Introduction
Mucosa-associated lymphoid tissue (MALT) lymphomas
were first described in 1983 by Isaacson and Wright, have since
been recognized as a separate entity and account for 8–10% of all
non-Hodgkin lymphomas (1).
Pulmonary MALT lymphoma (pMALToma), also referred to as
bronchial-associated lymphoid tissue lymphoma (2), is a rare disease. The development of
some pMALTomas has been reported to be associated with chronic
inflammation due to autoimmune or infectious diseases (3–4).
pMALToma usually has an indolent course and remains localized in
the lung for long periods prior to dissemination. The majority of
patients with pMALToma have a favorable prognosis. However, the
optimal therapy for this rare disease remains under debate and some
cases of pMALToma are managed with the watch-and-wait approach.
This is the case report of a 19-year-old patient with pMALToma
presenting with multiple pulmonary consolidations on computed
tomography (CT) scans. The patient received chemotherapy with a
chlorambucil-based regimen and achieved a complete remission.
Case report
A previously healthy 19-year-old female of Asian
ethnic origin, visited our hospital for an evaluation of abnormal
chest CT findings on routine physical examination. On admission the
patient was asymptomatic, without fever, cough, lymphadenopathy,
night sweats or weight loss. As shown in Table I, the patient had normal complete
blood count. Additional tests revealed minimally elevated
erythrocyte sedimentation rate. The antinuclear antibody test was
negative. The levels of lactate dehydrogenase, β2
microglobulin and liver function tests were normal. The tests for
tuberculosis, human immunodeficiency virus and other viruses,
including hepatitis A, B and C, were all negative. The spleen and
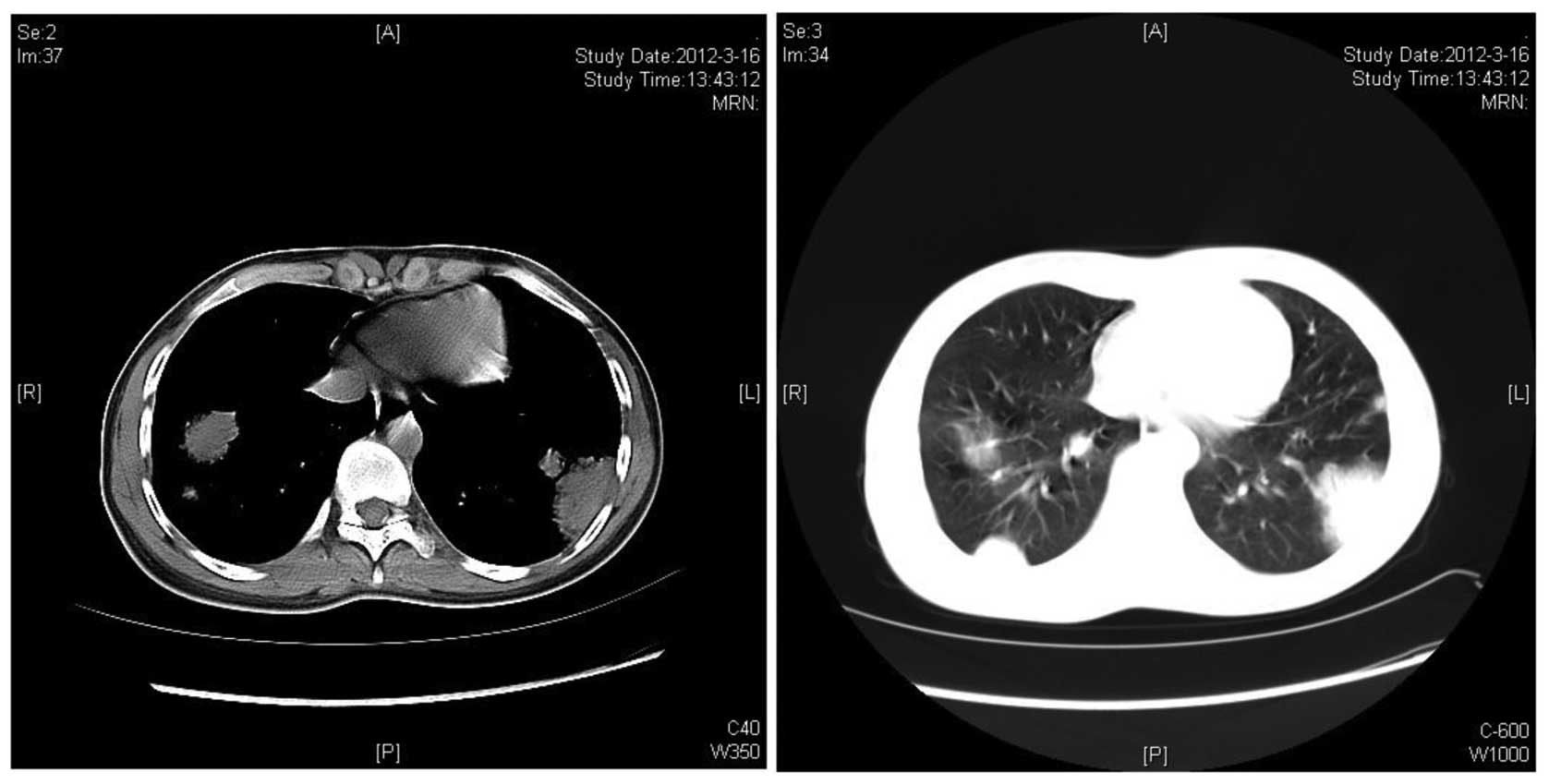
liver were of normal size and echogenicity. Spiral lung CT revealed
bilateral multiple pulmonary consolidations, with the largest
lesion sized ~4.5×4.5 cm (Fig. 1).
Bone marrow aspiration was negative for lymphoma involvement. The
patient underwent CT image-guided biopsy and the diagnosis of
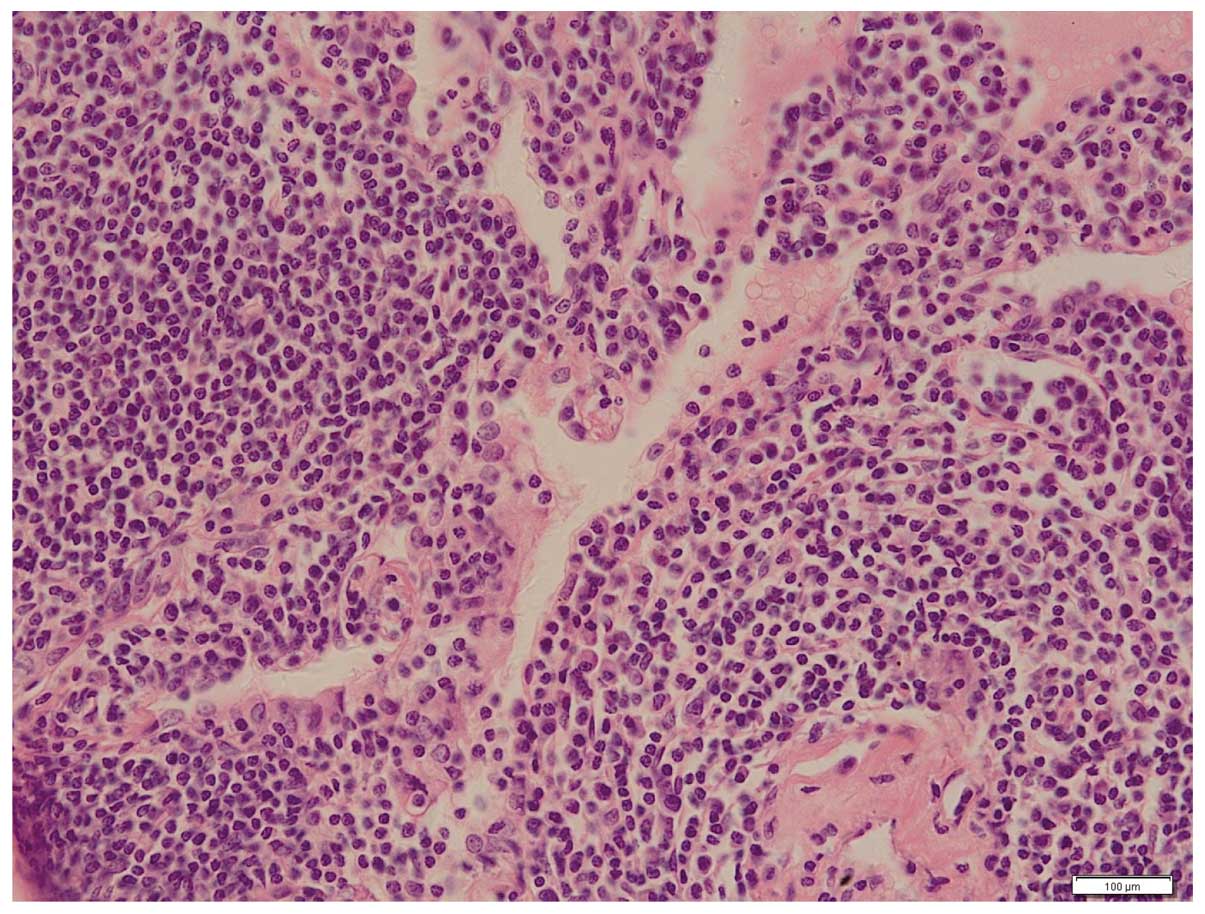
pMALToma was confirmed. The microscopic examination revealed
lymphoepithelial lesions characterized by diffuse infiltration of
the lung parenchyma by small lymphocytes, monocytoid cells and
plasma cells (Fig. 2).
Immunohistochemical staining demonstrated that the cells were
positive for CD20 and B-cell lymphoma 2 protein, with 10% nuclear
staining for Ki-67. According to the pathological findings, the
patient was treated with 6 courses of chemotherapy with 2 mg
vincristine, 4 mg/day chlorambucil plus 30 mg/day prednisolone for
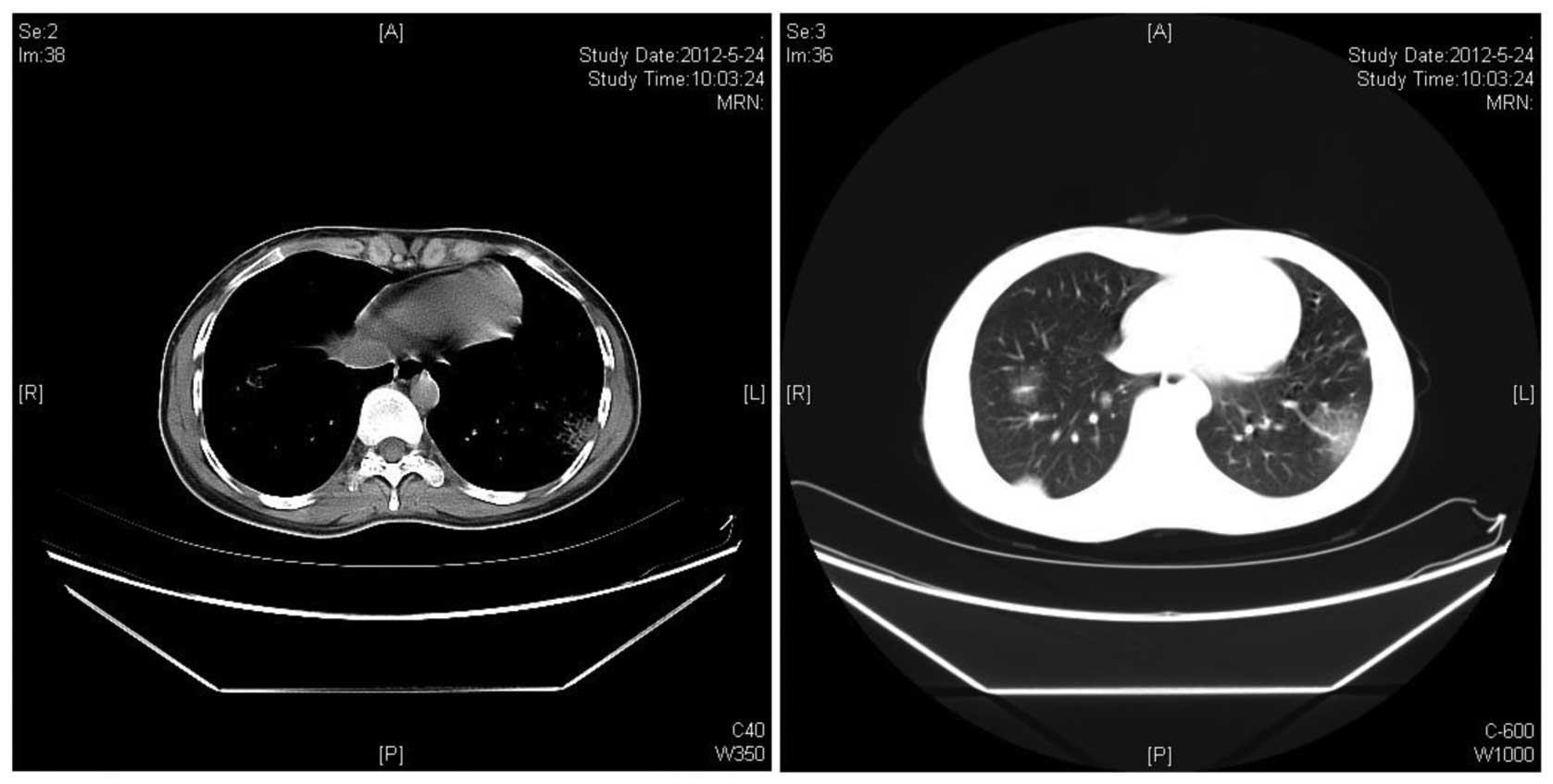
7 days. After 2 cycles of chemotherapy, all the pulmonary lesions
were significantly reduced in size (Fig. 3). Complete remission of the lung
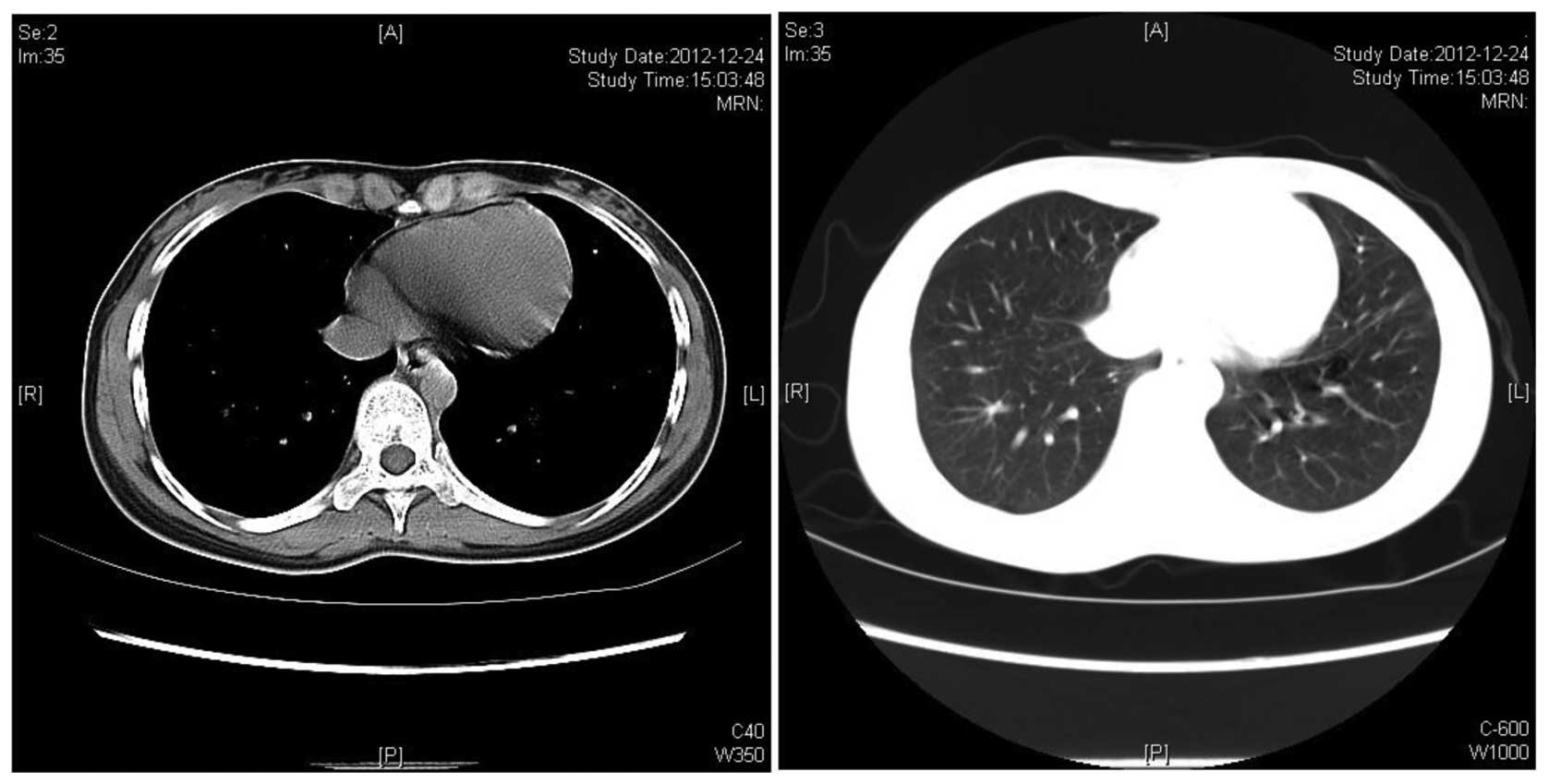
consolidations was observed following 6 cycles of treatment, as
visualized by CT scan (Fig. 4).
Fifteen months after the last course of chemotherapy, the patient
was in good condition without any evidence of relapse. The patient
remains in follow-up.
 | Table IPertinent laboratory data at the time
of the patient’s initial evaluation. |
Table I
Pertinent laboratory data at the time
of the patient’s initial evaluation.
| Laboratory data | Admission value | Reference range |
|---|
| White blood cell
count/μl | 5,100 | 4,000–10,000 |
| Erythrocyte
sedimentation rate, mm/h | 23 | 0–20 |
| Antinuclear antibody
titer | Negative | Negative |
|
β2-microglobulin, mg/l | 1.44 | 0.91–2.2 |
| Lactate
dehydrogenase, IU/l | 121 | 91–180 |
| Aspartate
aminotransferase, IU/l | 22 | 0–40 |
| Alanine
aminotransferase, IU/l | 23 | 0–40 |
Discussion
Although pMALToma is the most common form of primary
pulmonary lymphoma, it is a rare disease. The median age at
diagnosis for pMALToma is 50–60 years (5), with only few patients aged <30
years (6). pMALToma may be
comorbid with autoimmune or infectious diseases (3,4). It
has been reported that pMALToma is associated with Sjögren’s
syndrome, rheumatoid arthritis, dysgammaglobulinemia, amyloid
deposits, collagen vascular diseases, Helicobacter pylori
infection and acquired immune deficiency syndrome (7–10).
While the clinical manifestations are usually non-specific,
including cough, mild dyspnea, chest pain and occasionally
hemoptysis, the majority of the patients are asymptomatic, as in
the presently reported case. Therefore, symptoms and physical signs
contribute little to diagnosis (3). Patients with pMALTomas typically
present with single or multiple radiologically detected pulmonary
nodules or consolidations (8). A
definitive diagnosis of pMALToma may be achieved following
histological examination of biopsy specimens obtained via minimally
invasive procedures, including transbronchial biopsy or
radiologically-guided transthoracic core-needle biopsy. However,
the tissue sample is occasionally inadequate for diagnosis,
particularly in patients with atypical CT findings. Therefore,
several patients are diagnosed based on the results of surgical
biopsies (7,11). In addition, immunohistochemical
staining findings are crucial for accurate diagnosis.
pMALTomas typically have an indolent course and a
good prognosis, although systemic dissemination and transformation
into high-grade B-cell lymphoma may occur. Borie et al
(5) reported the clinical
characteristics and prognostic factors of 63 cases with pMALToma;
in that cohort of patients, the estimated 5- and 10-year overall
survival rates were 90 and 72%, respectively. The optimal therapy
for this rare disease remains under debate, mainly due to the
limited availability and heterogeneity of the data reported in the
literature (11–13). Various therapeutic regimens
including radiotherapy, surgery and chemotherapy have been
proposed. Troch et al (14)
suggested that a watch-and-wait policy may be adopted as the
initial management of pMALToma in the absence of symptoms. By
contrast, Ahmed et al (11)
reported 22 cases of biopsy-proven pMALTomas. Among those cases, 6
patients were initially observed for a median duration of 18 months
(range, 10–53 months), 4 of whom ultimately received treatment due
to disease progression. In addition, lung surgery may not be the
optimal option, as thoracic pain and lung function impairment are
observed in ~10–15% of the cases (15,16).
Radiation therapy for pMALToma is also avoided, in order to prevent
potential problems with lung function (16). Chemotherapy with chlorambucil
currently appears to be the optimal treatment option for cases with
disseminated disease (5). The
therapeutic role of rituximab remains unclear, since thus far there
is only a limited number of case reports with conflicting results
(14,16–17).
However, as pMALToma cells express the CD20 antigen, the
therapeutic possibility of rituximab appears to be promising,
either in combination with chemotherapy or as a single agent.
Recently, Zinzani et al (12) reported an observational
retrospective study on homogeneous clinical data from 17 patients
with pMALToma; all the patients were treated with fludarabine and
mitoxantrone-containing regimens. The immunochemotherapy mentioned
above achieved a high response rate: 82.3% of the patients achieved
a complete response and 11.8% a partial response. Furthermore, a
remarkable progression-free survival (71%) and overall survival
(100%) were reported at 14 years. The approach to the case
presented here was treatment with chlorambucil combined with
prednisolone and vincristine, which was effective in achieving a
complete remission.
In conclusion, pMALToma is a rare disease and its
progression is generally indolent. pMALToma may be accompanied by
autoimmune diseases. We presented the case of a 19-year-old patient
with biopsy-proven diagnosis of pMALToma who had no history of
autoimmune disease. The patient was successfully treated with a
chlorambucil-based regimen. However, long-term follow-up is
required to establish the efficacy of this treatment strategy.
References
|
1
|
Isaacson P and Wright DH: Malignant
lymphoma of mucosa-associated lymphoid tissue. A distinctive type
of B-cell lymphoma. Cancer. 52:1410–1416. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Harris N, Jaffe E, Stein H, et al: A
revised European-American classification of lymphoid neoplasm: a
proposal from the International Lymphoma Study Group. Blood.
84:1361–1392. 1994.PubMed/NCBI
|
|
3
|
Cadranel J, Wislez M and Antoine M:
Primary pulmonary lymphoma. Eur Respir J. 20:750–762. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Klein TO, Soll BA, Issel BF, et al:
Bronchus-associated lymphoid tissue lymphoma and Mycobacterium
Tuberculosis infection: an unusual case and a review of the
literature. Respir Care. 52:755–758. 2007.PubMed/NCBI
|
|
5
|
Borie R, Wislez M, Thabut G, et al:
Clinical characteristics and prognostic factors of pulmonary MALT
lymphoma. Eur Respir J. 34:1408–1416. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Xu HY, Jin T, Li RY, et al: Diagnosis and
treatment of pulmonary mucosa-associated lymphoid tissue lymphoma.
Chin Med J. 20:648–651. 2007.
|
|
7
|
Bae YA, Lee KS, Han J, et al: Marginal
zone B-cell lymphoma of bronchus-associated lymphoid tissue:
imaging findings in 21 patients. Chest. 133:433–440. 2008.
View Article : Google Scholar
|
|
8
|
Kurtin PJ, Myers JL, Adlakha H, et al:
Pathologic and clinical features of primary pulmonary extranodal
marginal zone B-cell lymphoma of MALT type. Am J Surg Pathol.
25:997–1008. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lazar EB, Whitman GJ and Chew FS: Lymphoma
of bronchus-associated lymphoid tissue. AJR Am J Roentgenol.
167:1161996. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Boulanger E, Meignin V, Baia M, et al:
Mucosa-associated lymphoid tissue lymphoma in patients with human
immunodeficiency virus infection. Br J Haematol. 140:470–474. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ahmed S, Kussick SJ, Siddiqui AK, et al:
Bronchial-associated lymphoid tissue lymphoma: a clinical study of
a rare disease. Eur J Cancer. 40:1320–1326. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zinzani PL, Pellegrini C, Gandolfi L, et
al: Extranodal marginal zone B-cell lymphoma of the lung:
experience with fludarabine and mitoxantrone-containing regimens.
Hematol Oncol. 31:183–188. 2013. View
Article : Google Scholar
|
|
13
|
Jäger G, Neumeister P, Quehenberger F, et
al: Prolonged clinical remission in patients with extranodal
marginal zone B-cell lymphoma of the mucosa-associated lymphoid
tissue type treated with cladribine: 6 year follow-up of phase II
trial. Ann Oncol. 11:1722–1723. 2006. View Article : Google Scholar
|
|
14
|
Troch M, Streubel B, Petkov V, et al: Does
MALT lymphoma of the lung require immediate treatment? An analysis
of 11 untreated cases with long-term follow-up. Anticancer Res.
27:3633–3637. 2007.PubMed/NCBI
|
|
15
|
Ferraro P, Trastek VF, Adlakha H, et al:
Primary non-Hodgkin’s lymphoma of the lung. Ann Thorac Surg.
69:933–937. 2000. View Article : Google Scholar
|
|
16
|
Oh SY, Kim WS, Kim JS, et al: Pulmonary
marginal zone B-cell lymphoma of MALT type - what is a prognostic
factor and which is the optimal treatment, operation, or
chemotherapy?: Consortium for Improving Survival of Lymphoma (CISL)
study. Ann Hematol. 89:563–568. 2010. View Article : Google Scholar
|
|
17
|
Conconi A, Martinelli G, Thiéblemont C, et
al: Clinical activity of rituximab in extranodal marginal zone
B-cell lymphoma of MALT type. Blood. 102:2741–2745. 2003.
View Article : Google Scholar : PubMed/NCBI
|