra- and
inter-day accuracy and precision. |
Table I.
Extraction recoveries, intra- and
inter-day accuracy and precision.
| Extraction recoveries
(n=5)
| Accuracy (%, n=6)
| Intra-day precision
(n=6)
| Inter-day precision
(n=6)
|
|---|
| Concentration
(ng/ml) | Recoveries (%) | RSD (%) | Intra-day | Inter-day | Mean | RSD (%) | Mean | RSD (%) |
|---|
| 5 | 60.26 | 4.09 | 102.20 | 101.60 | 5.11 | 5.01 | 5.08 | 6.33 |
| 25 | 67.13 | 1.48 | 100.12 | 102.52 | 25.03 | 1.49 | 25.63 | 3.19 |
| 160 | 64.37 | 2.21 | 99.38 | 102.71 | 159.00 | 3.35 | 164.33 | 3.63 |
Matrix effect (ME)
The ME represents the potential ion suppression or
enhancement effects of co-eluting and undetected matrix components
in plasma. This was obtained by comparing the peak area of Oxo and
IS spiked into post-extracted blank plasma samples to that of Oxo
and IS spiked into the mobile phase at an equivalent concentration.
In this study, the ME was evaluated by three quality control
concentrations (5.0, 25.0 and 160.0 ng/ml) of Oxo and an IS
concentration level of 250.0 ng/ml. Six samples at each
concentration level were analyzed. The blank plasma used in this
study was obtained from six different batches. If the peak area
ratios were <85 or >115%, an endogenous ME was implied. The
ME of plasma at concentrations of 5.0, 25.0 and 160.0 ng/ml were
95.50, 100.45 and 97.61%, respectively. The ME of IS was 95.6%. The
results obtained were within the acceptable limit, suggesting that
there was no ME observed in this study.
Stability
The stability of Oxo in plasma under various
conditions was evaluated. The quality control plasma samples (5.0,
25.0 and 160.0 ng/ml) were stable when placed at room temperature
for 4 h, following three freeze/thaw (−40°C) cycles and when stored
at −40°C for 3 months. The processed sample, placed in the
autosampler at an ambient temperature (20°C) for 2 h, was also
stable. These results (Table II)
demonstrated that no significant degradation occurred under
different conditions.
| Table II.Stability test for Oxo (n=5). |
Table II.
Stability test for Oxo (n=5).
| Room temperature
| −40°C for 3 months
| Freeze/thaw
| Autosampler
|
|---|
| Concentration
(ng/ml) | Mean | RSD (%) | Mean | RSD (%) | Mean | RSD (%) | Mean | RSD (%) |
|---|
| 5 | 5.16 | 7.40 | 4.69 | 8.90 | 5.27 | 3.66 | 4.83 | 5.92 |
| 25 | 25.77 | 2.94 | 25.03 | 4.83 | 25.97 | 3.47 | 23.83 | 2.42 |
| 160 | 166.00 | 4.93 | 163.67 | 3.14 | 163.00 | 1.62 | 158.33 | 2.85 |
Pharmacokinetic application
The method was applied for the analysis of plasma
samples obtained from 12 tumor patients, following single oral
administration of a 40-mg S-1 capsule in the pharmacokinetics
study. The pharmacokinetic parameters were estimated by DAS version
2.1.1 software. Pharmacokinetic analysis of Oxo was performed using
the noncompartmental method. The concentration maximum
(Cmax) and the time to reach it (Tmax) were
recorded directly. The elimination rate constant (Ke)
was calculated using linear regression of the terminal points from
the semi-log plot of plasma concentration against time. The
elimination half-life (t1/2) was calculated as
0.693/Ke. The area under the plasma concentration-time
curve of Oxo, from time zero to infinity (AUC0-∞), was
determined using the linear trapezoidal rule to the last measurable
plasma concentration (Ct), plus the additional area from
time t to infinity, calculated as Ct/Ke.
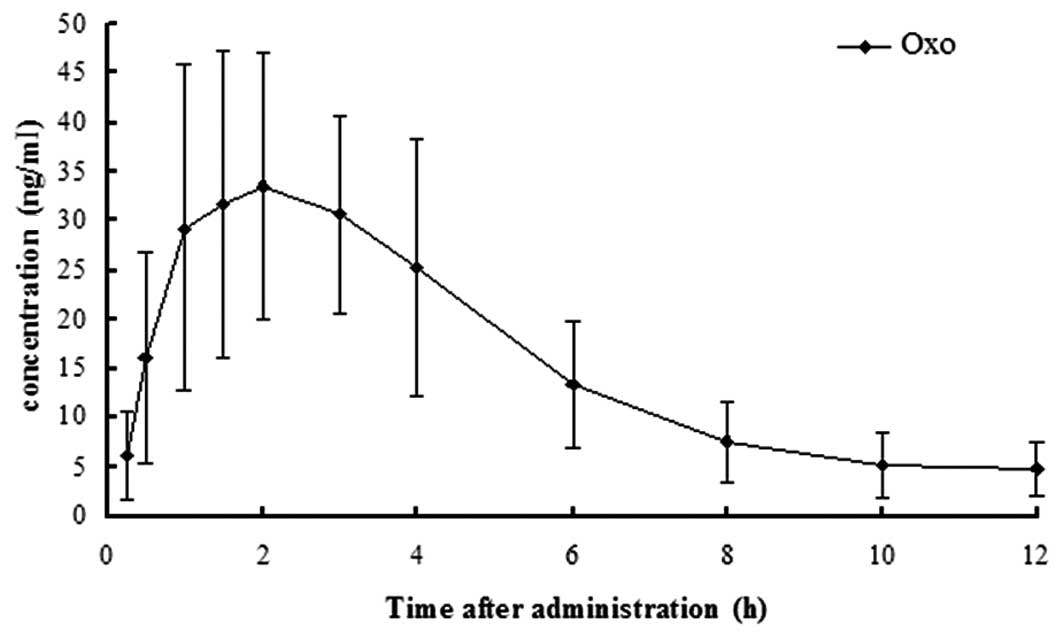
Finally, the mean plasma concentration versus time profile and
pharmacokinetic parameters of Oxo were obtained (Fig. 4; Table
III). The primary pharmacokinetic parameters of Oxo in our
study were similar to those as previously published (18–20).
The method developed in this study was demonstrated to be accurate
and sensitive when compared with the pharmacokinetic parameters and
concentration-time curves of previous studies.
| Table III.Pharmacokinetic parameters of Oxo
after single oral administration of a 40-mg S-1 capsule in 12 tumor
patients. |
Table III.
Pharmacokinetic parameters of Oxo
after single oral administration of a 40-mg S-1 capsule in 12 tumor
patients.
| Value | Cmax
(ng/ml) | Tmax
(h) | T1/2
(h) |
AUC(0–12) (ng/h/ml) | AUC(0-∞)
(ng/h/ml) |
|---|
| Mean | 39.56 | 2.29 | 2.78 | 184.96 | 202.61 |
| SD | 14.37 | 0.89 | 0.99 | 70.12 | 85.03 |
Conclusions
A HPLC-MS/MS method was developed for the
determination of Oxo in human plasma. There were numerous
advantages to this method, including low volumes of sample
requirement, simple sample processing, absence of the ME and a
short analysis time. The method was successfully applied to a
pharmacokinetic study after single oral administration of a 40-mg
S-1 capsule in 12 tumor patients.
References
|
1.
|
Kubota T: The role of S-1 in the treatment
of gastric cancer. Br J Cancer. 98:1301–1304. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Shirasaka T, Nakano K, Takechi T, et al:
Antitumor activity of 1 M tegafur-0.4 M
5-chloro-2,4-dihydroxypyridine-1 M potassium oxonate (S-1) against
human colon carcinoma ortho-topically implanted into nude rats.
Cancer Res. 56:2602–2606. 1996.PubMed/NCBI
|
|
3.
|
Blum M, Suzuki A and Ajani JA: A
comprehensive review of S-1 in the treatment of advanced gastric
adenocarcinoma. Future Oncol. 6:715–726. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Shimoyama S, Kiyokawa T, Nishida M and
Seto Y: S-1 monotherapy achieved twenty-month survival for
peritoneal lavage cytology-positive gastric cancer patient
undergoing noncurative resection. Gan To Kagaku Ryoho. 9:1411–1414.
2012.(In Japanese).
|
|
5.
|
Yamashita T, Arai K, Terashima T, et al: A
case report of S-1 monotherapy for advanced hepatocellular
carcinoma. Gan To Kagaku Ryoho. 9:1435–1437. 2012.(In
Japanese).
|
|
6.
|
Iwasa S, Yamada Y, Kato K, Goto A, Honma
Y, Hamaguchi T and Shimada Y: Long-term results of a phase II study
of S-1 plus irinotecan in metastatic colorectal cancer. Anticancer
Res. 9:4157–4161. 2012.PubMed/NCBI
|
|
7.
|
Kogashiwa Y, Nagafuji H and Kohno N:
Feasibility of concurrent chemoradiotherapy with S-1 administered
on alternate days for elderly patients with head and neck cancer.
Anticancer Res. 32:4035–4040. 2012.PubMed/NCBI
|
|
8.
|
Shirasaka T, Shimamato Y, Ohshimo H, et
al: Development of a novel form of an oral 5-fluorouracil
derivative (S-1) directed to the potentiation of the tumor
selective cytotoxicity of 5-fluorouracil by two biochemical
modulators. Anticancer Drugs. 7:548–557. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Shirasaka T, Shimamoto Y and Fukushima M:
Inhibition by oxonic acid of gastrointestinal toxicity of
5-fluorouracil without loss of its antitumor activity in rats.
Cancer Res. 53:4004–4009. 1993.PubMed/NCBI
|
|
10.
|
American Association for Cancer Research:
86th Annual Meeting: Proceedings; 36. American Association for
Cancer Research; Toronto: pp. 4061995
|
|
11.
|
Kitamura R, Satoh T, Maeda M and Tsuji A:
Enzyme immunoassay of potassium oxonate using specific antibody
isolated by immunosorbent gel. Yakugaku Zasshi. 114:171–175.
1994.(In Japanese).
|
|
12.
|
Matsushima E and Yoshida K, Kitamura R and
Yoshida K: Determination of S-1 (combined drug of tegafur,
5-chloro-2,4-dihydroxypyridine and potassium oxonate) and
5-fluorouracil in human plasma and urine using high-performance
liquid chromatography and gas chromatography-negative ion chemical
ionization mass spectrometry. J Chromatogr B Biomed Sci Appl.
691:95–104. 1997.
|
|
13.
|
Schwaninger AE, Meyer MR, Huestis MA and
Maurer HH: Development and validation of LC-HRMS and GC-NICI-MS
methods for stereoselective determination of MDMA and its phase I
and II metabolites in human urine. J Mass Spectrom. 46:603–614.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Liu K, Zhong D, Zou H and Chen X:
Determination of tegafur, 5-fluorouracil, gimeracil and oxonic acid
in human plasma using liquid chromatography-tandem mass
spectrometry. J Pharm Biomed Anal. 52:550–556. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Star-Weinstock M, Williamson BL, Dey S,
Pillai S and Purkayastha S: LC-ESI-MS/MS Analysis of Testosterone
at Sub-Picogram Levels Using a Novel Derivatization Reagent. Anal
Chem. 84:9310–9317. 2012.PubMed/NCBI
|
|
16.
|
Giorgianni F, Mileo V, Desiderio DM,
Catinella S and Beranova-Giorgianni S: Characterization of the
phosphoproteome in human bronchoalveolar lavage fluid. Int J
Proteomics. 2012:4602612012. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
de Mateo S, Estanyol JM and Oliva R:
Methods for the analysis of the sperm proteome. Methods Mol Biol.
927:411–422. 2013.
|
|
18.
|
Hirata K, Horikoshi N, Aiba K, et al:
Pharmacokinetic study of S-1, a novel oral fluorouracil antitumor
drug. Clin Cancer Res. 5:2000–2005. 1999.PubMed/NCBI
|
|
19.
|
Peters GJ, Noordhuis P, Van Kuilenburg AB,
et al: Pharmacokinetics of S-1, an oral formulation of ftorafur,
oxonic acid and 5-chloro-2,4-dihydroxypyridine (molar ratio
1:0.4:1) in patients with solid tumors. Cancer Chemother Pharmacol.
52:1–12. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Mende B, Krauss J, Thyssen D, et al:
Pharmacokinetic study of S-1. Int J Clin Pharmacol Ther. 47:65–67.
2009. View
Article : Google Scholar
|