Introduction
Corneal endothelium failure resulting from a
reduction in cell number or cellular dysfunction leads to blindness
that is treatable by the replacement of functional corneal
endothelial cells (CECs); however, this treatment is hampered by
the fact that human CECs have a poor or absent self-renewal
capacity (1). Pluripotent stem
cells (PSCs), such as embryonic stem cells (ESCs) and induced
pluripotent stem cells (iPSCs), have gained widespread attention
for their hallmark properties of extensive self-renewal capacity
and developmental pluripotency for all cell lineages under the
appropriate conditions (2,3). iPSCs hold great promise for
replacement therapies in regenerative medicine and the modeling of
numerous diseases that are currently unresponsive to traditional
clinical approaches because the generation of patient-specific
iPSCs directly from somatic cells renders the use of oocytes or
embryos unnecessary (4–7). Pluripotent cells represent a powerful
tool for tissue regeneration; however, until now, no protocol has
been established for the directed differentiation of PSCs into CECs
in vitro. This is likely due to a lack of knowledge about
the molecular mechanisms by which ESCs develop into CECs.
Development studies have revealed that CECs originate from
differentiation of the first cranial neural crest (NC) cell wave
during vertebrate embryo development (8). The NC is a transient structure in
vertebrate embryos that initially generates NC stem cells, which
then migrate throughout the body to produce a diverse spectrum of
differentiated tissue types, including CECs (9). Therefore, NC derivatives from iPSCs
have been considered for use in the first stage of the induction of
CEC differentiation. However, to the best of our knowledge, the
differentiation of NC stem cells after they reach their target
site, which is located beneath the primary stromal layer
synthesized by the corneal epithelium and adjacent to the lens
epithelial layer, has not been described. Knowledge of the
mechanisms governing NC cell differentiation to CECs remains
rudimentary. It is accepted that the microenvironment, or niche,
surrounding migrating cells, for example, lens epithelial cells
(LECs), determines their ultimate fate during differentiation
(10). However, the understanding
of CEC differentiation remains limited by the lack of defined
components that can be used as inducers of differentiation. The aim
of the present study was to evaluate the hypothesis that
reproducing the assembly of the CEC niche using a microenvironment
containing LECs and CECs may induce differentiation. Therefore,
co-culture with these potentially differentiation-inducing cells
was used during the second differentiation step to further guide
PSC-derived NC cells to the CEC fate.
Materials and methods
Preparation of differentiation-inducing
cells
A total of 20 New Zealand White rabbits and 40
C57BL/6J mice were obtained from the Shanghai Animal Experimental
Center (Shanghai, China), and all procedures were approved by the
Animal Research Committee of the Ninth People’s Hospital, Shanghai
Jiaotong University School of Medicine (Shanghai, China). Rabbit
CECs and LECs were isolated from rabbit eyes and cultured in fresh
growth medium (GM), which was Dulbecco’s modified Eagle’s medium
(MEM)/F12 (Invitrogen Life Technologies, Carlsbad, CA, USA) medium
containing 10% fetal bovine serum (FBS; Invitrogen Life
Technologies) and 100 U/ml penicillin-streptomycin (Invitrogen Life
Technologies). After eyeball enucleation and corneal dissection,
the intact Descemet’s membrane and lens anterior capsule tissues
were removed with micro-forceps and incubated in GM at 37°C
overnight. Tissues were then digested with 0.25% trypsin and 0.02%
EDTA (Sigma-Aldrich, St. Louis, MO, USA) to acquire a single-cell
suspension. After the cells were passed through a 100-μm mesh
strainer, they were centrifuged at 200 × g for 4 mins and
subsequently seeded in T25 flasks at a density of 2×105
cells/ml. When the cells reached 80% confluence after the second
passage, the GM was replaced with CEC defined medium (CEC-DM),
which was DMEM/F12 medium containing 20 ng/ml basic fibroblast
growth factor (recombinant murine bFGF; Invitrogen Life
Technologies), 1% N2 neural supplement (Invitrogen Life
Technologies), 2% B27 supplement (Invitrogen Life Technologies), 50
μg/ml ascorbic acid (Roche, Bromma, Sweden) and 100 U/ml
penicillin-streptomycin. After 12 h of culture, the conditioned
medium (CM) was collected and filtered using a 0.22-μm filter unit
and stored at −80°C for subsequent co-culture.
Culture of mouse ESCs and iPSCs
The mouse ESC line R1, a gift from Professor Yilin
Cao (Key Laboratory of Tissue Engineering, National Tissue
Engineering Center of China, Shanghai, China), and iPSCs derived
from mouse neural progenitor cells, a gift from Professor Jin Yin
(Key Laboratory of Stem Cell Biology, Institute of Health Sciences,
Shanghai Institutes for Biological Sciences, Chinese Academy of
Sciences, Shanghai, China), have been involved in previous studies
(1,11). Early-passage PSCs (less than P8)
were used in the present study. Mouse embryonic fibroblasts (MEFs)
were derived from mouse embryos at embryonic day 13.5, and
early-passage cells (P2–P3) were used as feeders for the culture of
ESCs and iPSCs. To maintain their undifferentiated state, R1 ESCs
and iPSCs were plated onto a feeder layer of MEFs treated with 10
μg/ml mitomycin-C (MMC) for 2 h, seeded onto T25 flasks pre-coated
with 0.1% gelatin and grown in embryonic stem medium (ESM), which
was Iscove’s modified Dulbecco’s medium (IMDM) supplemented with
15% FBS, 2 mM L-glutamine, 1% non-essential amino acids (NEAA), 1
mM pyruvate, 0.1 mM β-mercaptoethanol (all from Invitrogen Life
Technologies) and 1,000 U/ml leukemia inhibitory factor (LIF;
Millipore).
Step one: NC differentiation after EB
formation with RA treatment
To reduce the influence of the feeder layer on
differentiation, mouse R1 ESCs and iPSCs were trypsinized,
dissociated into single-cell suspensions and subcultured onto 10-mm
dishes pre-coated with 0.1% gelatin. After two days of culture in
ESM, the cells were trypsinized, centrifuged at 200 × g for 4 mins
and resuspended. The supernatant containing predominantly ESCs or
iPSCs was collected and resuspended in EB differentiation medium
(EBM) without LIF at a density of 1.0×105 cells/ml.
Low-adherence dishes were used to facilitate the suspension
culture. The medium was changed every 2 to 3 days after collecting
EBs by gravity sedimentation. All-trans retinoic acid (RA;
Roche, Sweden) was added to the EBM on day 4 of EB differentiation
at various final concentrations (0.1, 0.5, 1 or 10 μM), and the
cells were cultured for an additional 4 days (EBd4+4). The cells
were collected for total RNA extraction at multiple time points
during EB differentiation: on day 4 (EBd4) and after the addition
of RA, recorded as EBd4+n, where n = the number of RA exposure days
(for example, EBd4+2, EBd4+4 and EBd4+6). The mRNA expression
profile was obtained using reverse transcription-quantitative
polymerase chain reaction (RT-qPCR). Additionally, EBs were seeded
on gelatin-coated glass coverslips (VWR, West Chester, PA, USA) on
day EBd4+3 through EBd4+4 to allow cells to adhere and migrate, and
then fixed for analysis by immunocytochemistry (ICC).
Step two: Induced NC differentiation
towards CEC-like cells
Effects of different coatings on EB
migration and differentiation
To optimize the induction conditions, different
plate coatings were compared, as the coating serves as an
extracellular matrix (ECM) component and plays an important role in
cell differentiation. Gelatin, fibronectin (Fn) and laminin (Ln)
were assayed. The Fn and Ln (at 2 μg/cm2) coatings were
kept at 37°C for 2 h before use. The gelatin coating was incubated
at 37°C for >24 h. After treatment with 1 μM RA, EBs were plated
on various coatings and switched to LEC-CM to gain insight into the
impact of ECM on induced CEC differentiation.
Cell differentiation in different
co-culture environments
For the next stage of differentiation, following the
treatment with 1 μM RA on day 4 (EBd4+4), EBs were transferred to a
co-culture environment; various co-culture systems were compared.
EBs were first collected by gravity sedimentation for 10 min, after
which the supernatant was removed. After being washed in serum-free
DMEM/F12, EBs were plated on gelatin-coated 6- or 12-well plates in
one of the following media to induce CEC-like cell differentiation:
CEC-CM, LEC-CM and CEC-DM. The cells were cultured at 37°C in a 5%
CO2 humidified atmosphere. During the 7–10-day
co-culture experiment, media were changed every 2 to 3 days. At the
end of the induction, the cores of the endothelium-like colonies
were examined under a microscope (M620, Leica, Wetzlar, Germany)
and removed using needles to ensure that the majority of the
endothelium-like population was harvested for RNA extraction and
analysis.
RT-qPCR
Total RNA was extracted from the cultured cells
using TRIzol reagent (Invitrogen Life Technologies), and
first-strand cDNA synthesis was performed using the PrimeScript™ RT
reagent kit (Perfect Real Time; Takara, Dalian, China) (12). qPCR was performed in a 20-μl
reaction containing 10 μl reaction mixture, 1 μl cDNA, 2 μl primers
(Table I) and 7 μl
ddH2O. qPCR was conducted using a 7500 Real-time PCR
Detection System (Applied Biosystems, Irvine, CA, USA) after a hot
start at 95°C for 10 min and 40 cycles of amplification (15 sec at
95°C and 1 min at 60°C) (13). The
efficiency of the reaction primers was measured using serial
dilutions of the cDNA (1:1, 1:5, 1:25, 1:125, 1:625 and 1:3,125).
Each sample was tested in triplicate. The relative gene expression
was analyzed using the Pfaffl method (3). Data are expressed as a fold change
relative to untreated controls after normalizing to the β-actin
endogenous control.
 | Table IPrimers used for reverse
transcription-quantitative polymerase chain reaction. |
Table I
Primers used for reverse
transcription-quantitative polymerase chain reaction.
| Genes | Accession no. | Forward primer | Reverse primer | Product size
(bp) |
|---|
| Snail | NM_011427.2 |
ACACGCTGCCTTGTGTCTGC |
TGGAGCAAGGACATGCGGGAGA | 229 |
| Slug | NM_011415.2 |
ACGCCTCCAAGAAGCCCAACT |
TGGAGCTGCCGACGATGTCCATA | 161 |
| Twist | NM_011658.2 |
TTCAGACCCTCAAACTGGCGGC |
ATCCTCCAGACGGAGAAGGCGT | 139 |
| Sox10 | NM_011437.1 |
AGAAGGAACAGCAGGACGGCGA |
TGACGTGCGGCTTGCTCTTG | 150 |
| P0 | NM_008623.4 |
GGTGGTGCTGTTGCTGCTGT |
GCAGCTTTGGTGCTTCGGCT | 195 |
| P75 | NM_033217.3 |
AAAGCCTGCAACCTGGGCGA |
TAGGAGCATCGGCACACGGCAT | 200 |
| Nestin | NM_016701.3 |
AACTGGCACACCTCAAGATGT |
TCAAGGGTATTAGGCAAGGGG | 235 |
| β-actin | NM_007393.3 |
AGCCATGTACGTAGCCATCCA |
CTCAGCTGTGGTGGTGAA | 226 |
| Nanog | NM_028016.2 |
TTGGTTGGTGTCTTGCTCTTT |
CAGGAAGACCCACACTCATGT | 196 |
| AQP1 | NM_007472.2 |
TGCTGGCGATTGACTACA |
ACTGGTCCACACCTTCAT | 200 |
| ZO-1 | NM_001163574.1 |
ACGAGGTTATTTCCAGCGTTT |
GGTGGAACTTGCTCATAA | 237 |
|
Na+-K+ -ATPase | NM_178405.3 |
ATCAGCGAGCTCAGGACATT |
ACTACAGCCGCTAGCACGAT | 226 |
| Collagen VIII | NM_007739.2 |
CCATCACCCCAGGGAGAGTA |
CCGGTGGGAAAGGTACAGTC | 154 |
| N-cadherin | NM_007664.4 |
CCTTCTGTGTATCATCATCCT |
AGTCATAGTCCTGGTCTTCT | 176 |
| VE-cadherin | NM_009868.4 |
TGTGCTTGCCTATGAGAG |
ACAGATGCGTTGAATACCT | 177 |
| Vimentin | NM_011701.4 |
TGGTTGACACCCACTCAAAA |
GCTTTTGGGGTGTCAGTTGT | 268 |
| SLC4A4 | NM_018760 |
GGTCACCACACGATCTACATTG |
TTTGTCGGAGTAGTTCTCGGA | 129 |
ICC
Alkaline phosphatase (ALP) activity assay of PSCs
was performed using an ALP staining kit (Sigma-Aldrich) according
to the manufacturer’s instructions. Cells were seeded onto glass
coverslips pre-coated with gelatin that had been placed in 12-well
plates. At various differentiation time points, they were fixed in
4% (w/v) paraformaldehyde (Sigma-Aldrich) in 1× phosphate-buffered
saline (PBS; 2.68 mM KCl, 1.47 mM KH2PO4,
135.60 mM NaCl and 8.10 mM Na2HPO4) for 15
min at room temperature. The cells were then washed in PBS prior to
incubation with antibody blocking buffer [PBS containing 10% (v/v)
normal goat serum (Invitrogen Life Technologies), 0.3% TritonX-100
(Sigma-Aldrich) and 0.1% NaN3 (Sigma-Aldrich)] for 1 h
at room temperature. The incubation with primary antibodies
(Table II) was performed
overnight at 4°C. After three washes for 5 min in PBS, the
coverslips were incubated with fluorescently labeled secondary
antibodies (Alexa Fluor 546 or FITC 488 goat anti-mouse or goat
anti-rabbit IgG, 1:800 in PBS; Invitrogen Life Technologies) for 1
h at room temperature. After washing in PBS, the cell nuclei were
counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen
Life Technologies) for 5 min at room temperature. Negative control
samples were processed in parallel in the absence of primary
antibody. Immunoreactive cells were visualized and photographed
using a fluorescence microscope (Olympus BX51, Japan).
| Table IIPrimary antibodies used for
immunocytochemistry. |
Table II
Primary antibodies used for
immunocytochemistry.
| Antibodies | Type | Source | Dilution |
|---|
| AQP1 | Mouse
monoclonal | Abcam | 1:200 |
| ZO-1 | Rabbit
polyclonal | Abcam | 1:200 |
|
Na+-K+-ATPase | Mouse
monoclonal | Abcam | 1:200 |
| N-cadherin | Rabbit
monoclonal | Epitomics | 1:200 |
| VE-cadherin | Mouse
monoclonal | Abcam | 1:100 |
| Vimentin | Rabbit
monoclonal | Epitomics | 1:200 |
| Crystallin-αA | Rabbit
monoclonal | Santa Cruz | 1:100 |
| Oct4 | Mouse
monoclonal | Millipore | 1:200 |
| SSEA1 | Mouse
monoclonal | Abcam | 1:200 |
| P75 | Rabbit
polyclonal | Abcam | 1:300 |
| SOX10 | Rabbit
polyclonal | Abcam | 1:200 |
| AP2α | Rabbit
monoclonal | Epitomics | 1:200 |
Statistical analysis
The experimental statistics presented in this study
are expressed as the mean ± standard derivation (SD). All
experiments were performed ≥3 times unless otherwise specified.
Data were analyzed using Student’s two-tailed t-test to compare the
means of two groups or a one-way analysis of variance (ANOVA) for
comparison of the means of more than two groups using SPSS
Statistics version 17.0 software (SPSS, Inc., Chicago, IL, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
Culturing differentiation-inducing cells
(CECs and LECs) for the co-culture system
Rabbit CECs showed quick adherence and a positive
growth state; these cells reached confluence at approximately the
third day after the digestion from Descemet’s membrane and
displayed hexagonal or polygonal morphology (Fig. 1A). Positive immunostaining of ZO-1,
AQP1, Na+-K+-ATPase, VE-cadherin, N-cadherin
and vimentin, which have been used as general markers for CECs, was
used to identify the cultured cells as CECs (Fig. 1B–G).
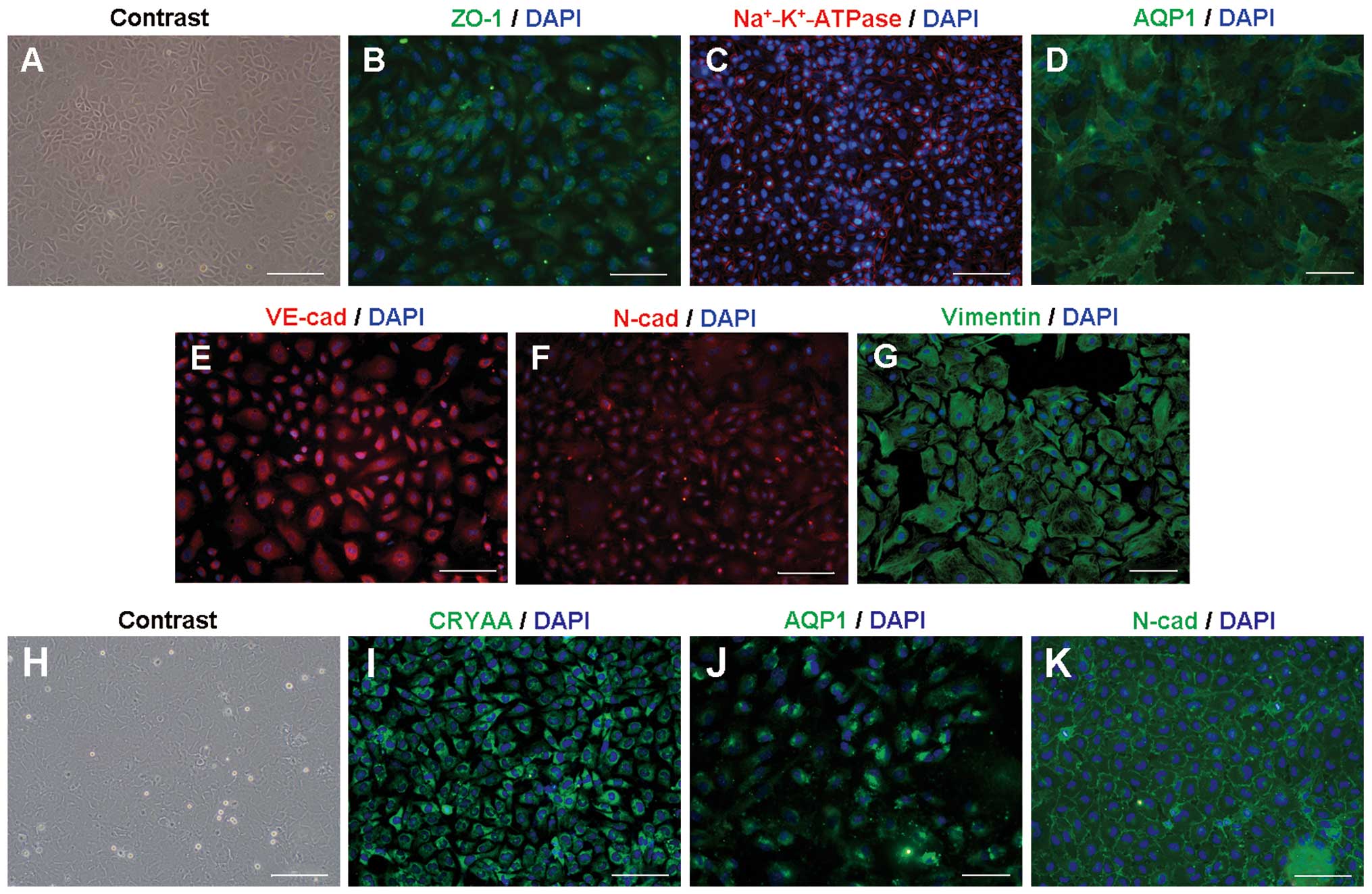 | Figure 1Characterization of the CEC and LEC
in vitro cultures. (A) In vitro cultured CECs from
rabbit presented as a hexagonal or polygonal monolayer and were
arranged in a cobblestone pattern that resembles the structure of
this tissue in vivo. When specific markers were analyzed by
immunocytochemistry, CECs stained positive for the expression of
(B) the tight junction marker ZO-1, (C)
Na+-K+-ATPase α-subunit, (D) AQP1, (E)
VE-cadherin, (F) N-cadherin and (G) vimentin. All nuclei were
counterstained with DAPI. Membrane localization was observed for
Na+-K+-ATPase, N-cadherin and AQP1.
Cytoplasmic localization was observed for vimentin and VE-cadherin.
ZO-1 was not well confined to the membrane. (H) The phenotype of
the LECs was similar to that of CECs with a polygonal monolayer,
and showed (I) high crystallin-α A (CRYAA) cytoplasmic expression
and (J) the expression of AQP1 and (K) N-cadherin at the membrane.
Scale bars: 100 μm in A, C and H; 50 μm in B, E, F, I and K; and 20
μm in D, G and J. CEC, corneal endothelial cell; LEC, lens
epithelial cell; DAPI, 4′,6-diamidino-2-phenylindole. |
Rabbit LECs adhered slowly to the plates and reached
confluence on approximately day 7 of culture (Fig. 1H). Positive identification of LECs
was made using immunostaining of LEC-specific crystallin-α A
(CRYAA) and two associated proteins, AQP1 and N-cadherin (Fig. 1I–K).
Analysis of iPSC and ESC culture and
differentiation based on EB formation
Mouse iPSCs and ESCs were cultured in the
undifferentiated state as well-shaped colonies on a feeder cell
layer in ESM containing LIF. Surface antigen SSEA1, regarded as a
marker of the undifferentiated state and ALP and Oct4, markers for
pluripotency in the mouse, were detected by immunofluorescent
staining and ALP staining (Fig.
2).
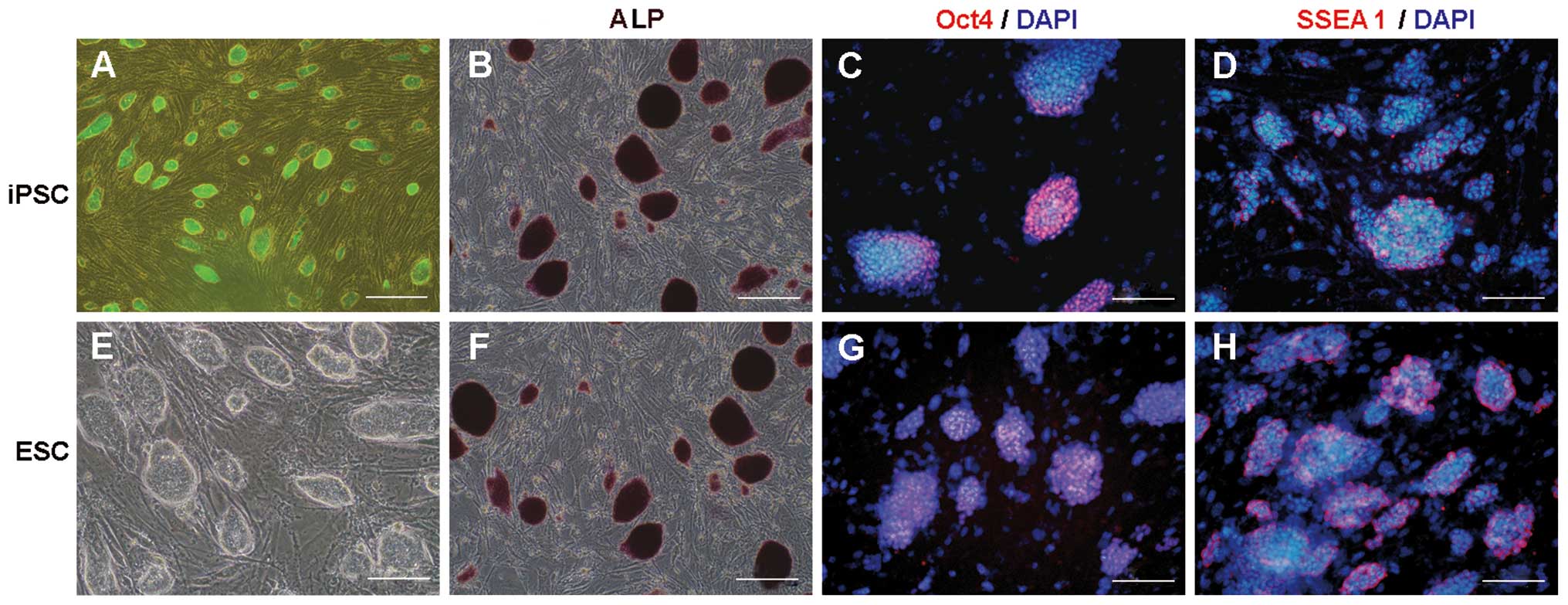 | Figure 2Pluripotency and the undifferentiated
state of mouse iPSCs and ESCs. Mouse iPSCs and ESCs maintained
their pluripotency and undifferentiated state in the presence of
MEF-feeder cells and LIF. The PSCs developed into colonies. iPSCs
expressed (A) EGFP, (B) alkaline phosphatase (ALP), (C) the
pluripotent marker Oct4 (nuclear localization) and (D) the
mouse-specific undifferentiated cell marker SSEA1 (membrane
localization). (E) Mouse ESCs appeared similar to the PSCs in terms
of colony morphology, and expressed (F) ALP, (G) Oct4 and (H)
SSEA1. Scale bars: A, 100 μm, B-H, 50 μm. iPSC, induced pluripotent
stem cell; ESC, embryonic stem cell; MEF, mice embryonic
fibroblast; LIF, leukemia inhibitory factor. |
Fig. 3A
demonstrates the procedure of PSC cell differentiation. Following
the subculture of PSCs on gelatin-coated plastic plates with a
reduced number of feeder cells for 2 days, the iPSC colony size was
increased (Fig. 3B). The complete
removal of LIF and feeder cells during suspension culture initiated
the spontaneous differentiation of iPSCs into embryoid bodies (EBs;
Fig. 3B–G). One day after the
plating of iPSCs, the cells congregated and proliferated to form
GFP-positive spheres, called induced pluripotent stem (iPS)-EBs
(Fig. 3C). The spheres continued
to grow until RA exposure on EBd4 (Fig. 3D). The addition of RA slowed the
growth of the EB spheres. In the presence of 1 μM RA, EBs were no
longer growing on EBd4+4 (Fig.
3E), in contrast to untreated EBs on EBd8 (Fig. 3G). Abundant apoptosis and cellular
debris were observed at a higher concentration of RA (10 μM) at
EBd4+4 (Fig. 3F).
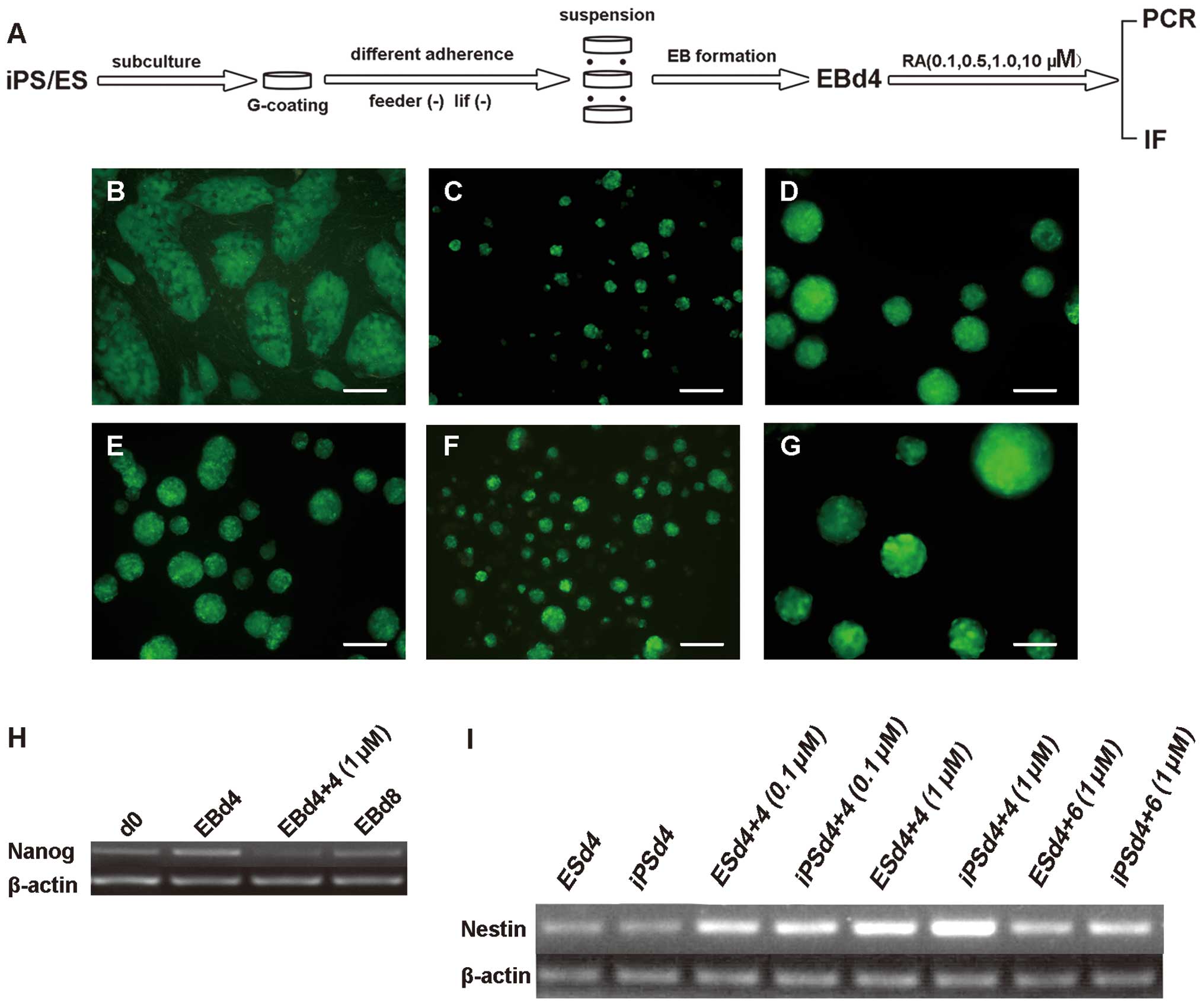 | Figure 3Neural crest differentiation of mouse
pluripotent stem cells relies on EB formation. (A) The procedure
for neural crest differentiation was based on EB formation and RA
treatment. Microscopic views of iPS-EBs at various time points: (B)
iPSCs subcultured on gelatin to reduce feeder cell numbers, (C) EBs
formed 1 day after the removal of feeder cells and LIF, (D) EB
spheres on EB differentiation day 4, (E) EB differentiation on day
4 (d4) and 1 μM RA treatment for another 4 days, (F) EB
differentiation on day 4 and 10 μM RA treatment for another 4 days,
and (G) EB differentiation on day 8 (d8) without RA treatment.
RT-qPCR for (H) Nanog mRNA expression in iPS-EBs. and (I) Nestin
mRNA expression in iPS-EBs and ES-EBs. Scale bars: A–F, 100 μm. EB,
embryoid body; RA, retinoic acid; iPS, induced pluripotent stem;
iPSC, induced pluripotent stem cell; ES, embryonic stem; LIF,
leukemia inhibitory factor; RT-qPCR, reverse-quantitative
transcription polymerase chain reaction. d4+n, day 4 plus the
number of RA exposure days. |
Effect of RA on the NC differentiation of
iPSC- and ESC-derived EBs
The addition of RA accelerated the differentiation
process, as shown by the severe reduction in the mRNA expression of
Nanog, a marker of the undifferentiated state, at EBd4+4 after 1 μM
RA treatment (Fig. 3H). The
expression level of the neuroectoderm marker Nestin varied based on
the duration and concentration of the RA treatment. The 1 μM RA
treatment promoted peak Nestin mRNA expression at EBd4+4 for both
iPS-EBs and ES-EBs (Fig. 3I).
However, Nestin was downregulated at EBd4+6 after treatment with 1
μM RA, which indicated that neuroectoderm began to predominate in
EBs at EBd4+4 following treatment with 1 μM RA in both types of
PSCs.
NC marker protein expression was examined using ICC
and gene expression was examined by RT-qPCR in the induced EBs.
Soon after EBs were seeded onto gelatin-coated plates, cells
migrated out of the EBs. The expression of the NC stem cell markers
P75, SOX10 and AP-2α in the RA-induced EBs (Fig. 4A–F) was detected in the migrating
cells around EB cores by ICC. The gene expression levels of Snail,
Slug, Twist, Sox10, P0 and P75 were upregulated in the induced
cells compared with the levels in undifferentiated cells. The
effect of RA was concentration- and time course-dependent. Based on
the gene expression profile of EBd4+4 EBs, RA at a concentration of
0.5–1 μM was better at promoting NC differentiation compared with
other concentrations that were investigated (Fig. 4G). The mRNA expression of these
genes was greatly upregulated in 0.5 μM and 1 μM RA but to a lesser
extent in 10 μM RA; indeed, Snail was downregulated in 10 μM RA.
All genes except SOX10, showed the highest mRNA expression level in
1 μM RA. The appropriate RA treatment time course was decided based
on the mRNA expression profile analysis and the comparison of
samples treated with 1 μM RA at the time points EBd4+2, EBd4+4, and
EBd4+6 and samples without RA treatment at day 0 and day 8
(Fig. 4H). Most genes were induced
to their relative highest expression levels at the EBd4+4 time
point. Slug, Twist, Sox10, and P75 showed the highest level of
expression at EBd4+4, while Snail and P0 showed higher levels of
expression at EBd4+4 and their peak expression level at EBd4+6.
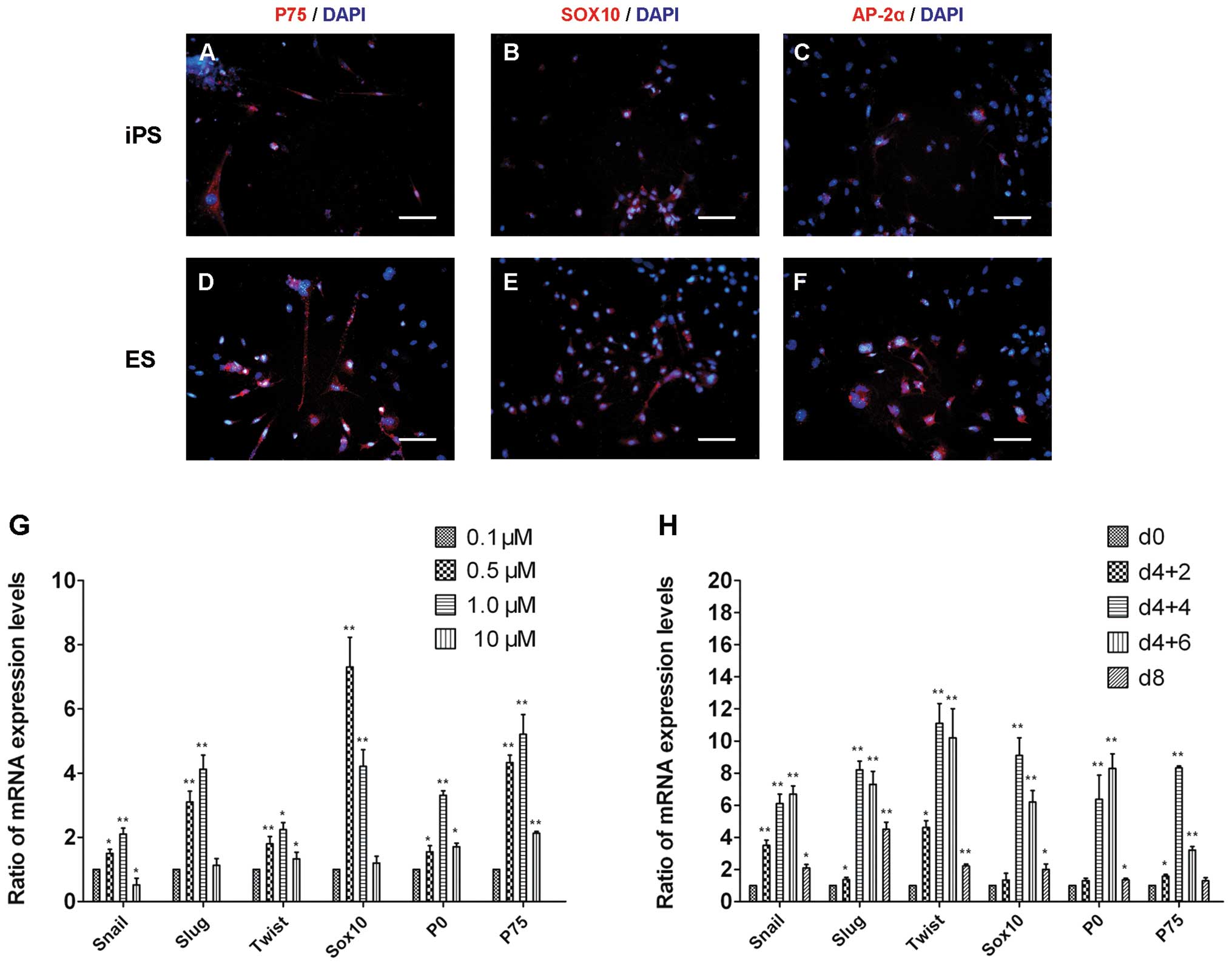 | Figure 4Characterization of neural crest
differentiation in RA-induced EBs. The cells migrating from iPS-EBs
were fixed and immunostained with antibodies against (A) P75, (B)
SOX10 and (C) AP-2α and the results indicated that the cells had
been induced toward neural crest differentiation. In ES-EBs, (D)
P75, (E) SOX10 and (F) AP-2 α protein expression were also
detected. Scale bars: A–F, 50 μm. (G) The gene expression profile
of EBd4+4 days of RA treatment at various concentrations was
determined by qPCR. (H) The mRNA expression levels of these genes
compared with neural crest differentiation in 1 μM RA at different
time points and undifferentiated iPSCs (day 0) cells were
determined in the same way. qPCR data from the iPS-EBs. The values
are expressed as means ± SEM (n=3). *P<0.05,
**P<0.01 vs. the 0.1 μM or d0 group, in G and H,
respectively. d0, iPSCs; d4+2, iPSCs after EB differentiation for 4
days plus 1 μM RA treatment for 2 days; d4+4, iPSCs after EB
differentiation for 4 days plus 1 μM RA treatment for 4 days; d4+6,
iPSCs after EB differentiation for 4 days plus 1 μM RA treatment
for 6 days; d8, iPSCs after EB differentiation 8 days without RA
treatment; RA, retinoic acid; EB, embryoid body; iPS, induced
pluropotent stem; iPSC, induced pluripotent stem cell; ES,
embryonic stem; qPCR, quantitative polymerase chain reaction. |
These data indicate that EB differentiation for 4
days followed by 1 μM RA treatment for another 4 days (EBd4+4, 1 μM
RA) could be used to induce NC differentiation as the first step of
CEC differentiation.
Effect of ECM components on EB cell
migration and differentiation
The effects of the ECM components gelatin, Ln and Fn
on EB cell migration and differentiation were investigated
(Fig. 5A–F). During the first few
days, cells on Ln (Fig. 5B)
migrated faster, but the difference was small. On day 7, the
superiority of gelatin in promoting cell migration (Fig. 5D) became clear. In comparison with
Ln (Fig. 5E) and Fn (Fig. 5F), gelatin supported greater EB
cell migration and formed stronger adherence of the
endothelium-like colonies. Therefore, gelatin was used as the
coating for cell culture and differentiation assays in subsequent
experiments.
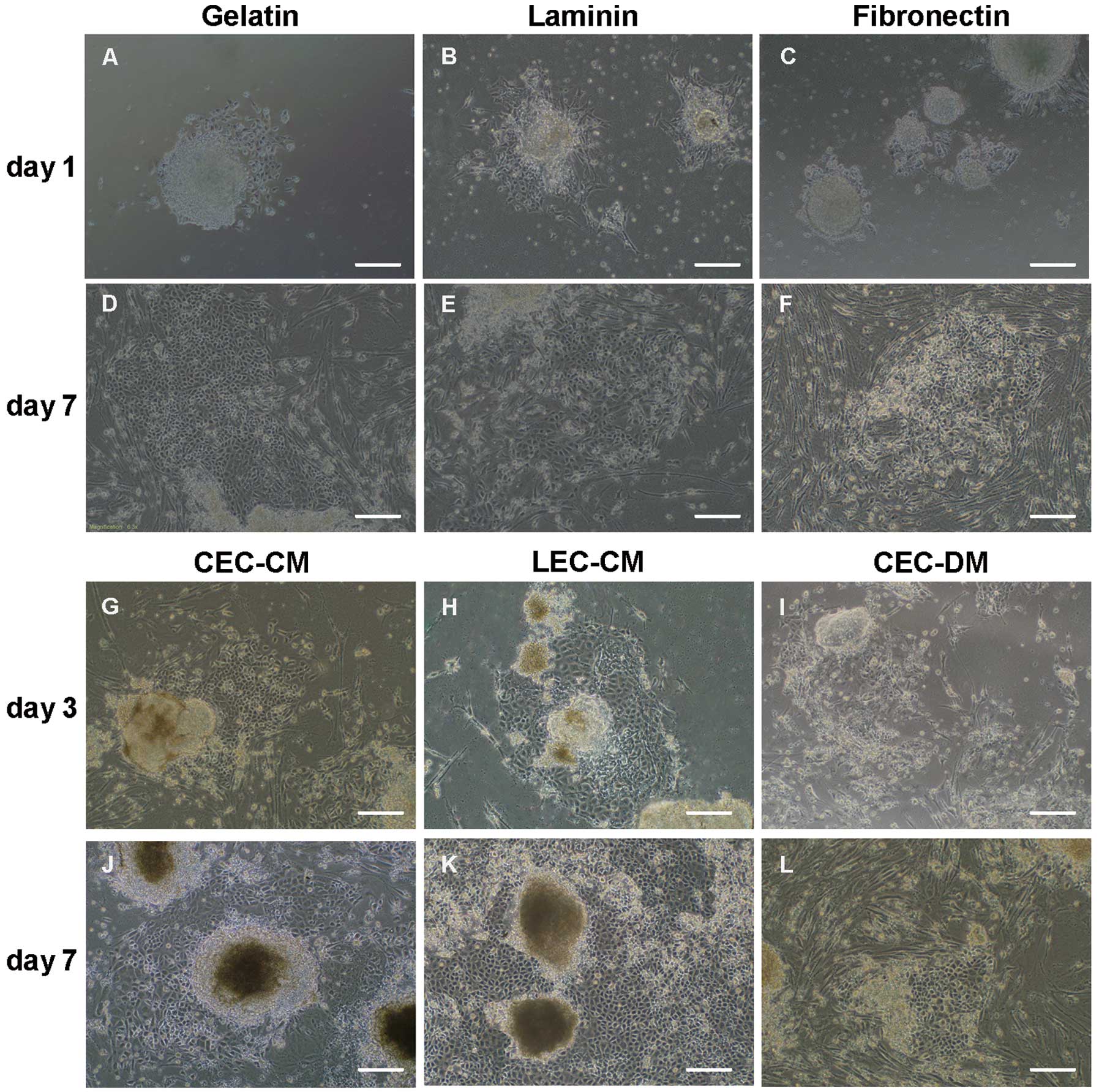 | Figure 5Morphology of EB cell migration and
differentiation in CM. Diverse effects of different coatings on EB
cell migration and differentiation were found. During early
migration on day 1 and late migration on day 7 after EB growth on
various coatings: (A, D) gelatin (B, E) laminin, and (C, F)
fibronectin. EBs continued to differentiate on gelatin-coated
plates in CEC-CM and LEC-CM with CEC-DM as a control. Cells
migrated out of the EBs and became endothelium-rich colonies.
Induced pluripotent stem cells (G) in CEC-CM, (H) LEC-CM and (I)
CEC-DM on day 3; (J) CEC-CM, (K) LEC-CM and (L) CEC-DM on day 7.
Scale bars: A–L, 100 μm. EB, embryoid body; CM, conditioned medium;
DM, differentiation medium; CEC, corneal endothelial cell; LEC,
lens epithelial cell. |
Comparison of different co-culture
systems for the second steps
Different co-culture models led to varied
differentiation results observed in the center of the EBs. The CM
induction cultures provided positive results (Fig. 5G–L); under these conditions, cells
from the EBs migrated out of the colonies early, re-adhered to the
gelatin coating, proliferated and differentiated into cells with a
polygonal shape, resulting in endothelium-like colonies. Following
cell migration and differentiation, classic-looking colonies
appeared containing an endothelium surrounding the core of the EBs.
Vigorous growth and significant endothelium differentiation, based
on morphology, was enhanced in the CM, and particularly in the
LEC-CM (Fig. 5H and K).
The expression of common CEC marker proteins,
including AQP1, ZO-1, Na+-K+-ATPase,
N-cadherin, VE-cadherin and vimentin, was examined using ICC after
LEC-CM induction for 7 days (Fig.
6). Positive expression of these markers was found in the
polygonal cells of both origins. Certain markers were expressed
differently between the pluripotent ES and iPS cell groups.
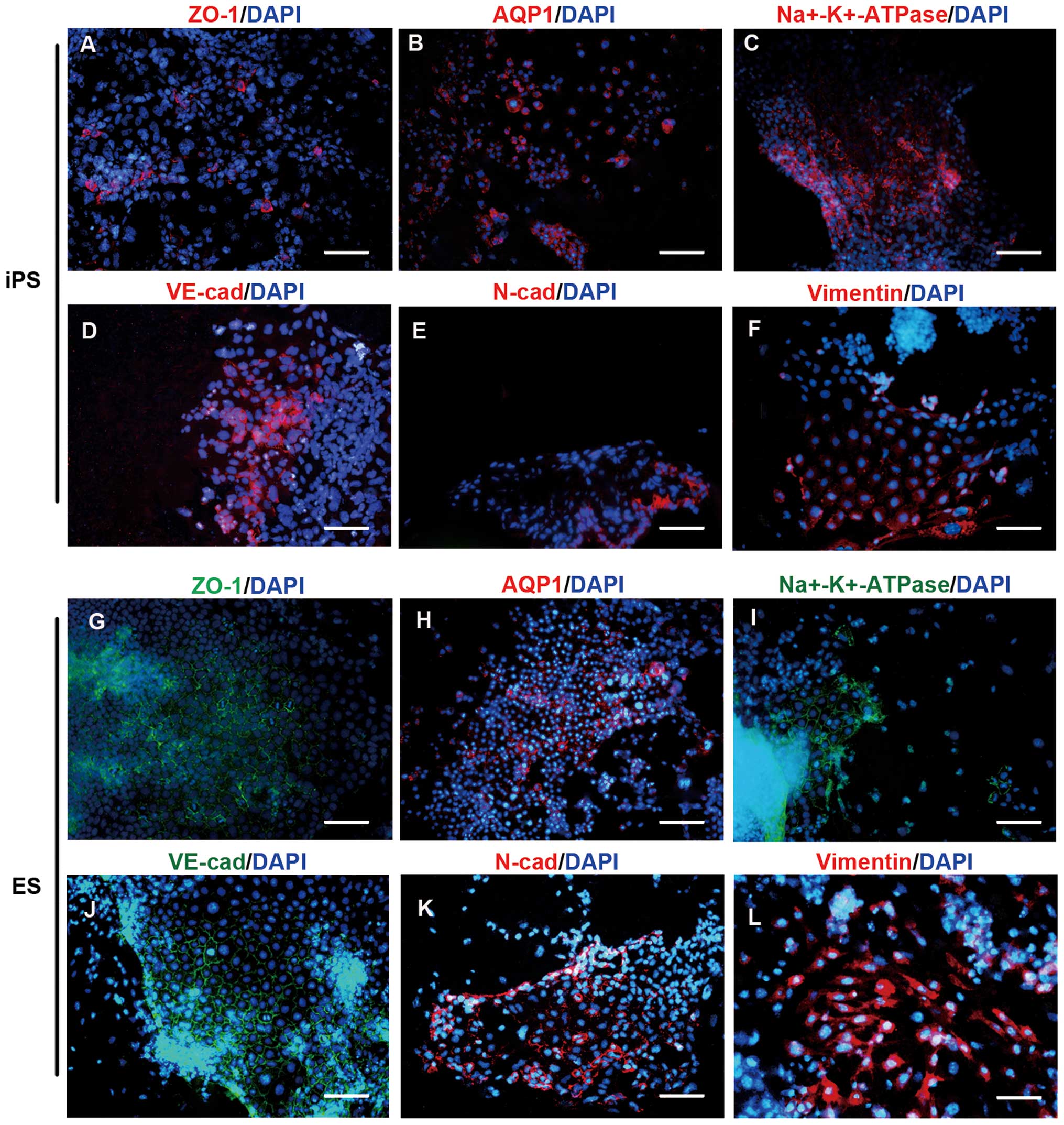 | Figure 6Protein expression of CEC-specific
markers in CEC-like colonies induced by LEC-CM. During the second
induction of differentiation by CM, endothelium-like colonies
developed, and CEC-specific markers were shown to be expressed in
the CEC-like populations by ICC after an LEC-CM induction of 7
days. Expression of (A) ZO-1, (B) AQP1, (C)
Na+-K+-ATPase, (D) VE-cadherin, (E)
N-cadherin and (F) vimentin was detected in iPSC derivatives. (G)
ZO-1, (H) AQP1, (I) Na+-K+-ATPase, (J)
VE-cadherin, (K) N-cadherin and (L) vimentin expression were
similar in ESC derivatives. Scale bars: A, C, D, E, G, I, J and K,
50 μm; B and H, 100 μm; F and L, 20 μm. CEC, corneal endothelial
cell; LEC, lens epithelial cell; CM, conditioned medium; ICC,
immunocytochemistry; iPSC, induced pluripotent stem cell: ESC,
embryonic stem cell. |
The gene expression levels of AQP1, ZO-1,
Na+-K+-ATPase, N-cadherin, VE-cadherin,
collagen VIII (Col8) and SLC4A4 in the differentiated cells were
determined by RT-qPCR analysis. All marker genes were strongly
upregulated in the differentiated cells compared with the
undifferentiated cells (Fig. 7).
At the mRNA level, in the iPSC derivatives and ESC derivatives,
there were increases of 13.9- and 12.12-fold in AQP1; 13.42- and
15.92-fold in ZO-1; 13.12- and 25.1-fold in
Na+-K+-ATPase; 15.2- and 12.1-fold in
vimentin; 50.1- and 43.6-fold in N-cadherin; 37.1- and 29.3-fold in
VE-cadherin; 41.22- and 50.73-fold in collagen VIII (Col8); and
7.36- and 10.09-fold in SLC4A4, respectively. Therefore, iPSCs and
ESCs showed a similar potential for differentiation in this
induction protocol, and LEC-CM was demonstrated to have the most
efficient induction effect, as shown by the mRNA expression
profile.
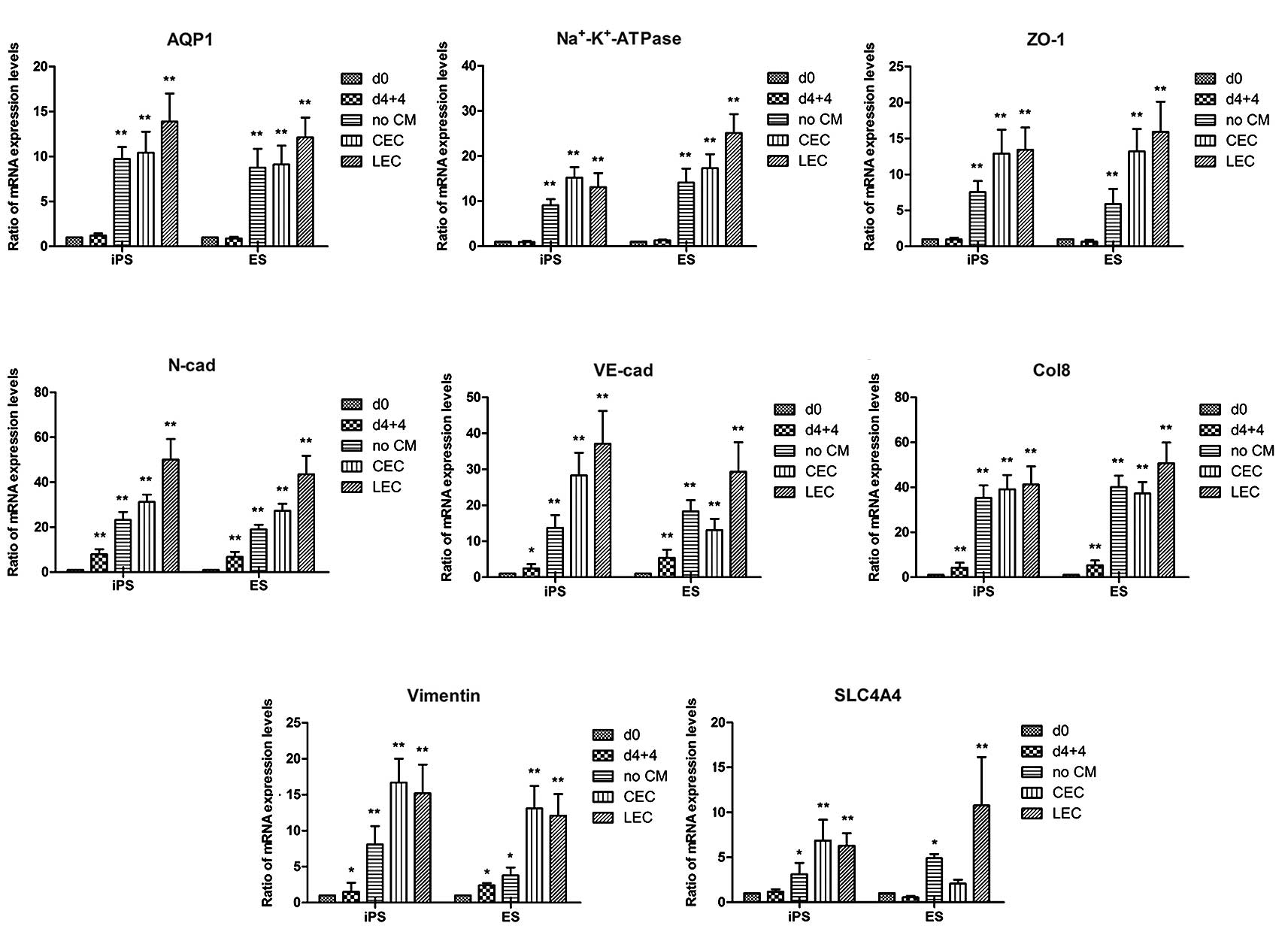 | Figure 7Induced mRNA expression profile of
differentiation-specific genes in CEC-like cells. (A–H) Gene
expression profiles of AQP1, Na+-K+-ATPase,
ZO-1, N-cadherin (N-cad), VE-cad, Col8, vimentin and SLC4 A4 at day
0, during the first stage of differentiation, and during the second
induction of differentiation using CEC-DM, CEC-CM or LEC-CM for 7
days. The error bars and statistical analysis show the standard
error of the mean (n=3). The values presented are the mean ± SEM
(n=3). *P<0.05, **P<0.01 compared with
day 0. d0, undifferentiated iPSCs or ESCs, d4+4, EB differentiation
for 4 days plus 1 μM RA treatment for 4 days; no CM, cells after
the second induction of EBd4+4 in CEC-DM without CM for 7 days;
CEC-CM, cells after the second induction of EBd4+4 in CEC-CM for 7
days; LEC-CM, cells after the second induction of EB d4+4 in LEC-CM
for 7 days. CEC, corneal endothelial cells; DM, differentiation
medium; CM, conditioned medium; LEC, lens epithelial cell; iPSC,
induced pluripotent stem cell; ESC, embryonic stem cell; RA,
retinoic acid; EB, embryoid body. |
Discussion
In this study, a protocol was developed for the
induction of differentiation towards CECs from two types of mouse
pluripotent stem cells (mPSCs): ESCs and iPSCs. The differentiation
protocol is based on a two-step induction that begins with the
formation of EBs and subsequent RA treatment followed by culture in
CM. This co-culture model is sufficient to induce the mPSCs after
~17 days.
During the first stage of the protocol, EB
formation, multiple cell lineages spontaneously differentiate but
do not yield pure populations of cells, which complicates the
characterization of the differentiated cells. RA is the active
metabolite derived from the liposoluble vitamin A (retinol), which
has extensive bioactivity and is increasingly recognized for its
role in guiding embryonic development. It is well-established that
exposure to RA for 4 days during embryoid body (EB) formation is
associated with neural differentiation, as shown in previous
studies (14–17). However, as this conventional
approach is inefficient in inducing the differentiation of many
different germ cell layers, including the ectoderm, mesoderm,
endoderm and NC, it was modified in the present study to induce NC
differentiation. The effective RA concentration was chosen based on
its ability to upregulate the expression of epithelial-mesenchymal
transition (EMT)-associated transcription factors and NC
differentiation marker genes (18,19).
In the present study, enrichment for NC cells by sorting or
isolation after the first stage of the differentiation protocol was
not carried out. Instead, the EBs were simply plated onto ECM. The
ICC and other results confirmed that without trypsinization or
sectioning, the NC stem cells residing in the EBs migrated out,
survived and differentiated during the second stage of induction,
which was treatment with CM and plating on gelatin-containing
ECM.
Several lines of ESC differentiation research
indicate that the ECM plays significant roles in cell
proliferation, cell death, cell survival and differentiation
(20,21); however, the role of the ECM in
NC-to-CEC differentiation has not been studied in detail. Plate
coatings such as gelatin, Fn and Ln, which are considered to be ECM
in cell culture, were evaluated in the present study. When the EBs
were grown on coated plates following RA exposure, differences were
found in cell migration and cell morphology. The data showed that
gelatin promoted cell migration and the generation of colonies with
an endothelium-like appearance.
The co-culture method of differentiation, a
simulation of cell-cell interaction, was used as a second induction
step for derived NC cells. The mechanisms governing cell fate
specification are not clear. LECs and CECs were selected as
inductive cells for co-culture based on the understanding of the
role of the lens epithelium in CEC organization during development.
LECs can affect neighboring cells by a paracrine mechanism,
although the signaling mechanisms are unclear. Mature CECs can also
contribute to the niche by autocrine regulation and were used as an
additional inducer. The findings of the present study are in
agreement with previous reports (22,23),
and demonstrate that CM from CECs and LECs can induce
differentiation. Significant CEC-like differentiation was observed
in the CM groups. CEC-CM and LEC-CM both enhanced CEC-like colony
formation, as demonstrated by the polygonal cells and gene
expression. CM from lens epithelium or corneal endothelium has been
demonstrated to be beneficial for cell survival (24,25).
Recent advances in generating functional corneal endothelium from
corneal stromal stem cells of NC origin were found to depend on
signaling by RA and Wnt/β-catenin (26). Whether the results in the present
study were due to the same signaling pathway is currently under
investigation.
To the best of our knowledge, this is the first
report of the generation of CEC-like cells from mouse ESCs and
iPSCs. As in most innovative work, clear morphological alterations
were observed and a number of problems remained unresolved.
Abundant polygonal-shaped cells that were arranged in
cobblestone-like clusters in CEC-CM- and LEC-CM-treated plates were
observed. The expression of CEC-specific proteins and mRNA was
enhanced, including ZO-1, Na+-K+-ATPase,
AQP1, N-cadherin, VE-cadherin, collagen VIII, vimentin and SLC4 A4
after inducement. However, the functions of the induced cells were
not evaluated in this study. The main function of mature corneal
endothelium is maintaining the stroma in a dehydrated state to
support corneal transparency. Chamber and clinical observations
made following transplantation in animal models of CEC failure are
required as the next step, and will be the focus of future
studies.
Acknowledgements
The authors are grateful to Professor Jin (Key
Laboratory of Stem Cell Biology, Institute of Health Sciences,
Shanghai Institutes for Biological Sciences, Chinese Academy of
Sciences) for providing mouse iPSCs and Professor Cao (Key
Laboratory of Tissue Engineering, National Tissue Engineering
Center of China) for ESCs R1. This study was supported by National
Natural Science Foundation (31000443, 81070737, 81000366), Shanghai
Science Committee Basic Research Leading Project (No. 11JC1407000)
and Shanghai Jiaotong University School of Medicine Doctor
Innovation Plan (BXJ201227).
Abbreviations:
|
PSC
|
pluripotent stem cell
|
|
ESC
|
embryonic stem cell
|
|
iPSC
|
induced pluripotent stem cell
|
|
EB
|
embryoid body
|
|
LEC
|
lens epithelial cell
|
|
CEC
|
corneal endothelial cell
|
|
MEF
|
mouse embryonic fibroblast
|
|
NC
|
neural crest
|
|
CM
|
conditioned medium
|
|
DM
|
defined medium
|
|
GM
|
growth medium
|
|
ESM
|
embryonic stem medium
|
|
ECM
|
extracellular matrix
|
|
Fn
|
fibronectin
|
|
Ln
|
laminin
|
References
|
1
|
Li C, Yu H, Ma Y, et al:
Germline-competent mouse-induced pluripotent stem cell lines
generated on human fibroblasts without exogenous leukemia
inhibitory factor. PloS One. 4:e67242009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Chen H, Fan X, Xia J, et al: Electrospun
chitosan-graft-poly
(epsilon-caprolactone)/poly(epsilon-caprolactone) nanofibrous
scaffolds for retinal tissue engineering. Int J Nanomedicine.
6:453–461. 2011.
|
|
3
|
Pfaffl MW: A new mathematical model for
relative quantification in real-time RT-PCR. Nucleic Acids Res.
29:e452001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kang L and Gao S: Pluripotency of induced
pluripotent stem cells. J Anim Sci Biotechnol. 3:52012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Grabel L: Prospects for pluripotent stem
cell therapies: into the clinic and back to the bench. J Cell
Biochem. 113:381–387. 2012. View Article : Google Scholar
|
|
6
|
Sugawara T, Nishino K, Umezawa A and
Akutsu H: Investigating cellular identity and manipulating cell
fate using induced pluripotent stem cells. Stem Cell Res Ther.
3:82012.PubMed/NCBI
|
|
7
|
Okita K, Yamakawa T, Matsumura Y, et al:
An efficient nonviral method to generate integration-free
human-induced pluripotent stem cells from cord blood and peripheral
blood cells. Stem Cells. 31:458–466. 2013. View Article : Google Scholar
|
|
8
|
Bard JB, Hay ED and Meller SM: Formation
of the endothelium of the avian cornea: a study of cell movement in
vivo. Dev Biol. 42:334–361. 1975. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gage PJ, Rhoades W, Prucka SK and Hjalt T:
Fate maps of neural crest and mesoderm in the mammalian eye. Invest
Ophthalmol Vis Sci. 46:4200–4208. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Genis-Galvez JM: Role of the lens in the
morphogenesis of the iris and cornea. Nature. 210:209–210. 1966.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yue W, Pi QM, Zhang WJ, et al: Platelet
endothelial cell adhesion molecule-1, stage-specific embryonic
antigen-1, and Flk-1 mark distinct populations of mouse embryonic
stem cells during differentiation toward hematopoietic/endothelial
cells. Stem Cells Dev. 19:1937–1948. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li C, Wu Q, Liu B, et al: Detection and
validation of circulating endothelial cells, a blood-based
diagnostic marker of acute myocardial infarction. PLoS One.
8:e584782013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yao Y, Wang L, Zhang H, et al: A novel
anticancer therapy that simultaneously targets aberrant p53 and
Notch activities in tumors. PLoS One. 7:e466272012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Akanuma H, Qin XY, Nagano R, et al:
Identification of stage-specific gene expression signatures in
response to retinoic acid during the neural differentiation of
mouse embryonic stem cells. Front Genet. 3:1412012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang X, Wang J, Huang V, Place RF and Li
LC: Induction of NANOG expression by targeting promoter sequence
with small activating RNA antagonizes retinoic acid-induced
differentiation. Biochem J. 443:821–828. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Paulsen BS, Souza CS, Chicaybam L, et al:
Agathisflavone enhances retinoic acid-induced neurogenesis and its
receptors alpha and beta in pluripotent stem cells. Stem Cells Dev.
20:1711–1721. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kumar M, Bagchi B, Gupta SK, Meena AS,
Gressens P and Mani S: Neurospheres derived from human embryoid
bodies treated with retinoic acid show an increase in nestin and
ngn2 expression that correlates with the proportion of tyrosine
hydroxylase-positive cells. Stem Cells Dev. 16:667–681. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Prasad MS, Sauka-Spengler T and LaBonne C:
Induction of the neural crest state: control of stem cell
attributes by gene regulatory, post-transcriptional and epigenetic
interactions. Dev Biol. 366:10–21. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Strobl-Mazzulla PH and Bronner ME:
Epithelial to mesenchymal transition: new and old insights from the
classical neural crest model. Semin Cancer Biol. 22:411–416. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brafman DA, Phung C, Kumar N and Willert
K: Regulation of endodermal differentiation of human embryonic stem
cells through integrin-ECM interactions. Cell Death Differ.
20:369–381. 2013. View Article : Google Scholar :
|
|
21
|
Rowland TJ, Blaschke AJ, Buchholz DE,
Hikita ST, Johnson LV and Clegg DO: Differentiation of human
pluripotent stem cells to retinal pigmented epithelium in defined
conditions using purified extracellular matrix proteins. J Tissue
Eng Regen Med. 2012.PubMed/NCBI
|
|
22
|
Shao C, Fu Y, Lu W and Fan X: Bone
marrow-derived endothelial progenitor cells: a promising
therapeutic alternative for corneal endothelial dysfunction. Cells
Tissues Organs. 193:253–263. 2011. View Article : Google Scholar
|
|
23
|
Ju C, Zhang K and Wu X: Derivation of
corneal endothelial cell-like cells from rat neural crest cells in
vitro. PLoS One. 7:e423782012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ishizaki Y, Voyvodic JT, Burne JF and Raff
MC: Control of lens epithelial cell survival. J Cell Biol.
121:899–908. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sugino IK, Rapista A, Sun Q, et al: A
method to enhance cell survival on Bruch’s membrane in eyes
affected by age and age-related macular degeneration. Invest
Ophthalmol Vis Sci. 52:9598–9609. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hatou S, Yoshida S, Higa K, et al:
Functional corneal endothelium derived from corneal stroma stem
cells of neural crest origin by retinoic acid and Wnt/beta-catenin
signaling. Stem Cells Dev. 22:828–839. 2013. View Article : Google Scholar
|














