Introduction
Proximal 10q duplication is a well-defined, but rare
syndrome and is often derived from a balanced translocation in a
parent (1,2). In this study, the case of a boy with
phenotypic abnormalities and duplication of the chromosomal region,
10q11.21→q11.22, characterized by the array-comparative genomic
hybridization (CGH) technique, is reported. The phenotypic findings
were compared with those in eight additional published cases with
proximal partial duplication of the long arm of chromosome 10q
(1–8). The partial proximal trisomy 10q
consists of mild to moderate developmental delay, postnatal growth
retardation, microcephaly, prominent forehead, small and deep set
eyes, epicanthus, upturned nose, bow-shaped mouth, micrognathia,
thick and flat helices of the ears and long, slender limbs
(8). The present study concerns an
8-year-old boy with severe central hypotonia in whom array-CGH
identified a de novo cryptic duplication of the proximal
10q.
Materials and methods
Cytogenetics
Conventional karyotyping based on GTG-banding
(600–800 bands) was performed using standard methods on metaphases
from blood leukocytes.
Array-CGH
Molecular karyotyping was performed on DNA extracted
from the whole blood of the patient and both parents. All the
experiments were performed through oligo-array platforms (Human
Genome CGH Microarray 44B Agilent kit; Agilent Technologies, Santa
Clara, CA, USA). Briefly, 500 ng of proband and of a gender-matched
pooled reference DNA (Promega Corporation, Fitchburg, WI, USA) were
processed according to the manufacturer’s instructions.
Fluorescence was detected in a dual-laser scanner (DNA Microarray
Scanner with Sure Scan High-Resolution Technology; Model G2565CA;
Agilent Technologies) and the images were extracted and analyzed
through Agilent Feature Extraction software (v10.5.1.1) and DNA
Analytics software (v4.0.73) (Agilent Technologies). Changes in
test DNA copy number at a specific locus were observed as the
deviation of the log2ratio value from the value of 0 of
at least three consecutive probes. The quality of each experiment
was assessed using a parameter provided by Agilent software (QC
metric) and on the basis of DNA quality. Copy number changes
identified in the samples were compared with the Database of
Genomic Variants (http://projects.tcag.ca/variation/) and also
visualized using the UCSC Genome Browser website (http://genome.ucsc.edu/). Moreover, DECIPHER
(http://decipher.sanger.ac.uk/) and
ECARUCA (http://umcecaruca01.extern.umcn.nl:8080/ecaruca/ecaruca.jsp)
databases were used for comparison with possible analogous reported
cases. The positions of oligomers referred to the Human Genome
March 2006 assembly (hg18).
Fluorescence in situ hybridization (FISH)
analysis
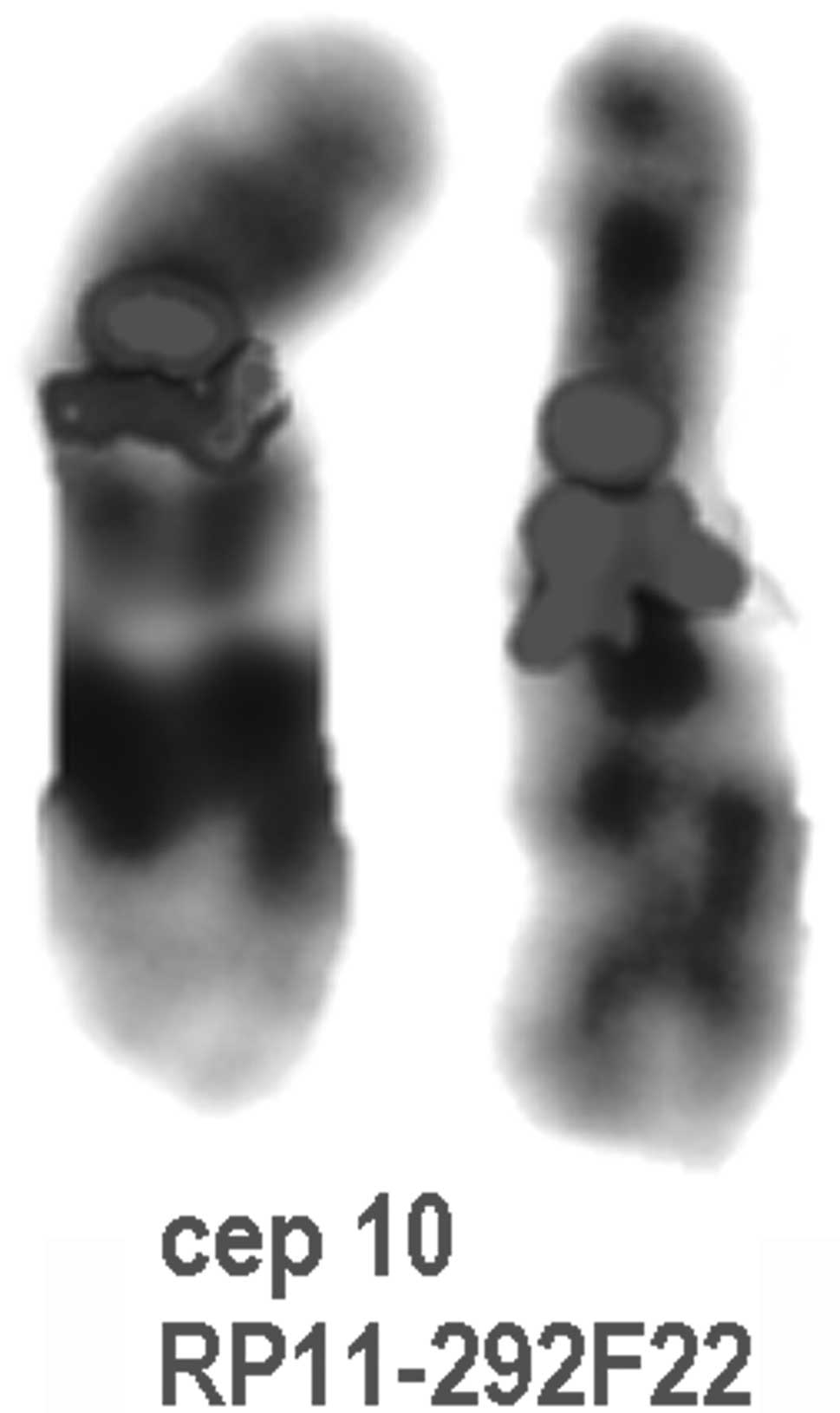
FISH experiments were performed on metaphase spreads
using the following as probes: Bacterial artificial chromosomes
(BACs) for the chromosomal region 10q11.22 (RP11-292F22 located in
47.075–47.114 Mb, as obtained from the Children’s Hospital Oakland
Research Institute, Oakland, CA, USA) and chromosome 10 centromeric
probe (cep 10; Kreatech Diagnostics, Amsterdam, Netherlands). Ten
metaphase spreads were analyzed for each FISH experiment. Analysis
was performed using a Zeiss epifluorescent Axioskop 2 Plus
microscope (Carl Zeiss Microscopy, LLC, Thornwood, NY, USA), and
images were captured, enhanced and analyzed using Cytovision (Leica
Biosystems, Wetzlar, Germany) software.
Case report
The patient was a 3-year-old boy, the only child of
healthy non-consanguineous parents of Greek origin (a 38-year old
father and 33-year old mother at the time of birth) with
unremarkable family history. The prenatal serial ultrasound
examinations were reported as normal and the pregnancy was
uneventful. The patient was born at 40 weeks of gestation by
vaginal delivery. The Apgar scores were 8 at 1 min and 9 at 5 min.
The birth weight was 2,950 g (10th centile), the birth length was
50 cm (50th centile) and the occipitofrontal circumference was 3 cm
(25th–50th centile).
At 8 months the patient responded normally to age
appropriate communication stimuli and parental overtures; however,
he exhibited severe central hypotonia, mild ataxia and mild
dysmorphic features, such as a triangular face, an enlarged cranium
cerebrale, a bifid scrotum, cryptorchidism, ulnar deviation of both
elbows, singe palmar creases of hands and syndactyly of the second
and third toes bilaterally. Brain magnetic resonance imaging
revealed hypoplasia of the corpus callosum and benign dilatation of
the subarachnoid areas, while G-banding karyotyping along with
detailed haematological, biochemical and metabolic examinations
showed no pathology. Physiotherapy was initiated and the patient
walked unaided at the age of 18 months. At 19 months, the
cryptorchidism was surgically corrected. The patient first uttered
words and two-word phrases at the ages of 23 and 27 months,
respectively.
At 2 years and 7 months, a referral for
developmental assessment was made due to language delay. The
patient was a sociable boy with mild dysmorphic features that could
easily have passed unnoticed, such as a triangular face, frontal
bossing, micrognathia, slight auricle abnormalities, a high-arched
palate, pectus carinatum, widely spaced nipples, a mild systolic
murmur and a clubfoot. The patient was 15 kg (75 centile) in weight
and 98 cm (90th centile) in height, and had a head circumference of
52 cm (90th centile). The developmental abilities of the patient
were equivalent to the level expected at 22 months, while the
patient’s speech consisted of 40 words and a few two-word phrases.
Visual, hearing and heart ultrasound examinations were normal and
speech therapy was initiated.
A re-evaluation was conducted at the age of 3 years.
Clinical examination revealed a sociable boy with mild facial
abnormalities (frontal bossing, moderate micrognathia, a gothic
palate and auricular abnormalities), pectus carinatum, widely
spaced nipples, a mild systolic murmur, single palmar crease and a
clubfoot. The developmental abilities of the patient were further
improved, being equivalent to the level at 2 years and 5 months.
According to Griffiths Scales of Mental Abilities his developmental
quotient was 82 (9). The patient
was 17 kg (75–90th centile) in weight, 98 cm (75th–90th centile) in
height and had a head circumference of 53 cm (90th–97th centile).
Neurological examination revealed truncal hypotonia, but without
focal signs. Informed consent was obtained from the patient’s
family and the study was approved by The Second Department of
Paediatrics, University of Athens, P&A Kyriakou Children’s
Hospital, Athens, Greece
Results
The karyotype analysis of the proband identified a
normal karyotype, 46XY, and chromosome analyses of both parents
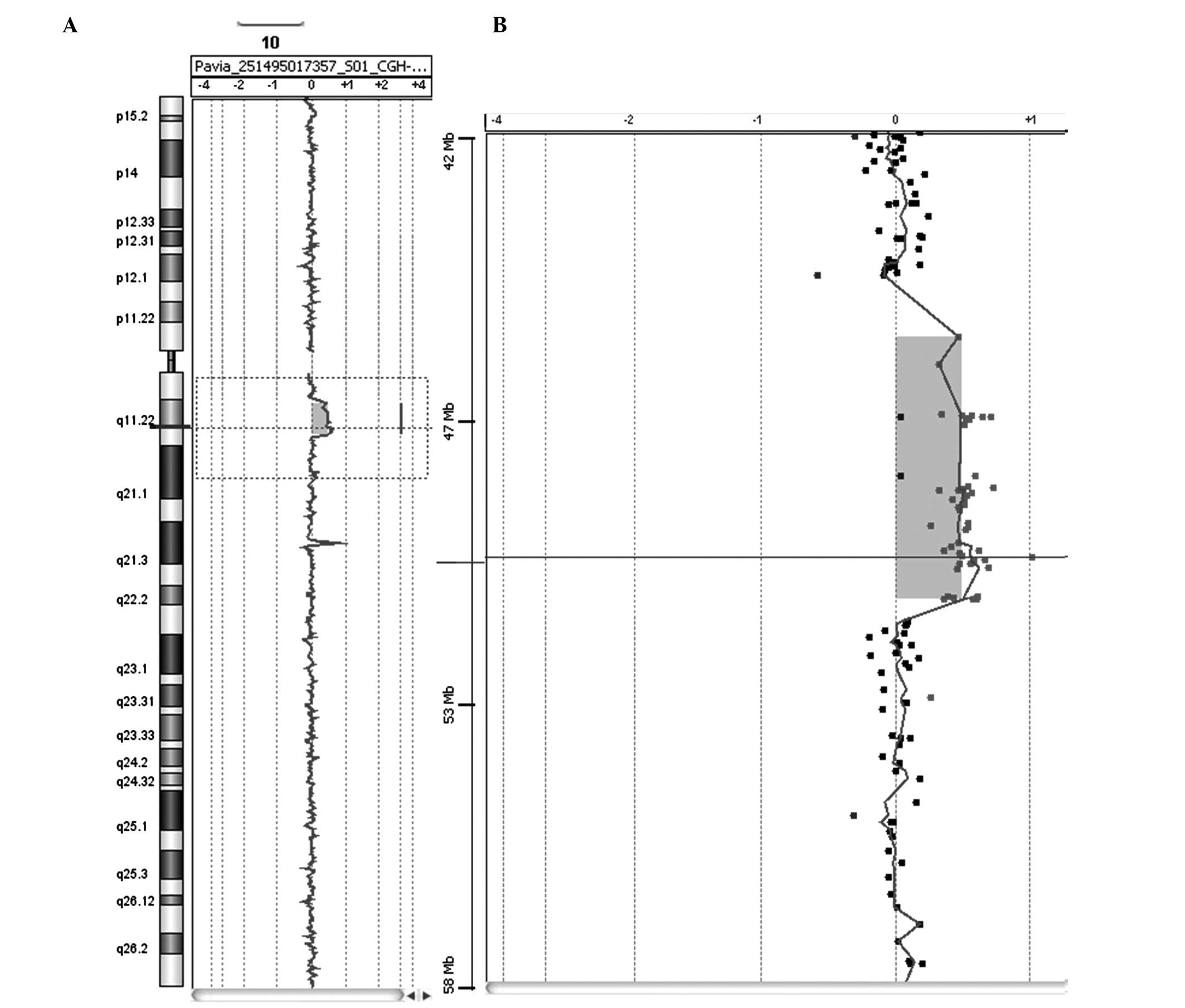
were also normal. Array-CGH analysis revealed a 5.6-Mb duplication
in the long arm of chromosome 10 located in the 10q11.21→q11.22
region (Fig. 1). The proximal
breakpoint was observed between 45.478 and 46.568 Mbp and the
distal breakpoint was between 51.264 and 51.676 Mbp. No abnormal
copy number variations were identified in either parent using
array-CGH technology. The DECIPHER and ECARUCA databases were used
as resources to aid the genotype-phenotype correlation analysis.
The duplication was confirmed by FISH analysis as shown in Fig. 2.
Discussion
Proximal 10q duplication is a well-defined but rare
genetic syndrome, often derived from a balanced translocation in a
parent. The partial proximal trisomy 10q consists of mild to
moderate developmental delay, postnatal growth retardation,
microcephaly, a prominent forehead, small and deep set eyes,
epicanthus, an upturned nose, a bow-shaped mouth, micrognathia,
thick and flat helices of the ears and long, slender limbs
(1–8).
The present study concerns the case of a 3-year-old
boy with phenotypic abnormalities and severe central hypotonia in
whom array-CGH resulted in the identification of a de novo
cryptic duplication of the proximal 10q (10q11.21→q11.22). This is
the first case of partial proximal trisomy 10q characterized by
array-CGH. To the best of our knowledge, only eight cases have
previously reported duplication at 10q11→10q22 (1–8). The
clinical characteristics of the eight previously published cases
with duplication at 10q11→q22 and the present case are outlined in
Table I. In concordance, the
patient of the present study showed severe central hypotonia,
ataxia, a triangular face, frontal bossing, moderate micrognathia,
slight auricular abnormalities, a high-arched palate, an enlarged
cranium cerebrale, pectus carinatum, widely spaced nipples, a mild
systolic murmur, a bifid scrotum, cryptorchidism, ulnar deviation
of both elbows, single palmar creases of the hand and foot, as well
as clubfoot and syndactyly of the second and third toes
bilaterally. The current case did not show growth retardation and
microcephaly, which have been considered common features of the
syndrome (1–5,7–8).
Similarly Lam et al (6)
reported a case with proximal trisomy 10q, but without the
phenotypic features of microcephaly.
 | Table ISummary of the clinical features in
previously published cases with partial proximal trisomy 10q
syndrome and the present case. |
Table I
Summary of the clinical features in
previously published cases with partial proximal trisomy 10q
syndrome and the present case.
| Variable | Vogel et al
(1) | Fryns et al
(2) | De Michelana and
Campos (3) | Aalfs et al
(4) | van Buggenhout et
al (5) | Lam et al
(6) | Nucaro et al
(7) | Lysy et al
(8) | Present case |
|---|
| Trisomic segment | 10q11→22 | 10q11.2→22 | 10q11→22 | 10q11.2→22.3 | 10q11→22.3 | 10q11→22 | 10q11.2→22.3 | 10q11.2→22.3 | 10q11.21→11.22 |
| Methods of
confirmation | Karyotype | Karyotype | Karyotype | Karyotype
FISH | Karyotype | Karyotype | Karyotype
FISH | Karyotype | Karyotype
FISH
Array-CGH |
| General |
| Birth weight
(g) | 2,200 | 2,650 | 2,400 | 3,750 | | 3,000 | 2,830 | 2,720 | 2,950 |
| Growth
retardation | + | + | + | + | − | − | + | + | + |
| Developmental
delay | + | + | + | + | | + | | + | + |
| Respiratory
distress | + | u | u | + | | | | | |
| Hypertonia | u | u | + | + | | | + | | |
| Craniofacial |
| Microcephaly | + | + | + | + | + | − | + | + | − |
| Prominent
forehead | − | + | − | + | + | + | − | − | + |
| Deep set, small
eyes | + | + | + | + | + | + | + | − | + |
| Epicanthus | − | − | − | + | − | + | − | − | + |
| Strabismus | − | + | + | + | + | + | − | − | − |
| Iris coloboma | u | + | − | − | − | + | − | − | − |
|
Blepharophimosis | − | − | − | − | − | u | − | + | − |
| Retinal
dysplasia | u | + | u | − | − | + | − | − | − |
| Upturned nose | − | + | + | + | + | + | + | − | − |
| Bow-shaped
mouth | + | + | + | + | + | + | + | − | + |
| Micrognathia | + | + | + | + | + | + | + | − | − |
| Highly arched
palate | + | + | + | − | + | + | + | + | − |
| Flat, thick ear
helix | + | + | + | + | + | + | + | − | + |
| Skeletal |
| Slender limbs | + | + | − | − | − | + | + | − | + |
| Finger
syndactyly | u | + | u | − | + | + | + | + | + |
| Hypermobile
joints | u | + | u | − | | | | | |
| Rib
abnormalities | + | − | u | u | | | | | |
The patient of the current case presented with brain
malformations, which have not been identified thus far, while Lysy
et al (8) reported a
patient who presented with biliary atresia, anal anteposition and
cardiac malformations.
Notably, these cases may be clearly distinguished
from the cases with duplications of 10p12.1 to 10q11.22 (10). Liehr et al (10) identified a novel unbalanced region
of chromosomal abnormalities in the pericentromeric region of
chromosome 10 (10p12.1 to 10q11.22), which did not have negative
phenotypic consequences.
The duplicated region identified in the present case
spans only ~5.6 Mb of genomic DNA and contains ~36 genes with
variable functions. Specifically these genes encode regulators of
membrane organization and membrane trafficking [annexin A8 (ANXA8),
ANXA8L1 and ANXA8L2], morphogenetic proteins and regulators of cell
growth and differentiation [growth differentiation factor 2 (GDF2)
and GDF10), transcription factors and modulators [dorsal root
ganglia homeobox (DRGX)], signal transducers [mitogen-activated
protein kinase 8 (MAPK8)], enzymes [choline acetyltransferase
(CHAT) and poly(ADP-ribose) glycohydrolase (PARG)] and DNA repair
proteins [excision repair cross-complementing rodent repair
deficiency, complementation group 6 (ERCC6)]. It is worth taking
into consideration that studies in rodents have suggested that the
proteins, GDF2 and GDF10 are important in the differentiation of
cholinergic central nervous system neurons and in skeletal
morphogenesis, respectively (11,12).
It is possible that the emerged severe phenotype is due to the fact
that the region includes major genetic information affecting
physical development, or that the duplication significantly alters
the expression pattern of the corresponding genes.
Further studies are required to elucidate the
pathogenic mechanisms as more cases are likely to be identified
with 10q proximal duplication, which may be characterized by the
advanced methodology of array-CGH. The critical region of the 10q
duplication syndrome will be identified more specifically by
including benign cases with duplication of 10p12.1 to 10q11.22.
References
|
1
|
Vogel W, Back E and Imm W: Serial
duplication of 10 (q11 leads to q22) in a patient with minor
congenital malformations. Clin Genet. 13:159–163. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fryns JP, Kleczkowska A, Igodt-Ameye L and
Van den Berghe H: Proximal duplication of the long arm of
chromosome 10 (10q11.2 → 10q22): a distinct clinical entity. Clin
Genet. 32:61–65. 1987.PubMed/NCBI
|
|
3
|
De Michelana MI and Campos PJ: A new case
of proximal 10q partial trisomy. J Med Genet. 28:205–206.
1991.PubMed/NCBI
|
|
4
|
Aalfs CM, Hoovers JM, Nieste-Otter MA,
Mannens MM, Hennekam RC and Leschot NJ: Further delineation of the
partial proximal trisomy 10q syndrome. J Med Genet. 32:968–971.
1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
van Buggenhout G, Decock P and Fryns JP: A
distinct phenotype associated with partial trisomy 10q due to
proximal direct duplication 10q11→q223? Genet Couns. 7:53–59.
1996.PubMed/NCBI
|
|
6
|
Lam FW, Chan WK, Lam ST, Chu WP and Kwong
NS: Proximal 10q trisomy: a new case with anal atresia. J Med
Genet. 37:E242000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nucaro A, Faedda A, Cao A and Boccone L:
Partial proximal trisomy 10q syndrome: a new case. Genet Couns.
13:411–416. 2002.PubMed/NCBI
|
|
8
|
Lysy PA, Sibille C, Gillerot Y, Smets F
and Sokal EM: Partial proximal 10q trisomy: a new case associated
with biliary atresia. Hereditas. 144:191–194. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Griffiths R: The Abilities of Young
Children. Bucks: Association for Research in Infant and Child
Development. A comprehensive system of measurement for the first
eight years of life. The Test Agency; Thames: pp. 101–172. 1984
|
|
10
|
Liehr T, Stumm M, Wegner RD, Bhatt S,
Hickmann P, Patsalis PC, Meins M, Morlot S, Klaschka V, Ewers E,
Hinreiner S, Mrasek K, Kosyakova N, Cai WW, Cheung SW and Weise A:
10p11.2 to 10q11.2 is a yet unreported region leading to unbalanced
chromosomal abnormalities without phenotypic consequences.
Cytogenet Genome Res. 124:102–105. 2009. View Article : Google Scholar
|
|
11
|
López-Coviella I, Berse B, Krauss R, Thies
RS and Blusztajn JK: Induction and maintenance of the neuronal
cholinergic phenotype in the central nervous system by BMP-9.
Science. 289:313–316. 2000.
|
|
12
|
Cunningham NS, Jenkins NA, Gilbert DJ,
Copeland NG, Reddi AH and Lee SJ: Growth/differentiation factor-10:
a new member of the transforming growth factor-beta superfamily
related to bone morphogenetic protein-3. Growth Factors. 12:99–109.
1995. View Article : Google Scholar : PubMed/NCBI
|