Introduction
Velocardiofacial syndrome/DiGeorge syndrome
(VCFS/DGS) is caused by a 1.5–3-Mb microdeletion of chromosome
22q11.2, and is frequently known as 22q11.2 deletion syndrome
(22q11.2DS) [Mendelian Inheritance in Man (MIM) no. 188400/192430].
This syndrome is typically characterised by conotruncal heart
defects, a cleft palate, thymic and parathyroid dysplasia with
resulting immune defects, hypocalcaemia and learning disabilities
(1).
Although 22q11.2DS is the most frequent interstitial
deletion in humans, this syndrome presents a wide phenotypic
spectrum with >180 clinical manifestations. The estimated
prevalence of the syndrome is one in 4,000 live births (2–4);
however, the actual occurrence may be higher due to the variation
in severity. Individuals present with symptoms on a spectrum from
life-threatening to nearly asymptomatic (5,6). The
diagnosis can be missed due to subtle dysmorphic facial features.
Numerous studies have focused on defining patients eligible for
screening (7–9). Agergaard et al (7) found that clinical assessment
identified fewer than three-quarters of the patients with a 22q11.2
deletion. In excess of one-quarter of patients are likely to remain
undiagnosed if genetic tests are not performed on a routine basis.
Oh et al (8) reported that
characteristic facial expressions and a small stature correlated
only with 22q11.2 microdeletions. Furthermore, Mikhail et al
(9) suggested that the recurrent
distal 22q11.2 microdeletions do not represent a single clinical
entity, and they proposed categorising these deletions into three
types according to their genomic position. The 22q11.2
microdeletion types share certain presenting features; however they
each have their own unique features and risks. Recently, based on a
clinical and dysmorphologic evaluation of 194 individuals and a
review of the literature, Monteiro et al (10) defined new guidelines for screening
the 22q11.2 deletion and divided patients into four groups: Group
I, clinical suspicion of 22q11.2DS with palatal anomalies; group
II, clinical suspicion without palatal anomalies; group III,
cardiac malformations associated with 22q11.2DS; and group IV,
juvenile-onset schizophrenia.
In the present study, three previously undiagnosed
22q11.2DS families were described and clinical and molecular
cytogenetic studies were performed. The aim of the study was to
provide additional data for prenatal counselling and for the
clinical diagnosis of 22q11.2DS.
Subjects and methods
Patient consent
Ethical approval was obtained for this study from
the Ethics Committee of the First Affiliated Hospital of Sun
Yat-Sen University (Guangzhou, China). Photographs of the
individuals were taken and used for clinical assessments. Samples
and photographs from the patients and their families were obtained
following informed consent.
Patient one
A 31-year-old male without underlying disease sought
genetic counselling due to adverse reproductive outcomes. The
patient and his wife were normal and non-consanguineous, and there
was no familial history of congenital malformations. His wife had
one foetus with a ventricular septal defect (VSD) and a normal
chromosome karyotype analysis; however, the foetus was not tested
for the 22q11.2 microdeletion. Two years later, a second foetus
exhibited cardiac anomalies (VSD, transposition of conducting
arteries and pulmonary artery stenosis), nuchal fold thickening and
bilateral renal pelvis separation on an ultrasound scan. Prenatal
diagnosis was performed using umbilical cord blood. The results
showed that the chromosome karyotype analysis was normal but
fluorescent in situ hybridisation (FISH) for a 22q11.2
microdeletion (probes for TUPLE1 and N25) indicated that a 22q11.2
microdeletion was present.
The father of the patient was 33 years old and his
mother was 30 years old at the time of his birth. The patient had a
middle school level of education and was frequently ill prior to
primary school. His height was 173 cm and his weight was 70 kg
(body mass index, 23.4 kg/m2). The blood pressure of the
patient was 100/60 mmHg and his pulse was 80 bpm. His abdominal
examinations were ordinary, and facial features included a long
face, pharyngeal abnormalities (two uvulas), bulbous nose, broad
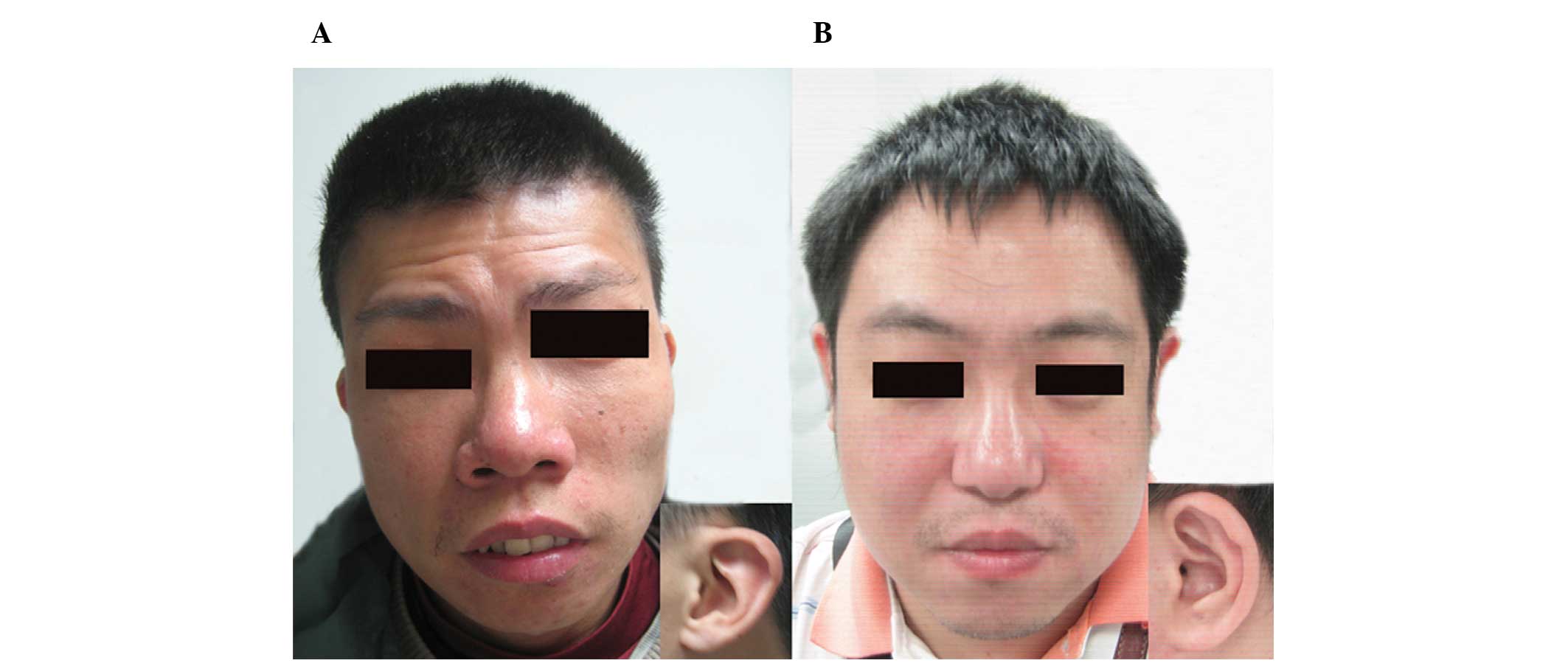
mouth, thin upper lip and low-set, dysplastic ears (Fig. 1A).
Patient two
A 38-year-old male was referred for evaluation as he
had a history of two children with congenital heart defects. The
patient and his wife were normal and non-consanguineous with no
familial history of congenital malformations. The first infant was
a term, natural-labour male with a birth weight of 3 kg and without
choking-rescue history. When the baby was eight months old, he was
diagnosed with tetralogy of Fallot and subsequently passed away.
The second child was also a term, natural-labour male. The child
had a birth weight of 2.5 kg and no choking-rescue history. A few
days after his birth, he was also diagnosed with tetralogy of
Fallot. At the age of one year, he succumbed during heart
surgery.
The mother of the patient was 39 years old at the
time of his birth. The patient has three older brothers and two
older sisters, all of whom had a normal pregnancy history. At the
age of eight years, the patient caught a high fever (40°C for 24 h)
that his parents thought harmed his brain and resulted in a
learning disability. No further genetic tests were performed. The
height of the patient was 175 cm and his weight was 65 kg (body
mass index, 21.2 kg/m2). His blood pressure was 110/75
mmHg and his pulse was 85 bpm. The patient had a history of
tuberculosis and was often ill before the age of 12. His ears were
high set, he had no earlobe and he exhibited auricle reversal
(photo not provided).
Patient three
A 39-year-old male presented with a history of
adverse reproductive outcomes. The patient and his wife had two
babies with congenital heart defects, were normal and
non-consanguineous and had no familial history of congenital
malformations. The first child was a term, natural-labour female
who was diagnosed with tetralogy of Fallot at birth. The baby
succumbed aged nine months. Four years later, a second female was
born and diagnosed with pulmonary artery stenosis, atrial septal
defect and patent ductus arteriosus at one week old. The baby
passed away at 11 months.
The mother of the patient recalled that, during her
pregnancy with him, polyhydramnios was found during the second
trimester; however no other abnormalities were noted. Until the
patient was seven years old, he was susceptible to disease and
often caught colds, including tonsillitis. Subsequent to his
seventh birthday, the patient’s physique improved. At the time of
the study, his height was 162 cm and his weight was 75 kg (body
mass index, 28.6 kg/m2). The blood pressure of the
patient was 150/90 mmHg and his pulse was 80 bpm. Physical features
included a bulbous nose and a high-set, small ear with no earlobe
(Fig. 1B). With the exception of
his hypertension, the health of the patient was unremarkable.
Molecular studies
Conventional cytogenetic analysis and
FISH
Peripheral blood samples were collected from the
three families, including their spouses and their parents. Foetal
blood samples were obtained by cordocentesis. Cytogenetic analysis
was performed following the standard harvesting of blood
lymphocytes. Metaphase chromosomes were G-banded at 550 bands of
resolution.
Metaphase FISH analysis on cultured peripheral blood
lymphocytes was performed using a Vysis DiGeorge region probe [LSI
TUPLE 1, SpectrumOrange/LSI ARSA SpectrumGreen, fluroescein
isothiocyanate (FITC); Abbott Laboratories, Abbott Park, IL, USA]
and a Cytocell DiGeorge region probe (TBX1, red spectrum/22qter,
green spectrum, FITC; N25, red spectrum/22qter, green spectrum,
FITC; Cytocell, Cambridge, UK). A minimum of 20 metaphase cells
were assessed under a fluorescence microscope (Leica Microsystems,
Wetzlar, Germany).
Single nucleotide polymorphism
(SNP)-array hybridisation profiling and data analysis
Genomic DNA was isolated from peripheral blood
samples using a QIAamp DNA Blood Mini kit (Qiagen, Valencia, CA,
USA). DNA concentrations were measured with a NanoDrop
spectrophotometer (ND-1000 V.3.1.2; NanoDrop, Thermo Fisher
Scientific Inc., Wilmington, DE, USA). The DNA samples (250 ng)
were hybridised to CytoScan HD arrays (Affymetrix®,
Santa Clara, CA, USA) according to the manufacturer’s instructions.
The Affymetrix CytoScan HD array includes >2.7 million copy
number markers, of which 750,000 are SNPs that can be used for
genotyping and 1.9 million are non-polymorphic probes. The
Chromosome Analysis Suite software package (Affymetrix) was used
for all analyses. Copy number variations (CNVs) were detected by
visual inspection of the normalised log2 intensity plots and
numerical analysis of the SNP log2 intensity ratios. Copy number
changes observed were compared with the CNVs catalogued in the
Database of Genomic Variants and the University of Santa Clara in
California (UCSC) genome browser. The gene content of the CNVs of
interest was determined using the UCSC browser based on the Genome
Reference Consortium human genome (GRCH; build 37; http://genome.ucsc.edu/). For putative candidate
regions containing at least one gene, each assessment included
searches for similar patients in the Database of Chromosomal
Imbalance and Phenotype in Humans using Ensembl Resources and
PubMed.
Results
Conventional cytogenetic analysis
Karyotyping of the cultured lymphocytes from all of
the patients indicated a normal karyotype. In addition, the spouses
and parents of the patients also had normal karyotypes, based on
G-banding techniques with a resolution of 550 bands.
FISH
The deletion of 22q11.2 was detected by FISH in the
three patients. Metaphase FISH analysis of the cultured
lymphocytes, using a Vysis DiGeorge region probe and a Cytocell
DiGeorge region probe, was used to detect the lack of 22q11.2
signal on chromosome 22, which revealed a deletion at 22q11.2.
SNP-array analysis
The SNP-array narrowed down the deletion size of
22q11.2 for the patients, and revealed that all four patients
(three adults and one fetus) shared the same breakpoints (Table I). The molecular details and
phenotypic features of the three patients with 22q11.2 deletion are
shown in Table I. The deletions
were approximately 2.5 Mb, with the identical breakpoint from
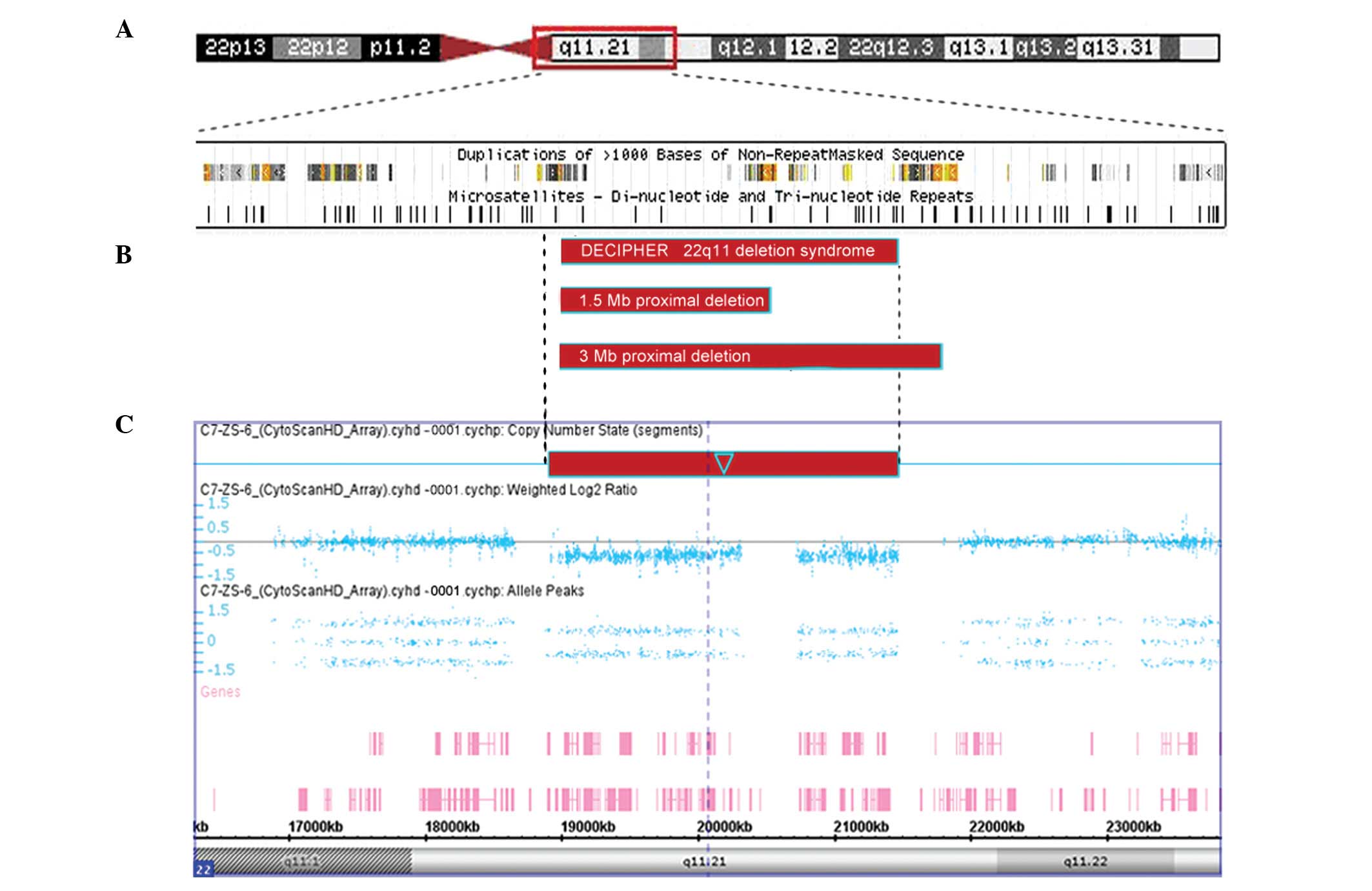
22:18,916,842 to 22:21,465,659 (Fig.
2C). This deletion region includes 58 RefSeq and eight Online
MIM genes (Fig. 2) encompassing
the genes TBX1, COMT, DGCR2, GP1BB, RTN4R, PRODH, SNAP29 and
SERPIND1. The results of both the FISH and SNP-array of the
parents of the three patients were normal, however the three
patients demonstrated a de novo 22q11.2 deletion.
 | Table IMolecular details and phenotypic
features of individuals with a 22q11.2 deletion. |
Table I
Molecular details and phenotypic
features of individuals with a 22q11.2 deletion.
| Patient | Deleted band | Start site; stop site
(bp)b | Size (Mb) | Origin | Age (years) | DD | SD | HD | Velopharyngeal
abnormalities | Facial
dysmorphology | Others |
|---|
| 1 | 22q11.21 | 18,916,842;
21,465,659 | 2.549 | Dn | 31 | Mild | + | - | Two uvulas | Long face, bulbous
nose, broad mouth, thin upper lip, low-set dysplastic ears | - |
| 1a | 22q11.21 | 18,916,842;
21,465,659 | 2.549 | Pat | Fetus | Unk | Unk | VSD TCA, PAS | - | Unk | Unk |
| 2 | 22q11.21 | 18,916,842;
21,465,659 | 2.549 | Dn | 38 | Mild | + | - | - | High-set ears with no
lobes, auricle reversal | Two fetuses with
congenital heart defect delivered |
| 3 | 22q11.21 | 18,916,842;
21,465,659 | 2.549 | Dn | 39 | No | + | - | - | Bulbous nose,
high-set ears with no lobes | Hypertension |
The karyotype of all four patients was therefore
46,XY.ish del(22)(q11.2q11.2)(TUPLE1-,N25-,TBX1-).arr
22q11.2(18,916,842–21,465,659)x1, according to the GRCH 37
(International System for Human Cytogenetics Nomenclature
2009).
Discussion
The chromosome 22q11.2 region contains eight
different chromosome-22-specific low-copy repeats (LCRs) that are
known as LCR22s (LCR22-A to LCR22-H). These LCR22s mediate
recurrent microdeletions and microduplications by non-allelic
homologous recombination (11).
Molecular characterisation of the patients in the current study
found that the deletion extended from LCR-A to LCR-E, which
overlapped with the 3-Mb common typically deleted region or 1.5-Mb
DiGeorge critical region (DGCR) observed in VCFS/DGS (Fig. 2).
The 22q11.2 microdeletion was initially detected
using FISH analysis for microdeletion/microduplication syndromes
during prenatal diagnosis and genetic consultation. This syndrome
is usually diagnosed in early childhood in the presence of a
typical facial appearance, congenital heart defects, a cleft palate
and early-onset hypocalcaemia. By contrast, the patients in the
present study were without any cardiac defects and with mild
phenotypes (mild developmental delays and dysmorphic features,
first diagnosed at the age of >30 years old). Shared traits
included two foetuses/infants with a heart defect and decreased
childhood immunity. Genomic DNA analysis using an Affymetrix
CytoScan HD microarray showed that these patients had identical
22q11.2 deletions of ≥2.5 Mb, including 58 RefSeq and eight Online
MIM genes (Fig. 2) encompassing
the genes TBX1, COMT, DGCR2, GP1BB,
RTN4R, PRODH, SNAP29 and SERPIND1.
Although up to 58 genes are deleted, it is the
haplo-insufficiency of the transcription factor TBX1 that is
believed to be the primary contributing factor in this disorder
(12–14). T-box transcription factor (Tbx1)
belongs to an evolutionarily conserved T-box family of
transcription factors, whose expression is precisely regulated
during embryogenesis, and appears to regulate the proliferation and
differentiation of various progenitor cells during organogenesis
that can be clinically affected in this syndrome (15). In addition to other
dosage-sensitive genes observed in 22q11.2DS, the incomplete
penetration of Tbx1 is the underlying factor for adults without
heart defects and with mild dysmorphic features. The low immunity
in childhood described by these patients correlated with the defect
of cellular immunity in DiGeorge syndrome. Patients with this
syndrome present with a broad range of T-cell deficiencies. Foetal
thymus transplantation is an effective treatment for reconstituting
cellular immunity and normalising the imbalance of regulatory
T-cell functions in patients with DiGeorge syndrome (16). Since the patients develop a normal
repertoire of mature T-cells, however, the immune defect appears
mild in patients surviving the correction of cardiac anomalies
(17); therefore, the autoimmune
features of 22q11.2DS typically become apparent in early childhood
but are rarely diagnosed in adulthood (18,19).
It was found in the present study that the foetus of
patient one with a 2.5-Mb deletion presented with severe cardiac
defects, whereas his father, with the identical deletion, had no
heart defects and only presented with mild developmental delays,
bifid uvula and mild dysmorphic features. The other two families
did not have the confirmation of genetic detection of their
affected foetuses or babies; however, it is suspected that these
heart-defect siblings harboured the 22q11.2 deletions transmitted
by their fathers.
There are hypotheses regarding the phenotypic
variations, such as differences in the size of the deletion, CNVs,
epigenetic changes, modifying genetic factors, somatic mosaicism
and differences in the intrauterine environment (20); thus, a ‘second hit’ (mutation) in a
modifying genetic factor appears more likely for the patients
considered in the present study. There are certain mechanisms for
the second hit hypothesis, which include replication errors, base
changes and additional deletions. According to Stalmans et
al (21) variations in the
gene encoding vascular endothelial growth factor may have the
potential to modify the cardiovascular phenotype of hemizygous
22q11.2 deletions. Similarly, a study by Driscoll et al
(22) described modifiers for
palatal phenotypes with this syndrome; as such, the ‘second somatic
hit’ is not just restricted to a genetic change at the level of the
DNA sequence but may also involve epigenetic changes. It has been
demonstrated that the genetic background affects the severity of
cardiovascular, thymic and parathyroid anomalies in mice (23,24).
The ‘two-hit’ model proposed by Girirajan et al (25) stated the requirement for a
secondary event during foetal development to cause more severe
clinical manifestations. This second hit could be another CNV, a
disruptive single-base-pair mutation in a phenotypically relevant
gene or an environmental event causing alterations to the phenotype
or deletion size, as was observed in the patients from the present
study. The two-hit model additionally helps to explain the
underlying phenotypic variability that has been reported for
several recurrent microdeletions. The majority of second hits are
likely to be undetectable, even using high-resolution arrays.
Re-sequencing the whole genome, however, may reveal a number of
additional contributing loci (20,26).
High-density, whole-genome SNP arrays have an
important advantage over conventional karyotyping in
microdeletion/microduplication detection, microarray-based copy
number analysis and genotype analysis. The resolution is affected
by the genomic distance between the probes and their size.
Molecular karyotyping provides information directly associated with
the physical and genetic map of the human genome, and microarrays
enable the identification of small CNVs with a high accuracy.
Commercially available arrays are an invaluable tool for the
diagnosis of patients presenting with intellectual disabilities
and/or multiple congenital abnormalities (27). Array-comparative genomic
hybridization or SNP arrays have been demonstrated to represent a
cost-effective alternative to multiple FISH testing for the
identification of genomic imbalances (28).
In conclusion, three adult cases of 22q11.2DS with
mild dysmorphic features have been reported in the present study.
Although a range of autoimmune features associated with 22q11.2DS
have been described, the condition typically becomes apparent in
early childhood and is rarely diagnosed in adulthood. It is worth
clinicians considering the diagnosis of 22q11.2DS in an adult
patient with a past medical history of compromised immunity, and it
is necessary to carry out DNA microarray analysis on couples who
have had recurrent abnormal pregnancies to exclude the
microdeletion/microduplication syndrome in patients without severe
abnormalities or with normal phenotypic manifestations.
Acknowledgements
The authors would like to thank the participants and
their families; Hongning Xie and Yunxiao Zhu for foetal ultrasonic
examination; Junhong Chen and Ailan Huang for assistance with
patient recruitment; Baojiang Chen for helpful comments and
guidance; and Chenyu Gou, Xiaomei Shi and Hanjing Chai for their
support of this study. The authors were assisted by numerous
people, including those in the Department of Gynaecology and
Obstetrics and the Department of Ultrasonography of The First
Affiliated Hospital of Sun Yat-Sen University.
Abbreviations:
|
22q11.2DS
|
22q11.2 deletion syndrome
|
|
SNP
|
single nucleotide polymorphism
|
|
FISH
|
fluorescent in situ
hybridisation
|
|
VCFS
|
velocardiofacial syndrome
|
|
DGS
|
DiGeorge syndrome
|
|
VSD
|
ventricular septal defect
|
|
CNV
|
copy number variation
|
|
LCR
|
low-copy repeat
|
|
DGCR
|
DiGeorge critical region
|
References
|
1
|
Emanuel BS, McDonald-McGinn D, Saitta SC
and Zackai EH: The 22q11.2 deletion syndrome. Adv Pediatr.
48:39–73. 2001.PubMed/NCBI
|
|
2
|
Goodship J, Cross I, LiLing J and Wren C:
A population study of chromosome 22q11 deletions in infancy. Arch
Dis Child. 79:348–351. 1998. View Article : Google Scholar
|
|
3
|
Devriendt K, Fryns JP, Mortier G, van
Thienen MN and Keymolen K: The annual incidence of
DiGeorge/velocardiofacial syndrome. J Med Genet. 35:789–790. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Oskarsdóttir S, Vujic M and Fasth A:
Incidence and prevalence of the 22q11 deletion syndrome: a
population-based study in Western Sweden. Arch Dis Child.
89:148–151. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
McDonald-McGinn DM, Tonnesen MK,
Laufer-Cahana A, et al: Phenotype of the 22q11.2 deletion in
individuals identified through an affected relative: cast a wide
FISHing net! Genet Med. 3:23–29. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bassett AS, Chow EW, Husted J, et al:
Clinical features of 78 adults with 22q11 Deletion Syndrome. Am J
Med Genet A. 138:307–313. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Agergaard P, Hebert A, Sørensen KM,
Østergaard JR and Olesen C: Can clinical assessment detect 22q11.2
deletions in patients with cardiac malformations? A review. Eur J
Med Genet. 54:3–8. 2011. View Article : Google Scholar
|
|
8
|
Oh AK, Workman LA and Wong GB: Clinical
correlation of chromosome 22q11.2 fluorescent in situ hybridization
analysis and velocardiofacial syndrome. Cleft Palate Craniofac J.
44:62–66. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mikhail FM, Burnside RD, Rush B, et al:
The recurrent distal 22q11.2 microdeletions are often de novo and
do not represent a single clinical entity: a proposed
categorization system. Genet Med. 16:92–100. 2014. View Article : Google Scholar
|
|
10
|
Monteiro FP, Vieira TP, Sgardioli IC, et
al: Defining new guidelines for screening the 22q11.2 deletion
based on a clinical and dysmorphologic evaluation of 194
individuals and review of the literature. Eur J Pediatr.
172:927–945. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shaikh TH, O’Connor RJ, Pierpont ME, et
al: Low copy repeats mediate distal chromosome 22q11.2 deletions:
sequence analysis predicts breakpoint mechanisms. Genome Res.
17:482–491. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Vitelli F, Taddei I, Morishima M, Meyers
EN, Lindsay EA and Baldini A: A genetic link between Tbx1 and
fibroblast growth factor signaling. Development. 129:4605–4611.
2002.PubMed/NCBI
|
|
13
|
Vitelli F, Morishima M, Taddei I, Lindsay
EA and Baldini A: Tbx1 mutation causes multiple cardiovascular
defects and disrupts neural crest and cranial nerve migratory
pathways. Hum Mol Genet. 11:915–922. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen M, Yang YS, Shih JC, et al:
Microdeletions/duplications involving TBX1 gene in fetuses with
conotruncal heart defects which are negative for 22q11.2 deletion
on fluorescence in-situ hybridization. Ultrasound Obstet Gynecol.
43:396–403. 2014. View Article : Google Scholar
|
|
15
|
Scambler PJ: 22q11 deletion syndrome: a
role for TBX1 in pharyngeal and cardiovascular development. Pediatr
Cardiol. 31:378–390. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mayumi M, Kimata H, Suehiro Y, et al:
DiGeorge syndrome with hypogammaglobulinaemia: a patient with
excess suppressor T cell activity treated with fetal thymus
transplantation. Eur J Pediatr. 148:518–522. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pierdominici M, Marziali M, Giovannetti A,
et al: T cell receptor repertoire and function in patients with
DiGeorge syndrome and velocardiofacial syndrome. Clin Exp Immunol.
121:127–132. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shea YF, Lee CH, Gill H, et al: Delayed
diagnosis of 22q11.2 deletion syndrome in an adult Chinese lady.
Chin Med J (Engl). 125:2945–2947. 2012.
|
|
19
|
Nakada Y, Terui K, Kageyama K, et al: An
adult case of 22q11.2 deletion syndrome diagnosed in a 36-year-old
woman with hypocalcemia caused by hypoparathyroidism and
Hashimoto’s thyroiditis. Intern Med. 52:1365–1368. 2013. View Article : Google Scholar
|
|
20
|
Halder A, Jain M, Chaudhary I and Varma B:
Chromosome 22q11.2 microdeletion in monozygotic twins with
discordant phenotype and deletion size. Mol Cytogenet. 5:132012.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Stalmans I, Lambrechts D, De Smet F, et
al: VEGF: a modifier of the del22q11 (DiGeorge) syndrome? Nat Med.
9:173–182. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Driscoll DA, Boland T, Emanuel BS, et al:
Evaluation of potential modifiers of the palatal phenotype in the
22q11.2 deletion syndrome. Cleft Palate Craniofac J. 43:435–441.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Taddei I, Morishima M, Huynh T and Lindsay
EA: Genetic factors are major determinants of phenotypic
variability in a mouse model of the DiGeorge/del22q11 syndromes.
Proc Natl Acad Sci USA. 98:11428–11431. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Marshak H, Morrow AA and Morrow DM:
Biplanar temple lift for lateral brow ptosis: comparison with
uniplanar dissection technique. Aesthetic Plast Surg. 32:517–522.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Girirajan S, Rosenfeld JA, Cooper GM, et
al: A recurrent 16p12.1 microdeletion supports a two-hit model for
severe developmental delay. Nat Genet. 42:203–209. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rauch A, Zink S, Zweier C, et al:
Systematic assessment of atypical deletions reveals
genotype-phenotype correlation in 22q11.2. J Med Genet. 42:871–876.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Vermeesch JR, Brady PD, Sanlaville D, Kok
K and Hastings RJ: Genome-wide arrays: quality criteria and
platforms to be used in routine diagnostics. Hum Mutat. 33:906–915.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fix A, Lucchesi C, Ribeiro A, et al:
Characterization of amplicons in neuroblastoma: high-resolution
mapping using DNA microarrays, relationship with outcome, and
identification of overexpressed genes. Genes Chromosomes Cancer.
47:819–834. 2008. View Article : Google Scholar : PubMed/NCBI
|