Introduction
Gliomas are a type of tumor that arise from glial
cells, constituting ~30% of brain and central nervous system tumors
and 80% of malignant brain tumors (1). To date, the prognosis of high-grade
(stage III–IV) glioma cases remains poor. microRNAs (miRNAs) are
small non-coding RNAs, comprised of 18–22 nucleotides, that
regulate gene expression and control various biological processes
(2) through binding to the
3′-untranslated region of mRNAs. Subsequently, the stability and
translation of the target mRNAs are affected. miRNAs have recently
been identified as crucial factors in tumorigenesis, but also tumor
aggressiveness (3,4). In addition, miRNAs are hypothesized
to be widely dysregulated in cancer; thus, may serve as potential
markers for cancer diagnosis, prognosis and treatment (5). miRNAs have also been reported to be
associated with the pathogenesis and anticancer treatment
sensitivity of gliomas (6).
Therefore, the identification of differentially expressed miRNAs
and the determination of their regulatory role in high-grade glioma
patients may help to further the understanding into the underlying
etiology.
High-throughput microarray technology facilitates
the investigation of characteristics that underlie the progression
of cancer. Several studies have investigated the miRNA expression
signature in glioma patients (7,8).
However, these studies have generally implemented regression or
variance analysis, which is unable to evaluate unaccounted array
specific factors. Partial least squares (PLS) analysis has been
demonstrated to be effective and more sensitive in handling
microarray data (9,10). A previous study used this method on
other complex diseases and demonstrated the feasibility (11). Thus, identifying the miRNA
expression signature of glioma patients using this method may
conduce to a new understanding of the disease progression.
In the present study, to identify the differentially
expressed miRNAs and determine their regulatory characteristics in
high-grade glioma patients, PLS analysis was performed using
microarray data collected from the Gene Expression Omnibus (GEO)
database. Survival analysis of the selected miRNAs was conducted to
analyze the effect of these miRNAs on the prognosis of the
patients. In addition, dysregulated miRNAs and target mRNAs were
used to construct a regulatory network. Pathway and Gene Ontology
(GO) enrichment analysis of the dysregulated target genes was also
used to evaluate the biological effects of the differentially
expressed miRNAs.
Materials and methods
Microarray data
An expression profile data set from the GEO database
(GSE4412) was used, which included the transcription profile of 24
grade III and 50 grade IV glioma patients. All the RNA samples were
extracted from fresh frozen tumor tissues that had been collected
during surgical treatment. The data set was based on two Affymetrix
platforms (GPL96 and GPL97).
Identification of differentially
expressed miRNAs
Raw data of all the samples, including CEL and
simple omnibus format in text-formatted files, were obtained from
the GEO database. Following quality control, normalization of the
raw intensity values was conducted using a Robust Multi-array
Analysis (RMA) (12) procedure.
Neutralization of background noise effects and processing artifacts
was performed with model-based background correction, and
expression values of all the probes were aligned to a common scale
using quantile normalization. The log2-transformed RMA
values of all the probes were subsequently used in PLS analysis to
estimate their effects on the grade III and IV samples. Briefly,
PLS latent variables were initially calculated with the non-linear
iterative partial least squares algorithm (13); subsequently, the effects of the
probe expression values on the disease status were estimated using
variable importance in the projection (VIP) (14). Finally, the false discovery rate
(FDR) of each probe was calculated based on the empirical
distribution of the PLS-based VIP scores, generated by a
permutation procedure (n=10,000). miRNAs with a FDR of <0.01
were considered to be significant differentially expressed miRNAs.
The aforementioned procedures were performed with R software
(version 3.0.0; http://www.r-project.org/), including BioConductor
(http://www.bioconductor.org/.), limma
packages (3.12.1) and libraries (15).
Survival analysis
To investigate the contribution of the
differentially expressed miRNAs to the survival time following
surgery, survival analysis was performed. For each miRNA, the
samples were separated into two classes using a K-mean algorithm
based on the expression value. Using the survival time or last
follow-up time of the patients, the log-rank test was used to
investigate whether the two classes were significantly different
from each other. P<0.05 was considered to indicate a
statistically significant difference; thus, miRNAs with these
values were found to be significantly associated with the survival
rate.
Target gene prediction and network
construction
Target gene prediction for the differentially
expressed miRNAs was performed using currently available methods,
including microT (16), miRanda
(17), mircode (18) and TargetScan (19). Target genes supported by at least
two methods were used in further analysis. Differentially expressed
genes were selected using the same procedure as for the detection
of differentially expressed miRNAs. Target genes that were
identified to be differentially expressed in the grade III and IV
samples were subsequently used to construct a regulatory network of
selected miRNAs using Cytoscape software (V 2.8.3, http://www.cytoscape.org/) (20).
Enrichment analysis
To estimate the biological effects of the
differentially expressed miRNAs, enrichment analysis was performed
for the differentially expressed target genes. Target genes were
firstly annotated based on the Kyoto Encyclopedia of Genes and
Genomes (KEGG) pathways (http://www.genome.jp/kegg/) and GO database. A
hypergeometric distribution test was then used to identify the
pathways and GO items enriched with dysregulated miRNA target
genes.
Results
Identification of differentially
expressed miRNAs
As shown in Table
I, six miRNAs were identified to be differentially expressed in
the grade III and IV glioma patients, including two downregulated
miRNAs (hsa-miR-4680 and hsa-miR-1908) and four overexpressed
miRNAs (hsa-miR-4656, hsa-miR-4467, hsa-miR-612 and
hsa-miR-21).
 | Table IDifferentially expressed miRNAs. |
Table I
Differentially expressed miRNAs.
| miRNAs | Fold change | FDR |
|---|
| hsa-miR-4680 | −0.7602 | 0.0014 |
| hsa-miR-1908 | −0.6327 | 0.0074 |
| hsa-miR-4656 | 0.4312 | 0.0127 |
| hsa-miR-4467 | 0.3912 | 0.0164 |
| hsa-miR-612 | 0.9768 | 0.0220 |
| hsa-miR-21 | 1.4927 | 0.0273 |
Survival analysis
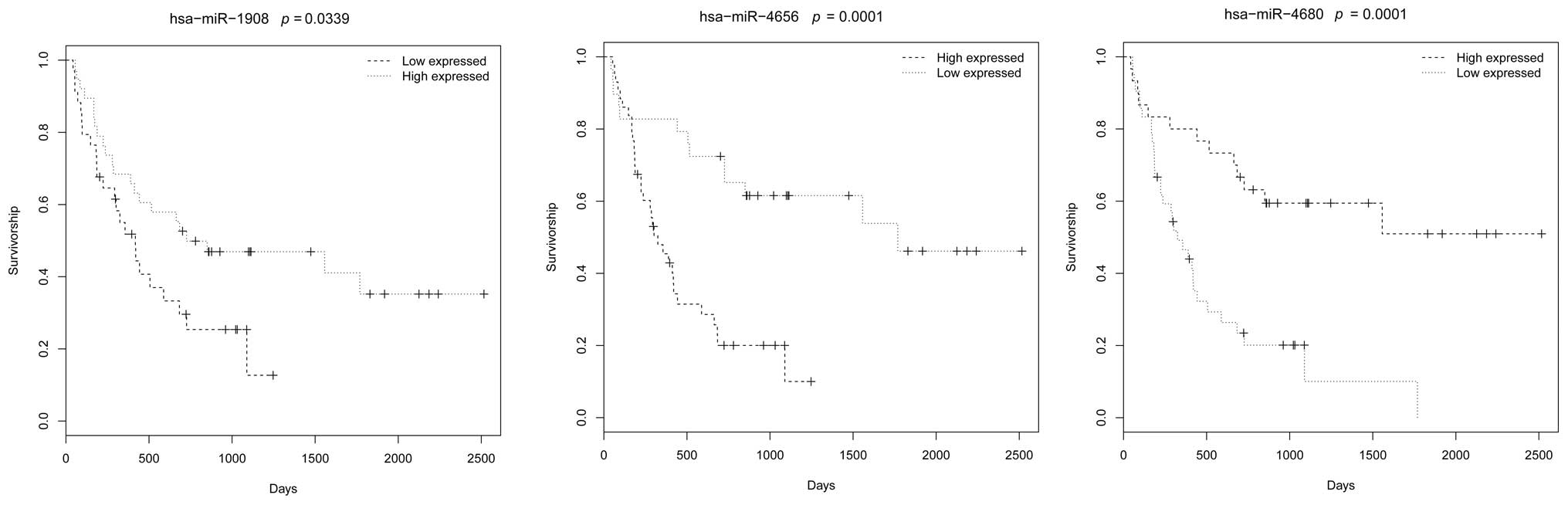
Survival analysis of these miRNAs revealed that
three miRNAs, hsa-miR-1908, hsa-miR-4656 and hsa-miR-4680, were
significantly associated with the survival rate of the patients,
with P-values of 0.0339, 0.0001 and 0.0001, respectively (Fig. 1).
Target gene prediction and network
construction
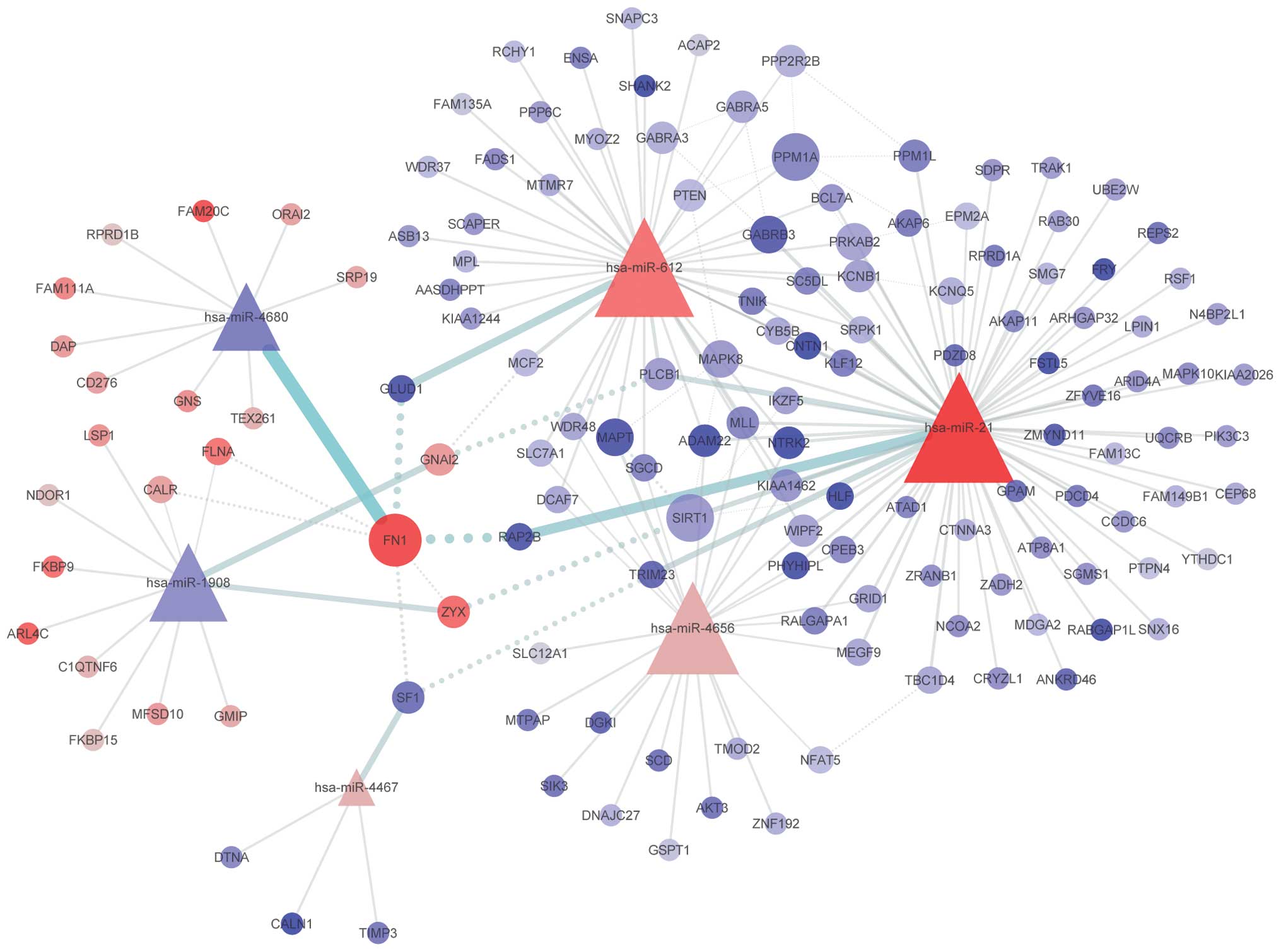
Target gene prediction and PLS analysis identified
139 differentially expressed target genes, and a network was
constructed using these miRNAs and target genes (Fig. 2). The degree of each node was
defined as the number of its interactions in the network. Nodes
with higher degrees are shown as a bigger size. Among the six
miRNAs, hsa-miR-21 was found to have a higher number of
interactions compared with the other miRNAs. In the three
overexpressed miRNAs, hsa-miR-21, hsa-miR-612 and hsa-miR-4656, a
number of the target genes were found to be shared. Among the
target genes, FN1 was detected to have the highest
degree.
Enrichment analysis
Enrichment analysis of the differentially expressed
target genes identified two KEGG pathways and four GO items
over-represented with dysregulated target genes (Table II). The two KEGG pathways were
retrograde endocannabinoid signaling and dopaminergic synapse,
which are both involved in the nervous system. In addition, one of
the GO items, the postsynaptic membrane (GO:0045211), is also
associated with the nervous system. The remaining three GO items
were shown to be associated with the protein phosphorylation
process, including protein serine/threonine phosphatase activity
(GO:0004722), JUN phosphorylation (GO:0007258) and protein
dephosphorylation (GO:0006470).
| Table IIPathways and GO items enriched with
differentially expressed genes. |
Table II
Pathways and GO items enriched with
differentially expressed genes.
| Parameter | Description | Class | P-value |
|---|
| KEGG pathway |
| hsa04723 | Retrograde
endocannabinoid signaling | Nervous system | 0.0054 |
| hsa04728 | Dopaminergic
synapse | Nervous system | 0.0308 |
| GO item |
| GO:0004722 | Protein
serine/threonine phosphatase activity | Function | 0.0012 |
| GO:0007258 | JUN
phosphorylation | Process | 0.0115 |
| GO:0006470 | Protein
dephosphorylation | Process | 0.0115 |
| GO:0045211 | Postsynaptic
membrane | Component | 0.0391 |
Discussion
To identify differentially expressed miRNAs and
determine their regulatory characteristics in high-grade glioma
patients, PLS analysis was performed. In total, six miRNAs were
identified to be dysregulated. Among them, hsa-miR-21 has been
previously reported to exhibit a significant correlation with tumor
grade and prognosis (21,22). In addition, the expression of
hsa-miR-612 has been reported to be associated with magnetic
resonance imaging features of glioblastoma multiforme (23). The results of the present study
further confirmed the involvement of the two miRNAs in the
progression of glioma. In addition, one of the target genes of
miR-612 is PTEN. PTEN is a tumor suppressor that negatively
regulates the protein kinase B/Akt-dependent cell survival pathway
(24). Therefore, depression of
PTEN may impact its negative regulation of tumor cell survival and
contribute to the deterioration of the disease. None of the
remaining four miRNAs have been previously reported to be
associated with glioma. However, hsa-miR-1908 has been reported to
be associated with the metastasis or tumorigenesis of other types
of tumor, including chordomas (25), hepatoma (26) and melanoma (27). In addition, hsa-miR-4680,
hsa-miR-4656 and hsa-miR-4467 have been hypothesized to be
associated with breast cancer (28). For miR-4680, one of its target
genes is FN1, which is a hub gene that was found to have the
highest degree among the target genes. The protein encoded by this
gene is fibronectin, which is involved in cell adhesion and
migration processes. A previous study reported a significant
correlation between this gene and malignant glioma (29), indicating the potential regulatory
mechanism of miR-4680 in the development of glioma. Survival
analysis also revealed that the expression levels of hsa-miR-1908,
hsa-miR-4656 and hsa-miR-4680 were significantly associated with
the survival rate of the patients (Fig. 1). Moreover, the constructed network
of dysregulated miRNAs and target genes revealed that hsa-miR-21,
hsa-miR-612 and hsa-miR-4656 share a number of target genes,
indicating that they may affect similar biological processes. Thus,
further investigation of these miRNAs is warranted.
Pathway enrichment analysis of the dysregulated
target genes revealed that the differentially expressed miRNAs
primarily affect pathways in the nervous system, including
retrograde endocannabinoid signaling and the dopaminergic synapse.
GO analysis additionally revealed the over-representation of
dysregulated target genes in the protein phosphorylation process.
The GO:0004722 item of protein serine/threonine phosphatase
activity exhibited the most significant enrichment. A previous
study reported the alteration of striatal dopaminergic function in
glioma development (30).
Furthermore, dysregulation of serine/threonine phosphatase
calcineurin has also been reported in grade IV astrocytoma tumor
tissue (31). The enrichment
analysis results further confirmed the effects of the dysregulated
miRNAs.
In summary, using an expression profile from the GEO
database, PLS-based analysis was performed to identify
differentially expressed miRNAs and evaluate their regulatory
characteristics in high-grade glioma patients. In total, six miRNAs
were identified to be dysregulated, including three miRNAs
significantly associated with the survival rate of the patients.
Enrichment analysis of the dysregulated target genes revealed that
the differentially expressed miRNAs predominantly affected
biological processes associated with the nervous system and the
protein phosphorylation process. Thus, the results may offer a new
understanding into the pathogenesis of high-grade glioma.
References
|
1
|
Goodenberger ML and Jenkins RB: Genetics
of adult glioma. Cancer Genetics. 205:613–621. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bartel DP: MicroRNAs: genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Calin GA and Croce CM: MicroRNA signatures
in human cancers. Nat Rev Cancer. 6:857–866. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Waldman SA and Terzic A: Translating
microRNA discovery into clinical biomarkers in cancer. JAMA.
297:1923–1925. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Garzon R, Marcucci G and Croce CM:
Targeting microRNAs in cancer: rationale, strategies and
challenges. Nat Rev Drug Discov. 9:775–789. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li M, Li J, Liu L, Li W, Yang Y and Yuan
J: MicroRNA in Human Glioma. Cancers (Basel). 5:1306–1331. 2013.
View Article : Google Scholar
|
|
7
|
Visani M, de Biase D, Marucci G, et al:
Definition of miRNAs expression profile in glioblastoma samples:
the relevance of non-neoplastic brain reference. PloS One.
8:e553142013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang C, Wang C, Chen X, et al:
Identification of seven serum microRNAs from a genome-wide serum
microRNA expression profile as potential noninvasive biomarkers for
malignant astrocytomas. Int J Cancer. 132:116–127. 2013. View Article : Google Scholar
|
|
9
|
Chakraborty S and Datta S and Datta S:
Surrogate variable analysis using partial least squares (SVA-PLS)
in gene expression studies. Bioinformatics. 28:799–806. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ji GL, Yang ZJ and You WJ: PLS-based gene
selection and identification of tumor-specific genes. IEEE Trans
Syst Man Cybern C Appl Rev. 41:830–841. 2011. View Article : Google Scholar
|
|
11
|
Gao QG, Li ZM and Wu KQ: Partial least
squares based analysis of pathways in recurrent breast cancer. Eur
Rev Med Pharmacol Sci. 17:2159–2165. 2013.PubMed/NCBI
|
|
12
|
Irizarry RA, Hobbs B, Collin F, et al:
Exploration, normalization, and summaries of high density
oligonucleotide array probe level data. Biostatistics. 4:249–264.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Martins JPA, Teófilo RF and Ferreira MMC:
Computational performance and cross-validation error precision of
five PLS algorithms using designed and real data sets. J Chemom.
24:320–332. 2010.
|
|
14
|
Gosselin R, Rodrigue D and Duchesne C: A
bootstrap-VIP approach for selecting wavelength intervals in
spectral imaging applications. Chemometr Intell Lab Syst.
100:12–21. 2010. View Article : Google Scholar
|
|
15
|
Smyth GK, Michaud J and Scott HS: Use of
within-array replicate spots for assessing differential expression
in microarray experiments. Bioinformatics. 21:2067–2075. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Maragkakis M, Alexiou P, Papadopoulos GL,
et al: Accurate microRNA target prediction correlates with protein
repression levels. BMC Bioinformatics. 10:2952009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
John B, Enright AJ, Aravin A, Tuschl T,
Sander C and Marks DS: Human MicroRNA targets. PLoS Biol.
2:e3632004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jeggari A, Marks DS and Larsson E:
miRcode: a map of putative microRNA target sites in the long
non-coding transcriptome. Bioinformatics. 28:2062–2063. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lewis BP, Shih IH, Jones-Rhoades MW,
Bartel DP and Burge CB: Prediction of mammalian microRNA targets.
Cell. 115:787–798. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shannon P, Markiel A, Ozier O, et al:
Cytoscape: a software environment for integrated models of
biomolecular interaction networks. Genome Res. 13:2498–2504. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hermansen SK, Dahlrot RH, Nielsen BS,
Hansen S and Kristensen BW: MiR-21 expression in the tumor cell
compartment holds unfavorable prognostic value in gliomas. J
Neurooncol. 111:71–81. 2013. View Article : Google Scholar
|
|
22
|
Wu L, Li G, Feng D, et al: MicroRNA-21
expression is associated with overall survival in patients with
glioma. Diagn Pathol. 8:2002013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li WB, Chen HY, Zhang W, et al:
Relationship between magnetic resonance imaging features and miRNA
gene expression in patients with glioblastoma multiforme. Chin Med
J (Engl). 126:2881–2885. 2013.
|
|
24
|
Stambolic V, Suzuki A, de Pompa JL, et al:
Negative regulation of PKB/Akt-dependent cell survival by the tumor
suppressor PTEN. Cell. 95:29–39. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Long C, Jiang L, Wei F, et al: Integrated
miRNA-mRNA analysis revealing the potential roles of miRNAs in
chordomas. PloS One. 8:e666762013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jin JC, Jin XL, Zhang X, Piao YS and Liu
SP: Effect of OSW-1 on microRNA expression profiles of hepatoma
cells and functions of novel microRNAs. Mol Med Rep. 7:1831–1837.
2013.PubMed/NCBI
|
|
27
|
Pencheva N, Tran H, Buss C, et al:
Convergent multi-miRNA targeting of ApoE drives LRP1/LRP8-dependent
melanoma metastasis and angiogenesis. Cell. 151:1068–1082. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Persson H, Kvist A, Rego N, et al:
Identification of new microRNAs in paired normal and tumor breast
tissue suggests a dual role for the ERBB2/Her2 gene. Cancer Res.
71:78–86. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wei KC, Huang CY, Chen PY, et al:
Evaluation of the prognostic value of CD44 in glioblastoma
multiforme. Anticancer Res. 30:253–259. 2010.PubMed/NCBI
|
|
30
|
Lonjon M, Quentien MH, Risso JJ, et al:
Alteration of striatal dopaminergic function induced by glioma
development: a microdialysis and immunohistological study in the
rat striatum. Neuroscience Lett. 354:131–134. 2004. View Article : Google Scholar
|
|
31
|
Brun M, Glubrecht DD, Baksh S and Godbout
R: Calcineurin regulates nuclear factor I dephosphorylation and
activity in malignant glioma cell lines. J Biol Chem.
288:24104–24115. 2013. View Article : Google Scholar : PubMed/NCBI
|