Introduction
Colorectal cancer (CRC) is one of the leading causes
of cancer mortality in the world. The incidence rate of CRC has
increased in a number of Asian countries, including China, during
the past few decades (1,2). Currently, the prognosis for CRC
remains poor, as little effective therapy has been developed
(3). Therefore, developing
effective therapeutic agents to treat CRC is an urgent
requirement.
Casticin is a primary component of the fruits of
Vitex rotundifolia L. that, for thousands of years, has been
extensively used as an anti-inflammatory agent in Traditional
Chinese Medicine (4). A number of
studies have shown that casticin inhibits the growth of various
cancer cells, including breast (5), lung (6) and colon cancer (7,8). In
addition, our previous studies demonstrated that casticin induced
apoptotic cell death of cervical cancer and hepatocellular
carcinoma cells (9–11), even without functional p53.
Therefore, investigations into the apoptosis-inducing effects and
the underlying molecular mechanisms of casticin in p53-mutated
human colon cancer cell lines are required.
The generation of reactive oxygen species (ROS)
occurs in a number of biological systems, and ROS are well known to
act as important determinants in the regulation of cell signaling
pathways associated with proliferation, apoptosis and senescence
(12). Other agents have also been
found to generate ROS in the mitochondria, including diallyl
trisulfide and several chemopreventive agents, such as benzyl
isothiocyanate, phenethyl isothiocyanate and sulforaphane, by
inhibiting complexes I or III of the mitochondrial respiratory
chain and disrupting the mitochondrial membrane potential (13). During mitochondrial respiration,
O2 acts as the terminal acceptor of electrons, with the
four-electron reduction of O2 yielding H2O.
In normal tissue, this has been estimated to occur 96–99% of the
time. However, the one-electron reduction of O2 can
yield superoxide; this is believed to occur in the electron
transport chain at Sites I (nicotinamide adenine
dinucleotide-dehydrogenase) or III (ubiquinone-cytochrome b)
(14). Manganese superoxide
dismutase catalyzes the dismutation of this superoxide to generate
H2O2 and O2. The
H2O2 is subsequently detoxified to
H2O and O2, either through the action of
glutathione (GSH) peroxidase in the mitochondria or, if it diffuses
into the cytosol, by catalase in the peroxisomes. We previously
reported that casticin induced apoptosis by causing ROS generation
in cervical cancer cells (9,11);
however, whether casticin stimulates ROS production in colon cancer
cells is unclear.
Apoptosis signal-regulating kinase 1 (ASK1) is a
multifunctional serine/threonine protein kinase that is involved in
a wide range of physiological processes, including cell
differentiation and apoptosis (15). ASK1 has been reported to be
activated by a number of stress signals, including ROS, tumor
necrosis factor-α and endoplasmic reticulum (ER) stress (16,17).
B-cell lymphoma 2 (Bcl-2)-interacting mediator of cell death (Bim)
is a member of the ‘BH3-only proteins’, a subgroup of Bcl-2
apoptotic regulators that contain only one of the Bcl-2 homologous
regions (BH3). In response to apoptotic stimuli, BH3-only proteins
undergo translocation from a number of cellular compartments to the
mitochondrial membranes, where they interfere with the function of
anti-apoptotic Bcl-2 family members, ultimately resulting in
apoptotic cell death (18,19). In the oxidizing environment created
by ROS, ASK1, an upstream protein in the c-Jun N-terminal kinase
(JNK)-associated signal transduction pathway phosphorylation,
causes activation of the JNK pathway (20). We previously showed that casticin
induced apoptotic cell death of cervical cancer cells through the
ROS-dependent activation of JNK (11). However, the role of the ASK1/JNK
signaling cascade in casticin-induced colon cancer cell apoptosis
remains unknown. In the present study, the anti-carcinogenic
effects of casticin on human colon cancer were investigated.
Materials and methods
Chemicals and materials
Casticin was purchased from Biopurify Phytochemicals
Ltd. (Chengdu, China). The compound has a molecular weight of
374.3, appears as yellow crystals and has a purity of 98.0%.
Casticin was prepared in dimethyl sulfoxide (DMSO) as a 10 mmol/l
stock solution and diluted in medium to the indicated concentration
prior to use. 2′,7′-Dichlorofluorescein diacetate (DCFH-DA) was
obtained from Molecular Probes (Eugene, OR, USA). Propidium iodide
(PI), ethidium bromide, N-acetylcysteine (NAC, an oxygen-free
radical scavenger) and SP600125 (a JNK inhibitor) were purchased
from Sigma-Aldrich (St. Louis, MO, USA). Anti-JNK1 and β-actin
antibodies were purchased from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA), and anti-phosphorylated JNK1/2,
-phosphorylated ASK1, -ASK1, -phosphorylated Bim-extra long (EL)
and -Bim-EL antibodies were purchased from Cell Signaling
Technology, Inc. (Beverly, MA, USA).
Cell lines and cell culture
The human colon cancer cell lines HT-29, HCT-116,
SW480 and Caco-2 were purchased from the China Center for Type
Culture Collection (Wuhan, China). The study was approved by the
ethics committee of Hunan Normal University (Changsha, China).
Cells were maintained in Dulbecco’s modified Eagle’s medium
supplemented with 10% fetal bovine serum, 4 mM glutamine, 100 U/ml
penicillin and 100 μg/ml streptomycin, and incubated at 37°C in a
humidified atmosphere of 5% CO2.
Flow cytometric analysis using PI
staining
Cells were seeded at a density of 4×106
cells/well in 250-ml culture flasks for 24 h and then treated with
the medium containing various concentrations of casticin or 0.1%
DMSO for 24 h, or 10.0 μM casticin for the indicated time periods.
PI staining for DNA content analysis was performed as described
previously (21). All analyses
were performed using a flow cytometer (Coulter Epics XL-MSL,
Beckman Coulter, Fullerton, CA, USA) with CellQuest™ software (BD
Pharmingen, San Diego, CA, USA).
Histone/DNA ELISA for the detection of
apoptosis
The cell apoptosis ELISA detection kit (Roche,
Basel, Switzerland) was used to detect apoptosis in cells treated
with casticin according to the manufacturer’s instructions.
Briefly, cells were seeded in a 96-well plate at a density of
1×104 cells/well for 24 h, the tested agents were added
and the cells were then cultured in RPMI-1640 medium containing 10%
fetal bovine serum. After 24 h, the cytoplasm of the control and
treatment groups was transferred to the 96-well plate peridiumed by
the streptavidin and incubated with the biotinylated histone
antibody and peroxidase-tagged mouse anti-human DNA (both from
Roche, Palo Alto, CA, USA) for 2 h at room temperature. The
absorbance at 405 nm was measured with an enzyme-linked
immunosorbent apparatus (ELX-800 type; Bio-Tek, Shanghai,
China).
DNA agarose gel electrophoresis
Cells were seeded at a density of 4×106
cells/well in 250-ml culture flasks for 24 h and treated with
medium containing various concentrations of the test/control agents
or vehicle and 10% fetal bovine serum for 24 h. This assay was
performed as previously described (21).
Measurement of ROS generation
ROS generation in the control and casticin-treated
cells was measured by flow cytometry (FCM) following staining with
the DCFH-DA. Briefly, cells were seeded in six-well plates
(1×105 cells per well), allowed to attach overnight and
exposed to DMSO (control) or the desired concentrations of casticin
for specified time periods. The cells were stained with 20 μM
DCFH-DA for 30 min at 37°C, and the fluorescence intensity of the
DCF in the cells was determined using the flow cytometer with
winMDI software (Microsoft Corp., Redmond, WA, USA). As a rule,
10,000 cells were counted in each determination.
Transfection of small interfering RNA
(siRNA)
Control siRNA and siRNA targeting ASK1 were obtained
from Santa Cruz Biotechnology, Inc. The sense sequences of the
siRNA reagents were 5′-GACGCGATCAGAGAGTAAT-3′ (siRNA control) and
5′-GGTGGCACAAGCAAGTTCT-3′ (siRNA ASK1). For transient siRNA
transfection, HT-29 cells were seeded at a density of
5×105 cells/ml into six-well plates. Cells were
transfected on the following day with the Lipofectamine®
LTX with Plus™ reagent (Invitrogen Life Technologies, Carlsbad, CA,
USA) containing 100 ng/well siRNA (ASK1 or control). Cells were
transfected with each siRNA and incubated for 48 h. The
interference of ASK1 protein expression was confirmed by western
blot analysis using the anti-ASK1 antibody.
Western blot analysis
Western blot analysis was performed as described
previously (22). Anti-JNK1,
-phosphorylated JNK1/2, -phosphorylated ASK1, -ASK1,
-phosphorylated Bim-EL, -Bim-EL and -β-actin antibodies were used
as the primary antibodies. The signals were detected using an ECL
Advance western blot analysis system (Amersham Pharmacia Biotech,
Inc., Piscataway, NJ, USA).
Statistical analysis
Data are presented as the mean ± standard deviation
for triplicate experiments and were analyzed using the Student’s
t-test. Differences from the controls were considered significant
when P<0.05.
Results
Casticin induces apoptosis in colon
cancer cells
It has been previously reported that casticin
significantly inhibits the proliferation of human colon cancer
cells (7,8). To further investigate its mechanisms,
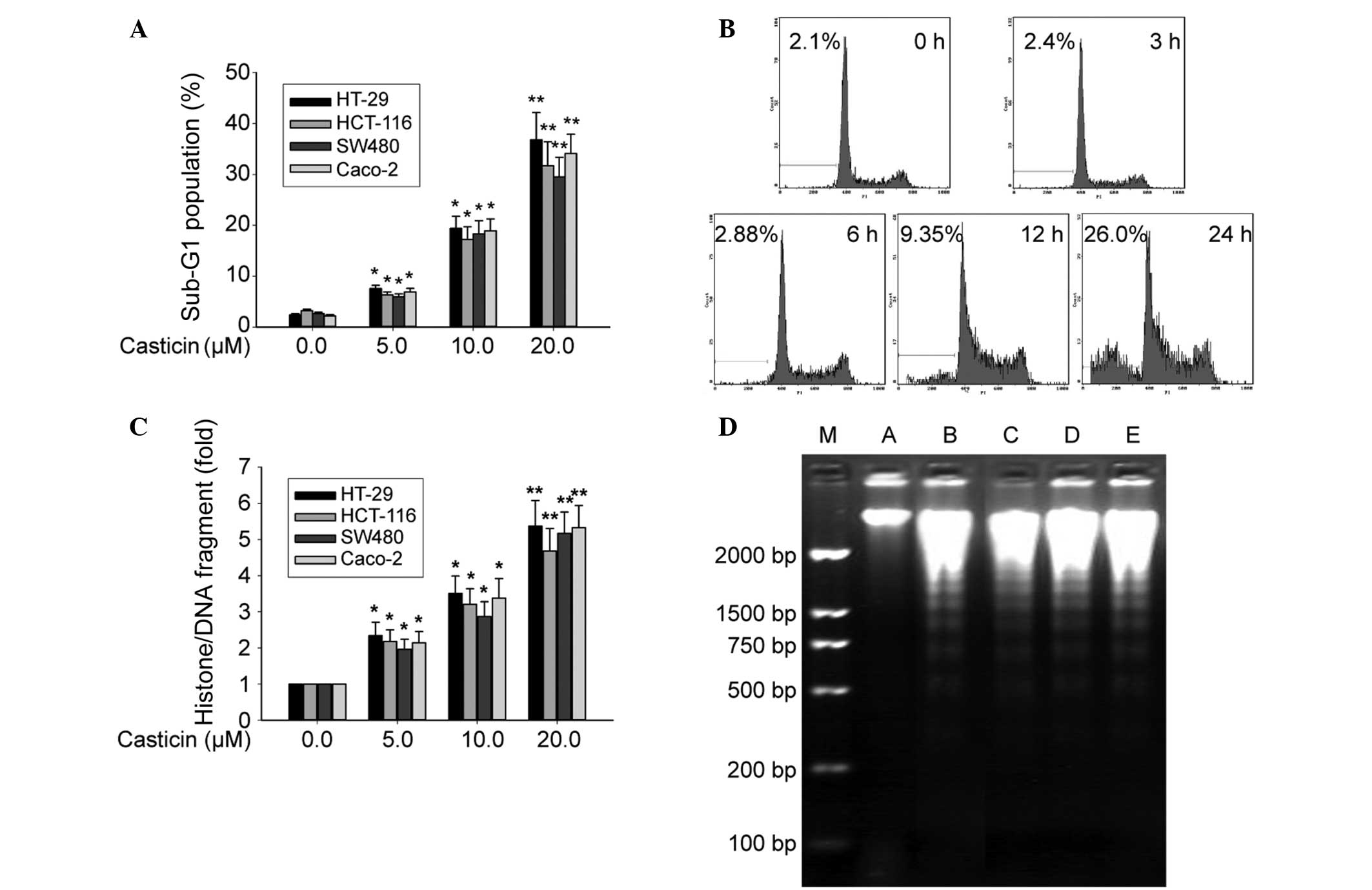
the hypodiploid cell populations were detected by FCM. Fig. 1A shows that casticin increased the
percentage of the sub-G1 population in a concentration-dependent
manner both in the p53 mutant cell line HT-29, and in the HCT116,
SW480 and Caco-2 cell lines (P<0.05). The sub-G1 population of
HT-29 cells treated with casticin was increased at 12 h and peaked
at 24 h (Fig. 1B). The histone/DNA
fragments of the HT-29, HCT-116, SW480 and Caco-2 cells, as
measured by the cell apoptosis ELISA detection kit, were increased
in a dose-dependent manner (P<0.05) following treatment with
casticin (Fig. 1C). Furthermore,
DNA fragmentation analysis by agarose gel electrophoresis revealed
a typical ladder pattern of internucleosomal DNA fragments in the
colon cancer cells that were treated with 10.0 μmol/l casticin for
24 h (Fig. 1D). These results
suggest that casticin inhibits colon cancer cell proliferation by a
mechanism involving the induction of apoptosis. The sequential
experiments in the study explored the molecular mechanism by which
casticin caused apoptosis using the p53-mutated human colon cancer
HT-29 cells.
Generation of ROS during treatment with
casticin
We have previously demonstrated that the
casticin-induced apoptosis of human cervical cancer cells is
associated with the induction of ROS generation (9,11).
Therefore, the present study investigated whether casticin also
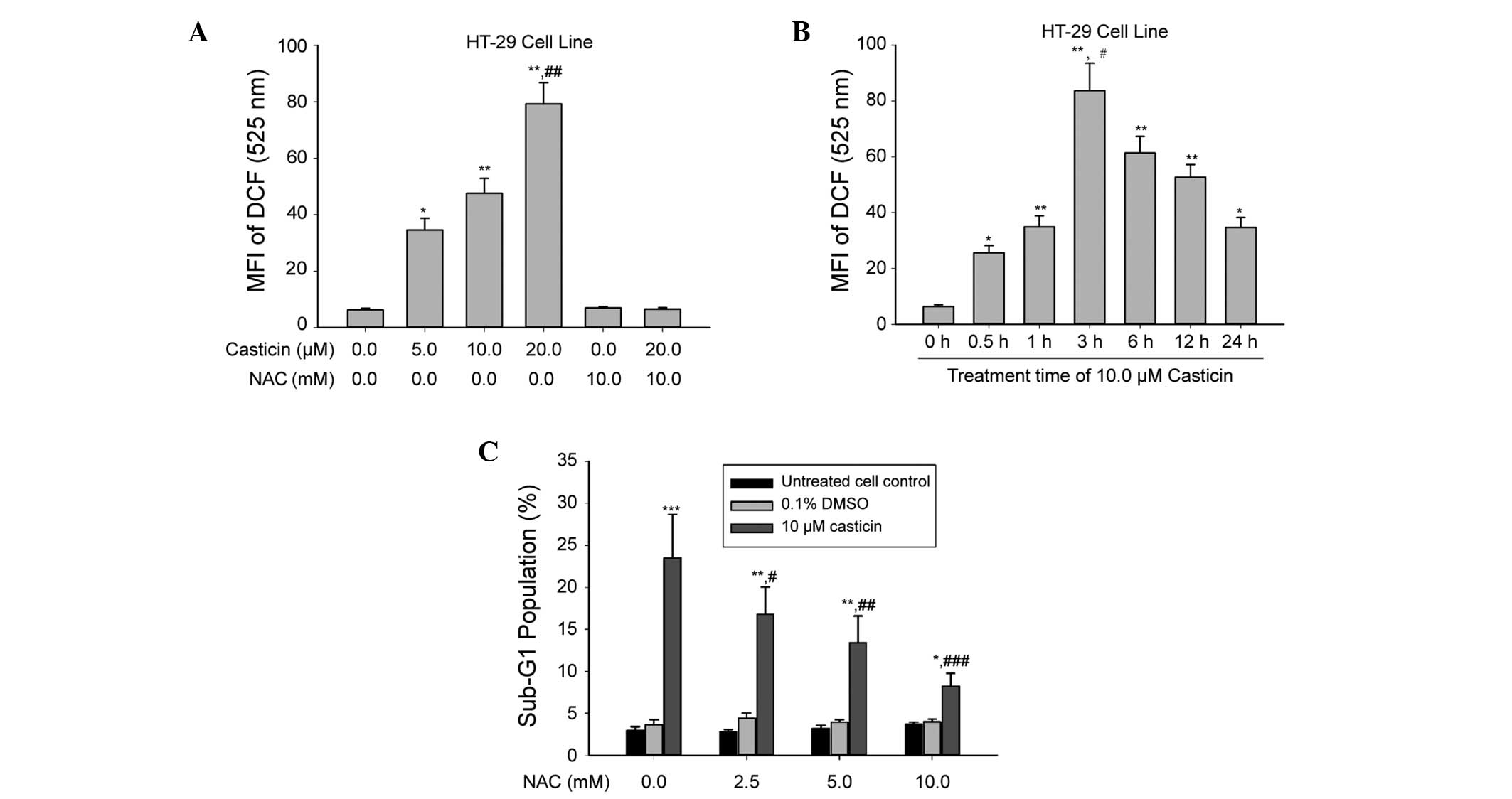
caused ROS production in colon cancer cells. Following the
treatment of HT-29 cells with 5.0, 10.0 and 20.0 μM casticin for 3
h, the levels of ROS increased in a dose-dependent manner (Fig. 2A). Time-course experiments revealed
that the levels of ROS increased initially at 0.5 h, reached a peak
at 3 h and persisted for up to 24 h after treatment with 10.0 μM
casticin (Fig. 2B).
To explore the role of ROS in casticin-induced colon
cancer cell apoptosis, the antioxidant NAC was used. As shown in
Fig. 2C, pretreatment of HT-29
cells with NAC (2.5, 5.0 and 10.0 mM) markedly attenuated
casticin-induced apoptosis in a concentration-dependent manner.
Furthermore, casticin-induced ROS production was almost completely
inhibited by treatment with 10.0 mM NAC (Fig. 2A). These results suggest that
casticin causes intracellular ROS generation, and the oxidative
stress further contributed to apoptosis of the human colon cancer
HT-29 cells.
Activation of the JNK signal transduction
pathway during treatment with casticin
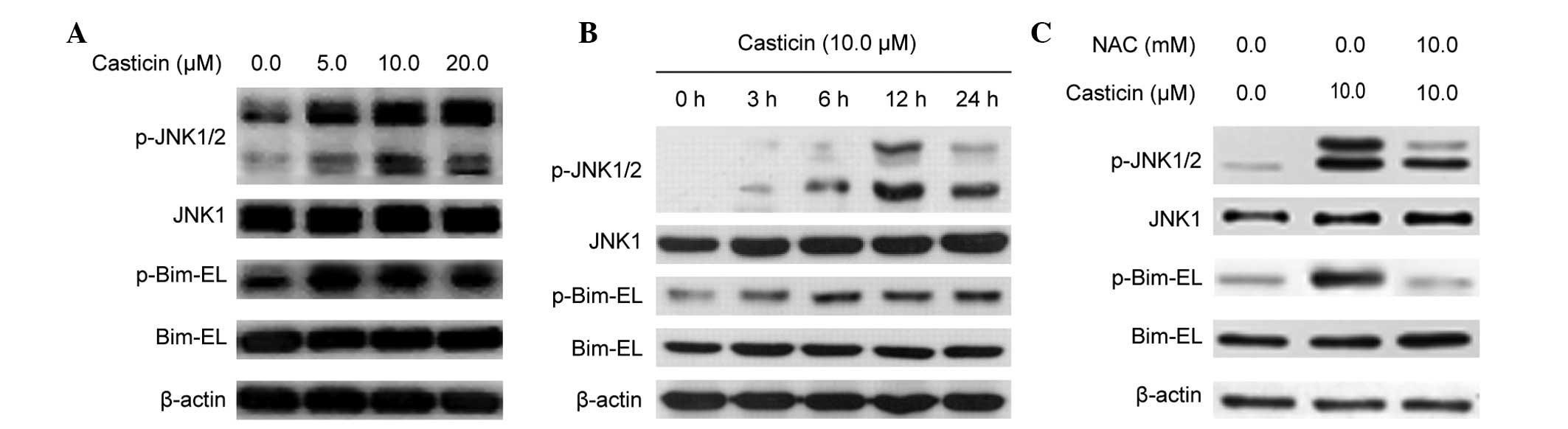
Our previous report indicated that ROS generation
and sustained JNK activation play a role in the casticin-induced
apoptosis of human cervical cancer cells (11). The present study examined whether
casticin activates the JNK pathway in human colon cancer cells.
Fig. 3A and B shows that casticin
treatment increased the phosphorylation levels of the JNK1/2
protein and its downstream molecule Bim-EL, and these
phosphorylation levels were attenuated in the presence of 10.0 mM
NAC (Fig. 3C) in the HT-29 cells.
These results suggest that casticin caused JNK activation through
intracellular ROS generation in the colon cancer HT-29 cells.
Activation of ASK1 during treatment with
casticin
ASK1 is a member of the mitogen-activated protein
kinase kinase kinase (MAPKK) family that activates the JNK pathways
by directly phosphorylating and thereby activating its respective
MAPKKs, mitogen-activated protein kinase kinase (MKK) 4/7 and
MKK3/6 (20). In the present
study, we next examined whether the casticin-induced ROS activated
ASK1. ASK1 is one of the upstream regulators of JNK, and is known
to be associated with cell death (21,23).
Fig. 4A and B shows that casticin
induced the phosphorylation of ASK1 in a concentration- and
time-dependent manner, and this was attenuated by pretreatment with
10 mM NAC in the HT-29 cells. These results suggest that, in the
colon cancer HT-29 cells, the activation of ASK1 caused by casticin
was dependent on intracellular ROS generation.
Knockdown of ASK1 by siRNA inhibits
casticin-induced JNK phosphorylation and apoptosis
To investigate the role of ASK1 in regulating JNK
and inducing apoptosis caused by casticin, siRNA that specifically
targeted ASK1 was used. The results of the western blot analysis
showed that ASK1 was downregulated following the transfection of
ASK1 siRNA in HT-29 cells (Fig.
5A). The effect of ASK1 siRNA on the activation of JNK caused
by casticin treatment was examined. Fig. 5A shows that ASK1 siRNA attenuated
the increased phosphorylation levels of JNK1/2 that were induced by
casticin, suggesting that ASK1 mediates casticin-induced JNK
activation.
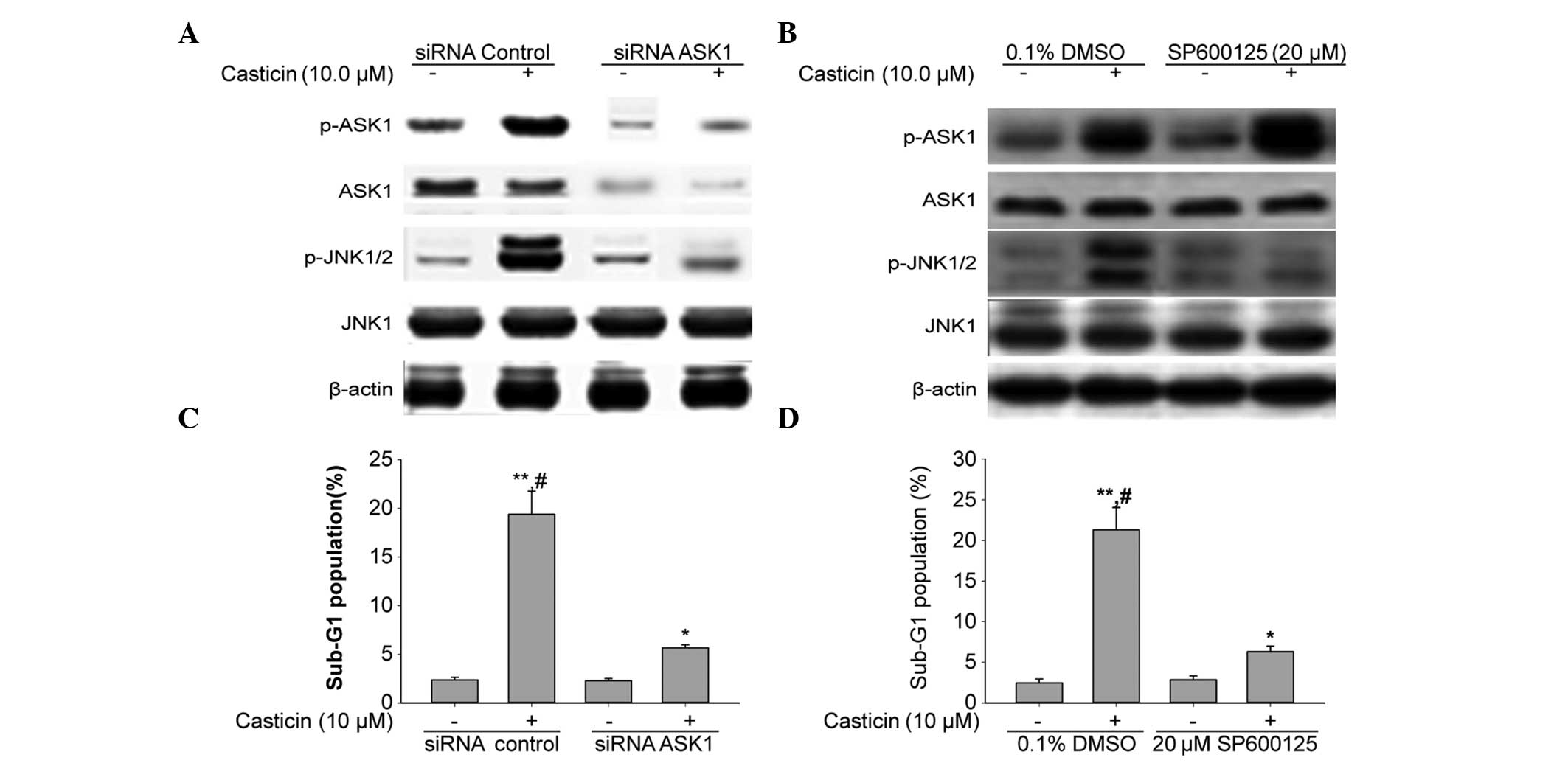 | Figure 5Downregulation of ASK1 using siRNA
inhibits JNK1/2 activation and apoptosis by casticin in HT-29
cells. (A) HT-29 cells were transfected with 100 nM siRNA
control or the siRNA duplexes against ASK1 mRNA. Forty-eight hours
after the transfection, the cells were treated with 10 μM casticin
for 12 h. (B) HT-29 cells were treated with 10 μM casticin for 12 h
in the presence or absence of 20 μM SP600125. (A and B) To assess
whether ASK1 activation was negatively regulated by JNK, western
blotting of p-ASK1, ASK1, p-JNK1/2 and JNK1 was completed following
the downregulation of ASK1 by (A) siRNA transfection and/or (B) the
JNK inhibitor. β-actin was used as the loading control. (C) HT-29
cells were transfected with 100 nM of siRNA control or the siRNA
duplexes against ASK1 mRNA. Forty-eight hours after the
transfection, the cells were treated with 10 μM casticin for 24 h.
*P<0.05 and **P<0.01 vs. siRNA control.
(D) HT-29 cells were treated with 10 μM casticin for 24 h in the
presence or absence of 20 μM SP600125. (C and D) The DNA contents
of the cells were analyzed by flow cytometry. Data are presented as
the mean ± standard deviation (n=3). *P<0.05 and
**P<0.01 vs. 0.1% DMSO; #P<0.05 and
##P<0.01 vs. the same concentration of casticin in
combination with siRNA ASK1 transfection or 20 μM SP600125
treatment. siRNA, small interfering RNA; DMSO, dimethyl sulfoxide;
ASK1, apoptosis signal-regulating kinase 1; p-, phosphorylated-;
JNK, c-Jun N-terminal kinase. |
In order to assess whether ASK1 activation is
negatively regulated by JNK, the cells were treated with a
pharmacological compound named SP600125, a known JNK inhibitor. The
results from the western blot analysis showed that activated ASK1
was not altered by pretreatment with SP600125 (Fig. 5B). These results suggest that ASK1
was activated prior to JNK in HT-29 cells treated with casticin.
Fig. 5C and D shows that the ASK1
siRNA and JNK inhibitor decreased the casticin-induced apoptosis in
the HT-29 cells. We can conclude that casticin may have caused
apoptosis by activating the ASK1-JNK signaling pathway in the HT-29
cells.
Effects of casticin on the generation of
ROS and phosphorylation of ASK1 and JNK in other human colon cancer
cells
We next investigated whether casticin induces
apoptosis by the same modality in other colon cancer cell lines,
including HCT-116, SW480 and Caco-2. As shown in Fig. 6A, FCM results obtained by
monitoring the DCFH-DA probe indicated that casticin significantly
induced ROS generation. Western blot analysis showed that casticin
treatment also caused an increase in ASK1, JNK and Bim-EL
phosphorylation levels in the HCT-116, SW480 and Caco-2 cells
(Fig. 6B). Together, these
findings suggest that casticin-induced apoptotic cell death, ROS
generation and the activation of ASK1, JNK and Bim were not
specific to human colon cancer cell types.
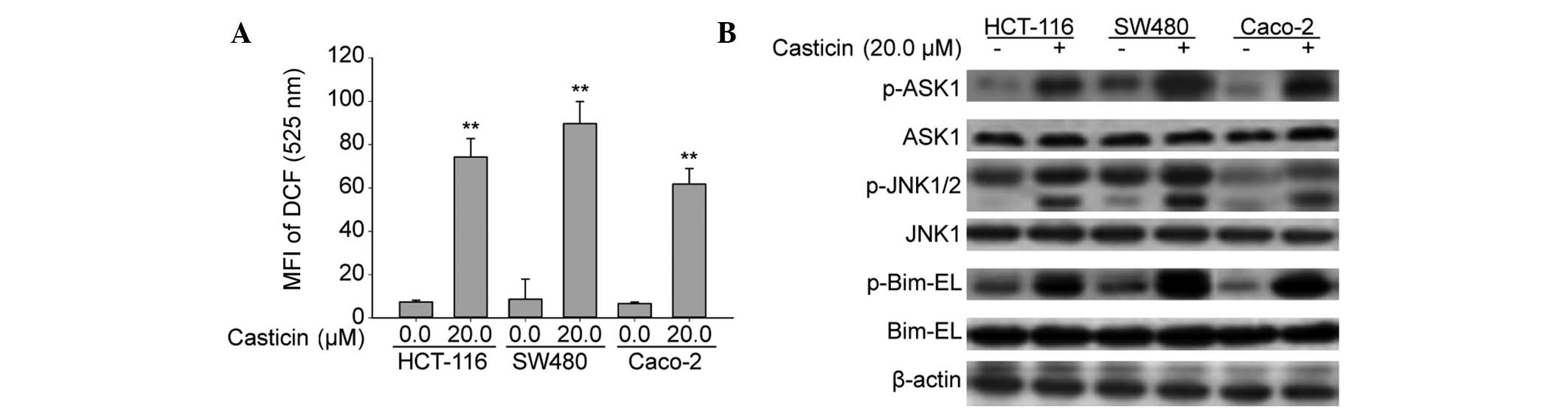 | Figure 6Casticin promotes the production of
ROS and activation of ASK1, JNK and Bim in HCT-116, SW480 and
Caco-2 cells. (A) Treatment of HCT-116, SW480 and Caco-2
cells with 20.0 μM casticin increased the intracellular ROS levels,
as determined by flow cytometry using a 2′,7′-DCF diacetate
fluorescence probe. **P<0.01 vs. 0 μM casticin. (B)
Treatment of HCT-116, SW480 and Caco-2 cells with 20.0 μM casticin
increased the phosphorylation levels of ASK1, JNK1/2 and Bim-EL.
β-actin was used as the loading control. MFI, mean fluorescence
intensity; DCF, dichlorofluorescein; p-. phosphorylated-; ASK1,
apoptosis signal-regulating kinase 1; JNK. c-Jun N-terminal kinase;
Bim, Bcl2-interacting mediator of cell death; Bim-EL, Bim-extra
long; ROS, reactive oxygen species. |
Discussion
The present study examined the apoptotic mechanism
of a potential chemopreventive agent, casticin, which is an active
ingredient of Fructus Viticis that has been widely used as an
anti-inflammatory drug in Traditional Chinese Medicine for
thousands of years (4). Casticin
significantly induced the apoptotic cell death of the human colon
cell lines HT-29, HCT-116, SW480 and Cacao-2 and has been shown to
exhibit similar effects on other cancer cells, including those of
the breast (5), prostate (24), cervix (9,11),
lung (6), liver (10) and hematological system (25). These observations suggest that
casticin can be used as a chemopreventive agent for a variety of
cancers.
Our present and previous studies have demonstrated
that casticin functions as a chemopreventive agent by inducing
apoptosis of tumor cells through the generation of ROS, which
creates oxidative stress (Figs. 1
and 2) (9,11,21,26).
We previously reported that the casticin-induced apoptosis of
hepatocellular carcinoma cells is involved in GSH depletion
(10). In present study, the
results showed that pretreatment of HT-29 cells with NAC markedly
attenuated casticin-induced apoptosis in a concentration-dependent
manner. Furthermore, casticin-induced ROS production was almost
completely inhibited by treatment with 10.0 mM NAC (Fig. 2). Therefore, it is possible that,
due to the disruption of mitochondrial electron transport chain
activity and/or the downstream GSH peroxidase/reductase system,
casticin elevated the intracellular level of ROS in colon cancer
cells; however, this mechanism requires further investigation in
future studies.
In the present study, it was also revealed that
casticin treatment activates the ASK1/JNK-associated signal
transduction pathway (Figs. 3 and
4) by ROS production. ASK1 is
activated by a variety of stresses, including calcium influx, ER
stress, lipopolysaccharide, ROS and tumor necrosis factor (27). These stresses induce the activation
of ASK1 by protein phosphorylation (28). A previous study revealed that the
activation of ASK1 plays a central role in a wide range of cellular
responses, including cell differentiation, apoptosis and the immune
response, with a particular focus on oxidative stress-induced
apoptosis (29). The present
results show that casticin treatment induced ASK1 phosphorylation,
which increased ASK1 activity. These findings suggest that
casticin-induced apoptosis mainly occurs through ASK1
phosphorylation. The activation of ASK1 can selectively activate
JNK, leading to apoptosis. The present study revealed that casticin
treatment caused JNK phosphorylation. To confirm that ASK1
regulates JNK, siRNA was utilized to specifically downregulate ASK1
expression. The phosphorylation of JNK by casticin stimulation was
revealed to be attenuated by siRNA targeting ASK1. However, the
inhibition of JNK with the pharmacological inhibitor SP600125 had
no effect on the phosphorylation of ASK1 protein, possibly
indicating that ASK1 cannot be negatively regulated by JNK.
Bcl-2 family proteins regulate
mitochondria-dependent apoptosis, with the balance of the anti- and
proapoptotic members regulating rates of cell survival and death.
Bim, a proapoptotic member of the Bcl-2 family, causes apoptosis by
disrupting mitochondrial integrity (30). Bim gene expression occurs in
response to selected stress signals. The present study revealed
that casticin induced Bim-EL phosphorylation, suggesting that
Bim-EL phosphorylation is causally associated with casticin-induced
HT-29 cell apoptosis. Furthermore, casticin-induced Bim
phosphorylation was inhibited by an antioxidant, the JNK inhibitor,
and siRNA targeting ASK1. Thus, it is plausible that casticin
activates the ROS-ASK1-JNK cascade causing Bim phosphorylation and
subsequent apoptotic cell death. A previous study indicated that,
in addition to Bim, other BH3-only members of the Bcl-2 family,
including Bcl-2-associated agonist of cell death and
Bcl-2-associated X protein, are involved in casticin-induced cancer
cell apoptosis (31). These
observations explain, at least in part, why blocking the ASK1
signaling cascade did not completely eliminate the casticin-induced
HT-29 cell apoptosis. In conclusion, the present study demonstrates
the possibility that the apoptotic mechanism of casticin is
effected by the production of ROS, which causes activation of the
ASK1-JNK-Bim pathway. The model used in the present study can also
be used in future investigations of these associations.
Acknowledgements
This study was supported by the Municipal Bureau of
Science and Technology of Changsha, Hunan, China (no. K1104060-31),
the Project of Scientific Research of Hunan Province the
Administration Bureau of Traditional Chinese Medicine (no. 201269),
the program for Excellent Talents of Hunan Normal University (no.
ET13107), the Construct Program of the Key Discipline of Basic
Medicine in Hunan Province and the Research Fund for the Doctoral
Program of Hunan Normal University (no. 110656).
References
|
1
|
Sung JJ, Lau JY, Goh KL and Leung WK; Asia
Pacific Working Group on Colorectal Cancer. Increasing incidence of
colorectal cancer in Asia: implications for screening. Lancet
Oncol. 6:871–876. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xiong F, Wu C, Bi X, et al: Risk of
genome-wide association study-identified genetic variants for
colorectal cancer in a Chinese population. Cancer Epidemiol
Biomarkers Prev. 19:1855–1861. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Laubert T, Habermann JK, Hemmelmann C, et
al: Metachronous metastasis- and survival-analysis show prognostic
importance of lymphadenectomy for colon carcinomas. BMC
Gastroenterol. 12:242012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lin S, Zhang H, Han T, Wu JZ, Rahman K and
Qin LP: In vivo effect of casticin on acute inflammation. Zhong Xi
Yi Jie He Xue Bao. 5:573–576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li WX, Cui CB, Cai B, Wang HY and Yao XS:
Flavonoids from Vitex trifolia L. inhibit cell cycle
progression at G2/M phase and induce apoptosis in mammalian cancer
cells. J Asian Nat Prod Res. 7:615–626. 2005.
|
|
6
|
Díaz F, Chávez D, Lee D, et al: Cytotoxic
flavone analogues of vitexicarpin, a constituent of the leaves of
Vitex negundo. J Nat Prod. 66:865–867. 2003.PubMed/NCBI
|
|
7
|
Haïdara K, Zamir L, Shi QW and Batist G:
The flavonoid Casticin has multiple mechanisms of tumor
cytotoxicity action. Cancer Lett. 242:180–190. 2006.PubMed/NCBI
|
|
8
|
Imai M, Kikuchi H, Denda T, Ohyama K,
Hirobe C and Toyoda H: Cytotoxic effects of flavonoids against a
human colon cancer derived cell line, COLO 201: a potential natural
anti-cancer substance. Cancer Lett. 276:74–80. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen D, Cao J, Tian L, Liu F and Sheng X:
Induction of apoptosis by casticin in cervical cancer cells through
reactive oxygen species-mediated mitochondrial signaling pathways.
Oncol Rep. 26:1287–1294. 2011.PubMed/NCBI
|
|
10
|
Yang J, Yang Y, Tian L, Sheng XF, Liu F
and Cao JG: Casticin-induced apoptosis involves death receptor 5
upregulation in hepatocellular carcinoma cells. World J
Gastroenterol. 17:4298–4307. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zeng F, Tian L, Liu F, Cao J, Quan M and
Sheng X: Induction of apoptosis by casticin in cervical cancer
cells: reactive oxygen species-dependent sustained activation of
Jun N-terminal kinase. Acta Biochim Biophys Sin (Shanghai).
44:442–449. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gamaley IA and Klyubin IV: Roles of
reactive oxygen species: signaling and regulation of cellular
functions. Int Rev Cytol. 188:203–255. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xiao D, Powolny AA and Singh SV: Benzyl
isothiocyanate targets mitochondrial respiratory chain to trigger
reactive oxygen species-dependent apoptosis in human breast cancer
cells. J Biol Chem. 283:30151–30163. 2008. View Article : Google Scholar
|
|
14
|
Boveris A: Mitochondrial production of
superoxide radical and hydrogen peroxide. Adv Exp Med Biol.
78:67–82. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kanamoto T, Mota M, Takeda K, et al: Role
of apoptosis signal-regulating kinase in regulation of the c-Jun
N-terminal kinase pathway and apoptosis in sympathetic neurons. Mol
Cell Biol. 20:196–204. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gotoh Y and Cooper JA: Reactive oxygen
species- and dimerization-induced activation of apoptosis
signal-regulating kinase 1 in tumor necrosis factor-alpha signal
transduction. J Biol Chem. 273:17477–17482. 1998. View Article : Google Scholar
|
|
17
|
Nishitoh H, Matsuzawa A, Tobiume K, et al:
ASK1 is essential for endoplasmic reticulum stress-induced neuronal
cell death triggered by expanded polyglutamine repeats. Genes Dev.
16:1345–1355. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Puthalakath H, Huang DC, O’Reilly LA, King
SM and Strasser A: The proapoptotic activity of the Bcl-2 family
member Bim is regulated by interaction with the dynein motor
complex. Mol Cell. 3:287–296. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Strasser A, Puthalakath H, Bouillet P, et
al: The role of bim, a proapoptotic BH3-only member of the Bcl-2
family in cell-death control. Ann NY Acad Sci. 917:541–548. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Song JJ, Rhee JG, Suntharalingam M, Walsh
SA, Spitz DR and Lee YJ: Role of glutaredoxin in metabolic
oxidative stress. Glutaredoxin as a sensor of oxidative stress
mediated by H2O2. J Biol Chem.
277:46566–46575. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang XH, Zheng X, Cao JG, Xiang HL, Liu F
and Lv Y: 8-Bromo-7-methoxychrysin-induced apoptosis of
hepatocellular carcinoma cells involves ROS and JNK. World J
Gastroenterol. 16:3385–3393. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhao XC, Tian L, Cao JG and Liu F:
Induction of apoptosis by 5,7-dihydroxy-8-nitrochrysin in breast
cancer cells: the role of reactive oxygen species and Akt. Int J
Oncol. 37:1345–1352. 2010.PubMed/NCBI
|
|
23
|
Lee BC, Park BH, Kim SY and Lee YJ: Role
of Bim in diallyl trisulfide-induced cytotoxicity in human cancer
cells. J Cell Biochem. 112:118–127. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ohyama K, Akaike T, Hirobe C and Yamakawa
T: Cytotoxicity and apoptotic inducibility of Vitex
agnus-castus fruit extract in cultured human normal and cancer
cells and effect on growth. Biol Pharm Bull. 26:10–18.
2003.PubMed/NCBI
|
|
25
|
Shen JK, Du HP, Yang M, Wang YG and Jin J:
Casticin induces leukemic cell death through apoptosis and mitotic
catastrophe. Ann Hematol. 88:743–752. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jing Y, Dai J, Chalmers-Redman RM, Tatton
WG and Waxman S: Arsenic trioxide selectively induces acute
promyelocytic leukemia cell apoptosis via a hydrogen
peroxide-dependent pathway. Blood. 94:2102–2111. 1999.PubMed/NCBI
|
|
27
|
Matsuzawa A, Saegusa K, Noguchi T, et al:
ROS-dependent activation of the TRAF6-ASK1-p38 pathway is
selectively required for TLR4-mediated innate immunity. Nat
Immunol. 6:587–592. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Tobiume K, Saitoh M and Ichijo H:
Activation of apoptosis signal-regulating kinase 1 by the
stress-induced activating phosphorylation of pre-formed oligomer. J
Cell Physiol. 191:95–104. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Matsuzawa A and Ichijo H: Redox control of
cell fate by MAP kinase: physiological roles of ASK1-MAP kinase
pathway in stress signaling. Biochim Biophys Acta. 1780:1325–1336.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jin HO, Park IC, An S, et al:
Up-regulation of Bak and Bim via JNK downstream pathway in the
response to nitric oxide in human glioblastoma cells. J Cell
Physiol. 206:477–486. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kuo CT, Hsu MJ, et al: Denbinobin induces
apoptosis in human lung adenocarcinoma cells via Akt inactivation,
Bad activation, and mitochondrial dysfunction. Toxicol Lett.
177:48–58. 2008. View Article : Google Scholar : PubMed/NCBI
|