1. Introduction
Subclinical hypothyroidism (SH) denotes a declined
thyroid activity without clear symptoms, but with elevated thyroid
stimulating hormone (TSH) and normal range free thyroxine (FT4) and
triiodothyronine (FT3) as the diagnostic indicators. Depending on
the extent of serum TSH elevation, SH can be divided into mild
(where the concentration of serum TSH is in the range of 4.5–9
mU/l) or severe (TSH≥10 mU/l) SH (1). Mild SH constitutes ~75% of the total
number of patients with SH. SH affects 4–20% of the adult
population, influenced by factors such as age, gender, race, body
mass index, dietary iodine intake and the inconsistency of the
boundary point of serum TSH for SH diagnosis amongst the studies
(2). In patients aged >60
years, the diagnosis poses a challenge as thyroid function test
results may be affected by certain physiological changes due to no
thyroidal illnesses following ageing (3).
Evidence supporting the correlation between SH and
atherosclerosis has been accumulating. The first study regarding
the associated cardiovascular risk in patients with SH was the
long-time large cross-sectional Rotterdam study in the Netherlands,
which showed an increased risk for atherosclerosis and prevalence
of myocardial infarction among female patients with SH aged >55
years (4). Subsequently, another
study also observed an increased risk of congestive heart failure
among older patients with SH that had a TSH level >7.0 mU/l, but
no significant correlation between SH and stroke, peripheral
arterial disease, cardiovascular-associated or total mortality was
revealed (5). Another
cross-sectional analysis further revealed SH to be an independent
risk factor for coronary heart disease, in parallel to
hypercholesterolemia, hypertension, smoking and diabetes (6). The association between SH and
ischemic heart disease and the associated mortality was also
confirmed by the unselected community-based, 20-year follow-up the
Whickham survey (7). In previous
years several meta-analyses have supported these conclusions from
the perspective of evidence-based medicine. One significant
meta-analysis based on the previous studies from 11 countries,
including in the Americas, Europe and Asia, showed that SH is
associated with the coronary heart disease incidence and mortality,
particularly for the group of TSH≥10 mU/l (8). A meta-analysis from Taiwan also
supported this conclusion (9).
Another study identified the original cohort studies with a
systematic review and obtained a more precise estimate of the risks
of the cardiovascular outcomes associated with SH (10). Since the association between SH and
cardiovascular disease is becoming increasingly convincing, it is
intriguing to understand the mechanisms for the association.
Atherosclerosis is characterized by extensive
porridge-like lipid deposit plaques in the great arterial wall, and
accounts for a vast majority of cardiovascular disease incidence.
Therefore, this association is the first to be assessed for the
link between SH and cardiovascular disease. Although a detailed
pathogenesis remains to be studied, atherosclerosis is believed to
be initiated by a combination of special shear flow condition,
sub-intimal lipoprotein deposition and modification, and
endothelial dysfunction (11).
Progression of atherosclerosis features a chronic unresolved
inflammatory response, leading to fibrosis, tissue necrosis and
thrombosis (12). Since
atherosclerosis is a chronic disease, to observe the whole
pathogenesis in one single investigation would require a long time
of study (13). Thus, endothelial
dysfunction, as one of the earliest signs for atherosclerosis,
could be most frequently observed in clinical investigations
(particularly prospective ones), prior to any overt manifestations
of cardiovascular disease (14).
Therefore, endothelial dysfunction would be a favorable initial
factor to investigate the correlation between SH and cardiovascular
disease.
Endothelium dysfunction contributes to
atherosclerosis in a number of ways. In normal conditions, vessel
smooth muscle cells are dependent on a variety of vasoactive agents
released by endothelial cells, including nitric oxide (NO),
thromboxane (TXA2) and prostacyclin (PGI2) to
dilate and constrict efficiently (15). Among these substances, NO,
synthesized by endothelial NO synthase (eNOS), has been recognized
as one of the most important vasodilating agents, since an array of
vasodilating substances have been found to be dependent on NO to
exert their function (16).
eNOS is constitutively expressed in endothelial
cells, however, inducible NOS (iNOS) is more powerful in NO
synthesis but is induced only by inflammatory stimuli (17). NO acts as a second messenger
activating guanylate cyclase in smooth muscle cells, which can lead
to protein kinase G activation leading to smooth muscle cell
hyperpolarization and subsequent relaxation, but the mechanism that
is independent of guanylate cyclase has also discovered (18). As NO has a short half-life, which
is approximately seconds, vascular smooth muscle cells require a
constant source of NO to maintain a normal function. During
increased shear stress condition, the change of blood velocity can
induce NO synthesis and lead to quickly adapted vasodilation, which
is the principle of mediating flow-mediated dilation (FMD) as an
indicator for endothelial function. Declined NO activity caused by
various factors impairs vasodilation in response to various
stimuli, accelerates recruitment of macrophages into the vascular
wall, promotes platelet adhesion, aggregation and thrombosis
(19). Besides vasodilation, NO
also regulates a number of diverse biological processes, such as
vascular permeability, neurotransmission, platelet adhesion and
mitochondrial respiration (20).
Under physiological conditions, the reduction in endothelial NO
bioactivity has been taken as a signature for endothelial
dysfunction. Using NO activity as the marker of endothelial
function can also be justified by another aspect of endothelial
function, which is the material exchange between blood and tissue.
Endothelial NOS function is also found to be regulated by
caveolin-1, the main component of the endocytosis structure
caveolae located in endothelium surface (21), which also mediates low density
lipoprotein (LDL) endocytosis contributing to atherosclerosis
(22). Therefore, NO activity
appears to be able to integrate various aspects of endothelial
function, and thus could be taken as a good indicator.
In clinical practice, endothelial dysfunction could
be represented by endothelium-dependent vasodilation dysfunction,
which reflects, in large part, the action of endothelial-derived
vasodilators, mainly NO. One of the most commonly used non-invasive
examinations of endothelium-dependent vasodilation dysfunction is
using vascular ultrasound to measure the FMD of the conduit
brachial artery. Other non-invasive methods to monitor arterial
stiffness as an indicator for endothelial function are also
available, including pulse wave velocity and arterial
distensibility measurement (23).
Carotid intima-media thickness (CIMT), which indicates the extent
of lipid deposition, is also a common marker of early
atherosclerosis.
Previously, microRNAs (miRNAs or miRs), a class of
short, single-stranded, small non-coding RNAs regulating gene
expression, emerged as a novel aspect in several diseases,
including endothelial dysfunction (24). Since miRNAs are highly expressed in
endothelial cells, circulation miRNA is mainly composed of
endothelial miRNA. Serum miRNA may be an extremely promising
indicator for endothelial function. The significance of different
miRNAs concerning endothelial function is undergoing increasing
investigations. For example, while miR-10a and miR18b appear to
suppress nuclear factor-κB (NF-κB) downstream signaling by
inhibiting NF-κB nuclear translocation, miR-146 has been shown to
promote NF-κB activation and eNOS expression. Shear stress, one of
the factors affecting endothelial dysfunction, has been found to
induce miR-10a and miR-92a (25).
miR-92a is also upregulated by oxLDL, which promotes endothelial
activation and the development of atherosclerotic lesions (26). There are also several miRNAs that
can reflect the change of endothelial function in atherosclerosis,
such as miR-34, miR-217, miR-146, miR-126, miR-92a and miR-21, and
these have been reviewed in a previous study (27). In addition to the classical
biomarkers, such as C-reactive protein (CRP), miRNAs may be
translated into novel therapeutic approaches and even be available
as approved drugs to humans in the future. As the association
between different miRNAs and atherosclerosis is becoming clear,
using miRNAs to understand the endothelial change in different
conditions would be feasible.
For example, the present study is trying to
establish novel indicators linking SH and endothelial function. Our
previous study (data not published) indicates that several miRNAs,
including miR-125a and miR-21-5p, all change in SH. The results
will be published in a forthcoming issue (28).
2. SH accelerates endothelial
dysfunction
Patients with SH have been found to be disposed to
endothelial dysfunction. In one clinical study, Turemen et
al (29) used brachial artery
responses to endothelium-dependent (FMD) and
endothelium-independent stimuli [sublingual nitroglycerin (NTG)] as
indicators for endothelial dysfunction, and found that when
confounding factors were excluded, patients with SH have a
statistically lower FMD and NTG response compared to the controls,
with FMD impairment correlating to serum TSH level. This study not
only revealed the increased endothelial dysfunction incidence in
patients with SH, but also indicated a possible role of serum TSH
in this phenomenon. Subsequently, the direct effect of TSH on
vessel dilation was observed in the study by Dardano et al
(30), which discovered impaired
FMD following recombinant human TSH (rhTSH) acute administration in
patients monitored for differentiated thyroid carcinoma. In this
study, the inflammation and oxidative stress indicators were also
assessed and implied as a possible interpretation for the link
between TSH and endothelial change.
In the past 10 years, several studies have indicated
that SH is associated with increased CIMT, but the data are
sometimes inconsistent. Recently a meta-analysis that included
eight observational studies with 3,602 SH patients fulfilling the
eligibility criteria made a conclusion that SH was associated with
an increase of CIMT correlated with TSH elevation, particularly
when TSH >10.0 mU/l. An increase of IMT was also found in
patients with TSH <10 mU/l, regardless of significant
heterogeneity (31).
The mechanism underlying the correlation remains
unknown. In SH, the only noticeable change is the elevation of TSH,
and it is possible that the elevation of TSH can bind extra TSH
receptor (TSHR) to exert its function. The discovery by Balzan
et al (32) that TSHR is
expressed by microvascular endothelial cells opens up a novel
prospective for understanding the association. The function of the
TSHR is further confirmed in a recently published study carried out
by Tian et al (33), which
indicates that elevated TSH can promote endothelial dysfunction in
human umbilical vein endothelial cells by attenuating eNOS and
prostacylin (PGI2) expression in a dose- and time-dependent
manner.
Recently, the association between cav-1 and thyroid
function has gained increasing attention. One study carried out by
Wang et al (34) found an
upregulation of caveolin-1 in the hippocampus and cerebella of
developmental hypothyroidism rat created by iodine deficient diet
or propylthiouracil (PTU) administration (35,35)
indicating an effect of hypothyroidism on cav-1 function. Another
study using adult hypothyroidism and age-matched euthyroid rats not
only confirmed the inductive effect of hypothyroidism on cav-1, but
also observed decreased eNOS activities and further revealed a
possible mechanism underlying the link between hypothyroid and
endothelial dysfunction (36).
However, since the above findings are all obtained from hypothyroid
animal models, whether or not SH has a similar effect on cav-1
requires investigation. However, as TSHRs are located and regulated
by constitutive multimerization within lipid micro-domains on the
plasma membrane (37), cav-1 and
TSHR signaling may be closely associated. As mentioned, TSH can
regulate endothelial function directly and therefore, the
interaction between cav-1 and TSH may be another significant
element to uncover the mechanism of endothelial dysfunction in SH
state.
The findings above support a direct effect of TSH
during the process. However, the body is an entity and therefore
there may be other noteworthy factors that could also impair
endothelial function in SH.
3. Mechanism of SH accelerating endothelial
dysfunction
Dyslipidemia
Hyperlipidemia is one of the common causal factors
of endothelial dysfunction. In endothelial cells, hyperlipidemia
can disturb the NO synthesis pathway by increasing levels of
asymmetric dimethylarginine (ADMA), the endogenous NO synthesis
inhibitor, possibly by reducing enzyme dimethylarginine
dimethylaminohydrolase (DDAH) activity (38). High density lipoprotein (HDL)
cholesterol has also been reported to improve the endothelial
dysfunction by stimulating NO release and inducing vasodilation in
the isolated aorta via Akt-mediated eNOS phosphorylation and
intracellular Ca2+ mobilization, and therefore is
protective to endothelial function (39). One of the major components of
plaques is lipid, elevation of serum LDL levels is recognized to
promote subintimal lipid deposition, and therefore more subintimal
modified LDL to aggravate endothelial dysfunction.
However, SH is also known for its correlation with
dyslipidemia. Therefore, SH may induce endothelial dysfunction by
increased lipid disorders. A study in the DaDong district of
Shenyang (China), showed that elevation of TSH, across the entire
TSH reference range, exhibits a positive correlation with serum
total cholesterol (TC), triglycerides, LDL cholesterol (LDL-C), and
a negative correlation to HDL cholesterol. Serum TSH was also found
to be positively correlated with the prevalence of obesity, which
also suggested that serum TSH may be a risk factor for metabolic
syndrome (40). Another
cross-sectional, population-based study further showed that
subsequent to excluding the possible interference of insulin
sensitivity, raised serum TSH remains a risk factor of dyslipidemia
(41). Data of the present authors
also supports a correlation between SH and atherosclerosis lipid
profile changes. The association between elevated TSH and
atherogenic lipid profiles (increase of TC, LDL-C and ox-LDL) is
observed even with a mild elevation of serum TSH, particularly in
postmenopausal females, a population with an increased risk of
atherosclerosis (42).
Recently, a clinical study performed by Xiang et
al (43) showed a close
association between increased postprandial lipaemia (PPL) and
impaired endothelial function in patients with SH by analyzing FMD
change prior and subsequent to an oral fat-loading in overt
hypothyroidism, SH and normal control groups. Oral fat-challenged
FMD was found to be impaired, while TC, LDL-C, CRP and
thiobarbituric acid reactive substance (TBARS) levels were also
found to be higher in patients with SH compared to the control
group, further indicating a role of dyslipidemia underlying the
bias to endothelial dysfunction in patients with SH.
The mechanism between SH and hyperlipidemia has been
interpreted to a certain extent. The present authors demonstrated
that TSH, acting on the TSHR in liver cells, upregulated the
expression of hepatic 3-hydroxy-3-methyl-glutaryl coenzyme A
reductase (HMGCR), a rate-limiting enzyme in cholesterol synthesis
in the liver. The results revealed the direct effect of TSH on
cholesterol levels in the liver form a novel perspective and
possibly partially explained hypercholesterolemia in SH (44).
Low grade chronic inflammation
factors
Chronic inflammation may initiate and promote
atherosclerosis or its complications by adverse effects on the
vascular endothelium, and it may be one of the contributing factors
that lead to the increased endothelial dysfunction in patients with
SH. The clinical study carried out by Turemen et al
(29) observed not only an
elevation of several inflammation indicators, including
interleukin-6 (IL-6), tumor necrosis factor-α (TNF-α) and CRP, in
patients with SH, but also a positive correlation of FMD between
these inflammation factors, indicating that low grade chronic
inflammation may be one of the factors that can promote endothelial
dysfunction in SH.
Another study by Taddei et al (45) revealed higher CRP and IL-6 values
and reduced vasodilation to Ach in the SH group compared to the
controls. The reduced vasodilation was resistant to N-mono methyl
arginine (L-NMMA), a NOS inhibitor, and normalized by vitamin C,
confirming an impaired NO availability. It was also normalized
following systemic but not local administration of indomethacin or
celecoxib, a selective COX-2 inhibitor, and therefore a
COX-2-dependent pathway may be involved. As COX-2 is the induced
type of COX that is upregulated during the inflammatory response,
inflammation is considered to play a role in endothelial
dysfunction.
Various inflammatory mediators, including IL-6,
TNF-α and CRP, have been found to be linked to SH, and thus require
more study. For example, IL-6, a pro-inflammatory cytokine that is
found to be detrimental to endothelium and atherosclerosis
(46) is also found to be induced
by TSH in preadipocytes (47).
TNF-α, another inflammatory cytokine that can impair NO activity in
endothelial cells by promoting oxidative stress (another aspect of
endothelial dysfunction) (48),
has been found to be induced in bone marrow cells by TSH (49). Furthermore, CRP, one of the ‘acute
phase proteins’ generated by liver under inflammatory challenge, a
traditionally used inflammatory marker later discovered to be
indicative of cardiovascular events, is observed to be increased in
patients with SH (50), male
patients with SH aged <50 years (51), or found to be in positive
correlation to TSH (52). CRP can
interfere with endothelial function directly by downregulation of
eNOS and upregulation of endothelin-1 (ET-1), which is a potent
vasoconstrictor that can antagonize NO action (53).
Oxidant stress
Oxidant stress indicates an imbalance between the
oxidant and antioxidant substance, during which reactive oxygen
species (ROS), a family of molecules including hydroxyl radical,
superoxide anion and their derivatives, exceeds endogenous
antioxidant defense mechanisms. Inflammation is an important cause
of oxidative stress as enzymatic systems producing a large amount
of ROS, including xanthine oxidase and nicotinamide adenine
dinucleotide (NADH)/NADH phosphate oxidase (NOX), are induced by
inflammatory stimuli. Since ROS reacts with NO extremely readily,
producing even more harmful reactive nitric intermediates, minimum
oxidative stress in endothelial cells can uncouple NO synthesis and
will be devastating to endothelial function (54). Induction of iNOS can therefore
aggravate oxidative stress in inflammatory conditions (55). ROS can also impair endothelial
function by activating NF-κB, which further increases the
expression of inflammation-associated genes (56), and thus act as a negative feedback
loop. As inflammation appears to link to SH, it is reasonable to
expect another link between oxidative stress and SH.
However, this link was not observed initially. In
the clinical study carried out by Coria et al (57), several serum indicators of
oxidative stress were utilized, including NO concentration, TBARS
(a by-product of lipid peroxidation) and paraoxonase (PON, a major
component of HDL), to represent the oxidative stress of each
individual, and denied a clear oxidative stress in the SH group.
However, this conclusion was inconsistent with the studies that
followed. Another study by Torun et al (58) found an altered level of
malondialdehyde (MDA; an indicator for lipid peroxidation) and
total antioxidant status (TAS, an indicator for overall
antioxidative activity) in SH and overt hypothyroidism (OHT) states
compared to normal control. Cebeci et al (59) also found lower activity of PON and
arylesterase (ARE) in patients with SH. These two studies are
supportive of an increased oxidative stress in patients with SH. In
the study carried out by Cebeci et al (59), while MDA, diene conjugate (DC),
protein carbonyl (PC), nitrotyrosine (NT) levels and ferric
reducing antioxidant power (FRAP) were all increased in overt
hypothyroid patients, only MDA levels were statistically increased
in patients with SH (60).
The different conclusions drawn from different
biomarkers may partially explain the inconsistencies of the above
studies, and also suggest an investigation of how these biomarkers
are different and which of them would represent a particular
process or stage of oxidative stress, and which would be the most
specific to be used as an indicator for body overall oxidation
condition. As a correlation between serum TSH and those oxidative
stress indicators was also observed (60), a direct effect of TSH to promote
oxidative stress is possible. This possibility is supported by the
study of Dardano et al (30), which found that rhTSH acute
injection can lead to oxidative stress. The contribution of
oxidative stress to the impaired endothelial function in patients
with SH is confirmed in a study (61), which observed a reduction of TBARS
levels and a marked improvement of FMD after three weeks of
α-lipoic acid antioxidant therapy, indicating a causal association
between oxidative stress and endothelial dysfunction in an SH
condition. In conclusion, there is growing evidence supportive to a
higher overall oxidative burden among patients with SH associated
to their impaired endothelial function.
Insulin resistance (IR)
IR is defined as decreased sensitivity and/or
responsiveness to metabolic actions of insulin. IR is associated
with dyslipidemia, chronic inflammation and oxidative stress, as
facets of metabolic syndrome. As discussed above, dyslipidemia,
chronic inflammation and oxidative stress can all directly promote
endothelial dysfunction. Metabolic syndrome has been taken as an
important underlying cause for the majority of the cardiovascular
diseases. The effect of IR on endothelial dysfunction was confirmed
by the observation that endothelium-dependent coronary vasodilation
is in association with the severity of IR in non-diabetic patients
(62). Despite the link with
metabolic syndromes, however, IR could also influence endothelial
function directly. Insulin stimulates the production of NO and
suppresses secretion of ET-1 in endothelium through a
phosphatidylinositol 3-kinase-dependent (PI-3K) pathway, leading to
vasodilation. IR can therefore lead to the imbalance between NO and
ET-1, manifested as endothelial dysfunction, which in turns leads
to decreased blood flow, and can worsen IR (63).
The evidence of a predisposition of IR among
patients with SH are accumulating. Patients with SH have been found
to have a higher homeostasis model assessment of IR (HOMA-IR) score
correlated with TSH level (64),
higher plasma insulin and increased HOMA-IR score (65), or a higher insulin level only
without affecting the HOMA-IR statistically (66). They are also supportive to a higher
cardiovascular risk in patients with SH, and the inconsistencies
between studies may possibly be due to the difference of insulin
level and HOMA-IR in their specificity and sensitivity.
Although it could be attributed to their link to
metabolic syndrome, the detailed mechanism underlying the
association between SH and IR remains uncertain. Peeters et
al (67) examined two common
polymorphisms of Tshr, Tshr-Pro52Thr and
Tshr-Asp727Glu, and concluded that Asp727Glu was
associated with IR in healthy elderly males. Taking this finding
together with the increased IR risk in SH, which is characterized
by elevated serum TSH, it is possible that individuals with
Tshr-Asp727Glu can express TSHRs with a higher affinity to
TSH, which can also result in increased TSHR signaling mimicking
the SH situation, and thus are predisposed to IR.
4. Levo-thyroxine (L-T4) replacement
Levo-thyroxine (L-T4) is the replacement therapy
drug used in hypothyroidism. Taking the proper amount of L-T4 aids
to increase serum FT4 and FT3 levels in SH, and can reduce TSH
secretion with negative feedback on the pituitary. The effect of
L-T4 replacement would be useful to verify the causal association
between endothelial dysfunction and SH. However, current evidence
remains insufficient to confirm the beneficial effect of L-T4
therapy on patients with SH.
In the study carried out by Taddei et al
(68), after six months of L-T4
replacement, a clear improvement in acetylcholine-induced
vasodilatation, associated with a restoration of the inhibitory
activity of L-NMMA, was observed. The study concluded that the
alteration of lipid profile, inflammatory status and the direct
effect of thyroid hormone in patients with SH jointly contributed
to their impaired endothelial dependent vasodilation and early L-T4
replacement therapy may be advisable to slow down atherogenesis.
Similarly, a double-blind, placebo-controlled L-T4 replacement
study, with 45 patients with SH and 32 controls aged <55 years,
also observed a significant improvement in lipoprotein profile and
IMT after six months of treatment (69).
Another similar clinical study also observed an
increased nitroglycerin-induced diameter (NID) following LT4
treatment in patients with SH, although the difference of NID
values between SH and the control group prior to the treatment was
not statistically significant (70). A randomized, double-blind, 12 weeks
crossover study by Razvi et al (71) also showed that L-T4 treatment can
reduce TC, LDL-C, waist-to-hip ratio and improve FMD in patients
with SH, which are all protective of cardiovascular events. The
study also noted that, although remaining within the normal range,
an increased level of free T4 concentration was associated to the
cardiovascular risk factor reduction (71). Another study by Adrees et al
(72), also reported an
improvement endothelial function characterized by an increase of
carotid artery baseline diameter, a decreased CIMT and an increased
endothelium-dependent vasodilatation. All these investigations
indicate that thyroid hormone replacement therapy may be beneficial
to endothelial dysfunction in patients with SH.
However, there were also contradictory opinions.
Another replacement study showed that although there was a
significant decrease in FMD in the SH compared to control group
prior to treatment, the improvement of 12 months L-T4 treatment on
FMD and mean CIMT were not statistically significant (73). Another study analyzing the United
Kingdom General Practitioner Research Database suggested that
treatment of SH with L-T4 was associated with fewer ischemic heart
disease events in younger individuals, but this was not evident in
older people. This finding is consistent with a recent review
conducted by Pasqualetti et al (74), concluding that the increased
cardiovascular risk in SH participants is more evident among the
young patients, and the oldest subjects (>85 years) should avoid
hormonal treatment due to negative effects of possible
overtreatment. An appropriately powered randomized controlled trial
of L-T4 in SH examining vascular outcomes is warranted,
particularly to value the effect for the older patients (75).
5. Conclusion
SH, associated with elevation of TSH while FT3 and
FT4 remain normal, is convincingly associated with increased
cardiovascular risk. As the earliest sign of atherosclerosis,
endothelial dysfunction is most frequently observed in clinical
studies. Weakened endothelium dependent vasodilation, together with
other indicators of endothelial dysfunction, have been found to be
associated with SH, supporting the correlation between SH and
cardiovascular disease. The emergence of miRNAs as an indicator for
endothelial function may help to further reveal this correlation.
From various clinical investigations, factors contributive to
endothelial dysfunction, dyslipidemia, chronic inflammation,
oxidative stress and IR also link with SH, which partly accounts
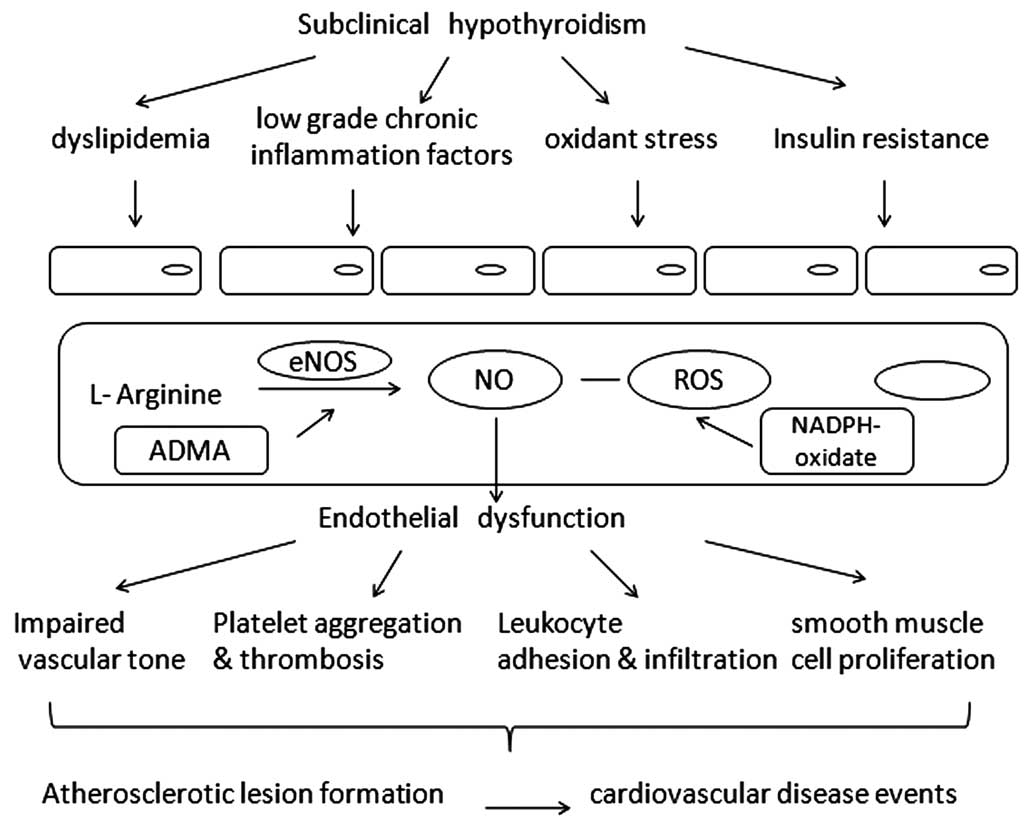
for the correlation (Fig. 1).
However, all these factors interacted with each other, with none
playing the decisive role alone. For example, IR is induced by
inflammation and oxidative stress, and oxidative stress also plays
a role in dyslipidemia. Inflammation and oxidative stress promote
each other. All the aforementioned induce endothelial dysfunction,
resulting in early stage atherogenesis.
Elevated TSH alone, independent of FT3 and FT4, has
been shown to modulate the whole process. It has been discovered
that TSH is able to bind hepatocyte TSHR to promote cholesterol
synthesis, bind adipocyte TSHR to induce IL-6 synthesis and bind
bone marrow cell TSHR to increase TNF-α secretion. A polymorphism
in Tshr has also been identified to be associated with IR,
indicating a possible role of TSH signaling in IR pathogenesis.
These actions of TSH are closely associated with altered
endothelial function, and therefore could be promising mechanisms
underlying the correlation between SH and endothelial function.
However, endothelial cells also express TSHR, therefore TSH could
also bind to endothelial TSHR to exert its effect, which remains a
largely limited area until recently. The study of the contribution
of endothelial TSHR to endothelial dysfunction would be noteworthy.
Studies on extra thyroidal TSHR function have been supportive to
clinical findings, and have complemented the understanding of the
observed correlation. The novel association between cav-1 and
thyroid function also suggests cav-1 may be another notable element
of the underlying mechanism.
Using the currently available data, L-T4 replacement
therapy is beneficial for the recovery of endothelial cells from
early stage injury in the majority of cases, particularly in the
severe SH (TSH≥10 mU/l) group. Early intervention can slow down the
progress of atherosclerosis and improve the condition that
long-term SH increases the risk of cardiovascular events. However,
L-T4 replacement therapy would also increase the risk of osteopenia
and atrial fibrillation, and there remain controversial opinions on
substitution treatment, particularly for the elderly (76). Therefore, more large-scale and
long-term evidence-based medical study data to further demonstrate
the feasibility of replacement therapy are required for a finalized
conclusion.
Acknowledgements
The present study was written by Dr Ming Lu. Dr
Chongbo Yang revised the manuscript and was involved the
discussion. The present study was supported in part by grants from
the National Basic Research Program (no. 2012CB524900), the
National Natural Science Foundation (nos. 81230018, 81170794 and
81270869 30971409) and the Department of Science and Technology of
Shandong Province of China (no. 2012GSF11824).
References
|
1
|
Surks MI, Ortiz E, Daniels GH, et al:
Subclinical thyroid disease: scientific review and guidelines for
diagnosis and management. JAMA. 291:228–238. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cooper DS and Biondi B: Subclinical
thyroid disease. Lancet. 379:1142–1154. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Boelaert K: Thyroid dysfunction in the
elderly. Nat Rev Endocrinol. 9:194–204. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hak AE, Pols HA, Visser TJ, Drexhage HA,
Hofman A and Witteman JC: Subclinical hypothyroidism is an
independent risk factor for atherosclerosis and myocardial
infarction in elderly women: the Rotterdam study. Ann Intern Med.
132:270–278. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Rodondi N, Newman AB, Vittinghoff E, et
al: Subclinical hypothyroidism and the risk of heart failure, other
cardiovascular events, and death. Arch Intern Med. 165:2460–2466.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Walsh JP, Bremner AP, Bulsara MK, et al:
Subclinical thyroid dysfunction as a risk factor for cardiovascular
disease. Arch Intern Med. 165:2467–2472. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Razvi S, Weaver JU, Vanderpump MP and
Pearce SH: The incidence of ischemic heart disease and mortality in
people with subclinical hypothyroidism: reanalysis of the Whickham
Survey cohort. J Clin Endocrinol Metab. 95:1734–1740. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rodondi N, den Elzen WP, Bauer DC, et al:
Subclinical hypothyroidism and the risk of coronary heart disease
and mortality. JAMA. 304:1365–1374. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tseng FY, Lin WY, Lin CC, et al:
Subclinical hypothyroidism is associated with increased risk for
all-cause and cardiovascular mortality in adults. J Am Coll
Cardiol. 60:730–737. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gencer B, Collet TH, Virgini V, Auer R and
Rodondi N: Subclinical thyroid dysfunction and cardiovascular
outcomes among prospective cohort studies. Endocr Metab Immune
Disord Drug Targets. 13:4–12. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lusis AJ: Atherosclerosis. Nature.
407:233–241. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Galkina E and Ley K: Immune and
inflammatory mechanisms of atherosclerosis. Ann Rev Immunol.
27:1652009. View Article : Google Scholar
|
|
13
|
Libby P, Ridker PM and Hansson GK:
Progress and challenges in translating the biology of
atherosclerosis. Nature. 473:317–325. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bonetti PO, Lerman LO and Lerman A:
Endothelial dysfunction: a marker of atherosclerotic risk.
Arterioscler Thromb Vasc Biol. 23:168–175. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Makino A and Kamata K: Possible modulation
by endothelin-1, nitric oxide, prostaglandin I2 and thromboxane A2
of vasoconstriction induced by an α-agonist in mesenteric arterial
bed from diabetic rats. Diabetologia. 41:1410–1418. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Beckman JS and Koppenol WH: Nitric oxide,
superoxide, and peroxynitrite: the good, the bad, and the ugly. Am
J Physiol. 40:C14241996.
|
|
17
|
Alderton WK, Cooper CE and Knowles RG:
Nitric oxide synthases: structure, function and inhibition. Biochem
J. 357:593–615. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hollenberg SM and Cinel I:
Bench-to-bedside review: nitric oxide in critical illness-update
2008. Crit Care. 13:2182009. View
Article : Google Scholar
|
|
19
|
Esper RJ, Nordaby RA, Vilariño JO,
Paragano A, Cacharrón JL and Machado RA: Endothelial dysfunction: a
comprehensive appraisal. Cardiovasc Diabetol. 5:4–22. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mudau M, Genis A, Lochner A and Strijdom
H: Endothelial dysfunction: the early predictor of atherosclerosis.
Cardiovasc J Afr. 23:222–231. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Frank PG, Pavlides S and Lisanti MP:
Caveolae and transcytosis in endothelial cells: role in
atherosclerosis. Cell Tissue Res. 335:41–47. 2009. View Article : Google Scholar
|
|
22
|
Sowa G: Caveolae, caveolins, cavins, and
endothelial cell function: new insights. Front Physiol. 2:1202012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Widlansky ME, Gokce N, Keaney JF Jr and
Vita JA: The clinical implications of endothelial dysfunction. J Am
Coll Cardiol. 42:1149–1160. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kim VN: MicroRNA biogenesis: coordinated
cropping and dicing. Nat Rev Mol Cell Biol. 6:376–385. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sun X, Belkin N and Feinberg MW:
Endothelial microRNAs and atherosclerosis. Curr Atheroscler Rep.
15:1–13. 2013. View Article : Google Scholar
|
|
26
|
Loyer X, Potteaux S, Vion AC, et al:
Inhibition of MicroRNA-92a Prevents Endothelial Dysfunction and
Atherosclerosis in Mice. Circ Res. 114:434–443. 2014. View Article : Google Scholar
|
|
27
|
Menghini R, Casagrande V and Federici M:
MicroRNAs in endothelial senescence and atherosclerosis. J
Cardiovasc Transl Res. 6:924–930. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang X, Shao S, Geng H, et al: Expression
profiles of six circulating microRNAs critical to atherosclerosis
in patients with subclinical hypothyroidism: a clinical study. J
Clin Endocrinol Metab. 99:E766–774. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Turemen EE, Cetinarslan B, Sahin T,
Canturk Z and Tarkun I: Endothelial dysfunction and low grade
chronic inflammation in subclinical hypothyroidism due to
autoimmune thyroiditis. Endocr J. 58:349–354. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dardano A, Ghiadoni L, Plantinga Y, et al:
Recombinant human thyrotropin reduces endothelium-dependent
vasodilation in patients monitored for differentiated thyroid
carcinoma. J Clin Endocrinol Metab. 91:4175–4178. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gao N, Zhang W, Zhang YZ, Yang Q and Chen
SH: Carotid intima-media thickness in patients with subclinical
hypothyroidism: a meta-analysis. Atherosclerosis. 227:18–25. 2013.
View Article : Google Scholar
|
|
32
|
Balzan S, Del Carratore R, Nicolini G, et
al: Proangiogenic effect of TSH in human microvascular endothelial
cells through its membrane receptor. J Clin Endocrinol Metab.
97:1763–1770. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tian L, Zhang L, Liu J, Guo T, Gao C and
Ni J: Effects of TSH on the function of human umbilical vein
endothelial cells. J Mol Endocrinol. 52:215–222. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang Y, Zhong J, Wei W, et al:
Developmental iodine deficiency and hypothyroidism impair neural
development, upregulate caveolin-1, and downregulate
synaptotagmin-1 in the rat cerebellum. Biol Trace Elem Res.
144:1039–1049. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gong J, Dong J, Wang Y, et al:
Developmental iodine deficiency and hypothyroidism impair neural
development, up-regulate caveolin-1 and down-regulate synaptophysin
in rat hippocampus. J Neuroendocrinology. 22:129–139. 2010.
View Article : Google Scholar
|
|
36
|
Sarati LI, Martinez CR, Artés N, et al:
Hypothyroidism: age-related influence on cardiovascular nitric
oxide system in rats. Metabolism. 61:1301–1311. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Latif R, Ando T and Davies TF: Lipid rafts
are triage centers for multimeric and monomeric thyrotropin
receptor regulation. Endocrinology. 148:3164–3175. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ito A, Tsao PS, Adimoolam S, Kimoto M,
Ogawa T and Cooke JP: Novel mechanism for endothelial dysfunction:
dysregulation of dimethylarginine dimethylaminohydrolase.
Circulation. 99:3092–3095. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Nofer J-R, van der Giet M, Tölle M, et al:
HDL induces NO-dependent vasorelaxation via the lysophospholipid
receptor S1P3. J Clin Invest. 113:569–581. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lai Y, Wang J, Jiang F, et al: The
relationship between serum thyrotropin and components of metabolic
syndrome. Endocr J. 58:23–30. 2011. View Article : Google Scholar
|
|
41
|
Åsvold BO, Vatten LJ, Nilsen TI and Bjøro
T: The association between TSH within the reference range and serum
lipid concentrations in a population-based study. The HUNT Study.
Eur J Endocrinol. 156:181–186. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Geng H, Zhang X, Wang C, et al: Even
mildly elevated TSH is associated with an atherogenic lipid profile
in postmenopausal women with subclinical hypothyroidism. Endocr
Res. Mar 28–2014.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xiang GD, Xiang LW, He HL and Zhao LS:
Postprandial lipaemia suppresses endothelium-dependent arterial
dilation in patients with hypothyroidism. Endocrine. 42:391–398.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tian L, Song Y, Xing M, et al: A novel
role for thyroid-stimulating hormone: Up-regulation of hepatic
3-hydroxy-3-methyl-glutaryl-coenzyme a reductase expression through
the cyclic adenosine monophosphate/protein kinase A/cyclic
adenosine monophosphate-responsive element binding protein pathway.
Hepatology. 52:1401–1409. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Taddei S, Caraccio N, Virdis A, et al:
Low-grade systemic inflammation causes endothelial dysfunction in
patients with Hashimoto’s thyroiditis. J Clin Endocrinol Metab.
91:5076–5082. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ridker PM, Rifai N, Stampfer MJ and
Hennekens CH: Plasma concentration of interleukin-6 and the risk of
future myocardial infarction among apparently healthy men.
Circulation. 101:1767–1772. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Antunes TT, Gagnon A, Bell A and Sorisky
A: Thyroid-stimulating hormone stimulates interleukin-6 release
from 3T3-L1 adipocytes through a cAMP-protein kinase A pathway.
Obes Res. 13:2066–2071. 2005. View Article : Google Scholar
|
|
48
|
Zhang C, Xu X, Potter BJ, et al: TNF-alpha
contributes to endothelial dysfunction in ischemia/reperfusion
injury. Arterioscler Thromb Vasc Biol. 26:475–480. 2006. View Article : Google Scholar
|
|
49
|
Whetsell M, Bagriacik EU, Seetharamaiah
GS, Prabhakar BS and Klein JR: Neuroendocrine-induced synthesis of
bone marrow-derived cytokines with inflammatory immunomodulating
properties. Cell Immunol. 192:159–166. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Christ-Crain M, Meier C, Guglielmetti M,
et al: Elevated C-reactive protein and homocysteine values:
cardiovascular risk factors in hypothyroidism? A cross-sectional
and a double-blind, placebo-controlled trial. Atherosclerosis.
166:379–386. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kvetny J, Heldgaard PE, Bladbjerg EM and
Gram J: Subclinical hypothyroidism is associated with a low-grade
inflammation, increased triglyceride levels and predicts
cardiovascular disease in males below 50 years. Clin Endocrinol
(Oxf). 61:232–238. 2004. View Article : Google Scholar
|
|
52
|
Tuzcu A, Bahceci M, Gokalp D, Tuzun Y and
Gunes K: Subclinical hypothyroidism may be associated with elevated
high-sensitive c-reactive protein (low grade inflammation) and
fasting hyperinsulinemia. Endocr J. 52:89–94. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Verma S, Li S-H, Badiwala MV, et al:
Endothelin antagonism and interleukin-6 inhibition attenuate the
proatherogenic effects of C-reactive protein. Circulation.
105:1890–1896. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Cai H and Harrison DG: Endothelial
dysfunction in cardiovascular diseases: the role of oxidant stress.
Circ Res. 87:840–844. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lubos E, Handy DE and Loscalzo J: Role of
oxidative stress and nitric oxide in atherothrombosis. Front
Biosci. 13:5323–5344. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
56
|
Higashi Y, Noma K, Yoshizumi M and Kihara
Y: Endothelial function and oxidative stress in cardiovascular
diseases. Circ J. 73:411–418. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Coria MJ, Pastran AI and Gimenez MS: Serum
oxidative stress parameters of women with hypothyroidism. Acta
Biomed. 80:135–139. 2009.PubMed/NCBI
|
|
58
|
Torun AN, Kulaksizoglu S, Kulaksizoglu M,
Pamuk BO, Isbilen E and Tutuncu NB: Serum total antioxidant status
and lipid peroxidation marker malondialdehyde levels in overt and
subclinical hypothyroidism. Clin Endocrinol (Oxf). 70:469–474.
2009. View Article : Google Scholar
|
|
59
|
Cebeci E, Alibaz-Oner F, Usta M, Yurdakul
S and Erguney M: Evaluation of oxidative stress, the activities of
paraoxonase and arylesterase in patients with subclinical
hypothyroidism. J Investig Med. 60:23–28. 2012.
|
|
60
|
Ozturk U, Vural P, Ozderya A, Karadag B,
Dogru-Abbasoglu S and Uysal M: Oxidative stress parameters in serum
and low density lipoproteins of Hashimoto’s thyroiditis patients
with subclinical and overt hypothyroidism. Int Immunopharmacol.
14:349–352. 2012. View Article : Google Scholar
|
|
61
|
GDX, JHP, HLS and LSZ: Alpha-lipoic acid
improves endothelial dysfunction in patients with subclinical
hypothyroidism. Exp Clin Endocrinol Diabetes. 118:625–629. 2010.
View Article : Google Scholar
|
|
62
|
Fujii N, Tsuchihashi K, Sasao H, et al:
Insulin resistance functionally limits endothelium-dependent
coronary vasodilation in nondiabetic patients. Heart Vessels.
23:9–15. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kim J-a, Montagnani M, Koh KK and Quon MJ:
Reciprocal relationships between insulin resistance and endothelial
dysfunction molecular and pathophysiological mechanisms.
Circulation. 113:1888–1904. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Gen R, Akbay E and Sezer K: Insulin
resistance and cardiovascular risk factors in patients with mild
and severe subclinical hypothyroidism. The Endocrinologist.
20:128–130. 2010. View Article : Google Scholar
|
|
65
|
Maratou E, Hadjidakis DJ, Kollias A, et
al: Studies of insulin resistance in patients with clinical and
subclinical hypothyroidism. Eur J Endocrinol. 160:785–790. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Al Sayed A, Al Ali N and Alfadhli E:
Subclinical hypothyroidism is associated with early insulin
resistance in Kuwaiti women. Endocr J. 53:653–657. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Peeters RP, Van Der Deure WM, Van Den Beld
AW, et al: The Asp727Glu polymorphism in the TSH receptor is
associated with insulin resistance in healthy elderly men. Clin
Endocrinol (Oxf). 66:808–815. 2007. View Article : Google Scholar
|
|
68
|
Taddei S, Caraccio N, Virdis A, et al:
Impaired endothelium-dependent vasodilatation in subclinical
hypothyroidism: beneficial effect of levothyroxine therapy. J Clin
Endocrinol Metab. 88:3731–3737. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Monzani F, Caraccio N, Kozakowa M, et al:
Effect of levothyroxine replacement on lipid profile and
intima-media thickness in subclinical hypothyroidism: a
double-blind, placebo-controlled study. J Clin Endocrinol Metab.
89:2099–2106. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Oner FA, Yurdakul S, Oner E, Uzum AK and
Erguney M: Evaluation of the effect of L-thyroxin therapy on
endothelial functions in patients with subclinical hypothyroidism.
Endocrine. 40:280–284. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Razvi S, Ingoe L, Keeka G, Oates C,
McMillan C and Weaver JU: The beneficial effect of L-thyroxine on
cardiovascular risk factors, endothelial function, and quality of
life in subclinical hypothyroidism: randomized, crossover trial. J
Clin Endocrinol Metab. 92:1715–1723. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Adrees M, Gibney J, El-Saeity N and Boran
G: Effects of 18 months of l-T4 replacement in women with
subclinical hypothyroidism. Clin Endocrinol (Oxf). 71:298–303.
2009. View Article : Google Scholar
|
|
73
|
Cabral MD, Teixeira P, Soares D, Leite S,
Salles E and Waisman M: Effects of thyroxine replacement on
endothelial function and carotid artery intima-media thickness in
female patients with mild subclinical hypothyroidism. Clinics (Sao
Paulo). 66:1321–1328. 2011.
|
|
74
|
Pasqualetti G, Tognini S, Polini A,
Caraccio N and Monzani F: Is subclinical hypothyroidism a
cardiovascular risk factor in the elderly? J Clin Endocrinol Metab.
98:2256–2266. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Razvi S, Weaver JU, Butler TJ and Pearce
SH: Levothyroxine treatment of subclinical hypothyroidism, fatal
and nonfatal cardiovascular events, and mortality. Arch Intern Med.
178:811–817. 2012.
|
|
76
|
Cooper DS: Clinical practice. Subclinical
hypothyroidism. N Engl J Med. 345:260–265. 2001. View Article : Google Scholar : PubMed/NCBI
|