Introduction
Primitive neuroectodermal tumor (PNET) is a rare
aggressive malignant small round cell tumour, which is most common
in children and young adults (1).
PNET belongs to the Ewing’s sarcoma family tumors (ESFT), due to
its specific chromosomal translocation: t(11;22)(q24;q12) (2). It is thought to be of neural crest
origin and is most commonly encountered in the soft tissue or bone
(3). Reports of cases arising in
the intestines are extremely rare. To the best of our knowledge,
there are 18 cases in the literature (4–21).
The present study reports three unusual cases of PNET arising from
the mesentery and ileocecum and describes the presenting symptoms,
imaging findings, pathological features and molecular genetics of
the tumors.
Case reports
Ethics
The present study was conducted in accordance with
the Declaration of Helsinki and was approved by the Institutional
Review Board of Jingling Hospital (Nanjing, China). Furthermore,
informed consent was obtained from the relatives of each
participating patient.
Case 1
In June 2005, a previously healthy 59-year-old male
presented to the Department of Pathology, Nanjing Jinling Hospital
(Nanjing, China) following 20 days of lower quadrant pain. Computed
tomography (CT) scanning revealed a 5×6 cm mass in the lower
quadrant. Tumor markers, such as carcinoembryonic antigen and
carbohydrate antigen 19–9, were normal. Explorative laparotomy
revealed a 6×5×3 cm mass in the mesentery of the terminal ileum,
without any macroscopic spread elsewhere in the abdomen.
Microscopically, the sections revealed the proliferation of small
round cells in nests with a finely distributed chromatin pattern
and rosette formation, hyperchromatic nuclei and scant cytoplasm,
and the rosettes were diffusely infiltrated with fat. The tumor
cells were positive for CD99, FLI1 and EMA, but negative for CD3,
CgA, CD20, CKpan, S100 and HMB45. As no other foci were detected in
extensive clinical and radiological investigations, the tumor was
considered to be primary. Molecular testing demonstrated the
expression of EWS/FLI1 fusion transcripts corresponding to the
t(11;22)(q24;q12) translocation. Based on the above
clinicopathological and genetic findings, a histopathological
diagnosis of PNET developing from the mesentery of the terminal
ileum was made. The patient was surgically treated for the tumor
and administered with adjuvant chemotherapy.
Case 2
In September 2005, a 22-year-old male presented with
abdominal intermittent abdominal pain of 20 days duration. Physical
examination revealed the presence in the lower abdomen of a ~10×8
cm mass, with an uneven surface and no tenderness. Abdominal CT
showed a mass (10×11 cm) in the lower abdomen and pelvis; it also
revealed multiple areas of intrahepatic cystic density. Explorative
laparotomy revealed that the mass adhered tightly to the sigmoid
colon, rectum and omentum. The left lateral lobe and right lobe of
the liver could touch two obvious nodules, 6 and 8 cm in diameter,
respectively. Due to massive intraoperative blood loss and
hypotension, the patient did not undergo liver tumor resection.
Macroscopic examination revealed a grayish-brown, soft tissue mass,
with varicose veins on the surface and areas of cystic changes.
Microscopically, the tumor featured a uniform, sheet-like
proliferation of small round tumor cells with high mitosis. Focally
the tumor cells also formed ribbon-like or rosette-like structures,
with areas of hemorrhage and necrosis, and infiltration into
adipose tissue. The mesenteric lymph nodes were free.
Immunohistochemically, the tumor cells showed diffuse membrane
positivity for CD99, Syn and FLI1. There was a punctate pattern of
positive staining for S100 and negativity for CKpan, CD3, CD34,
SMA, HMB45, MelanA and CgA. EWS/FLI1 fusion gene translocation
demonstrated the translocation t(11;22)(q24;q12). The final
diagnosis was intestinal PNET with liver metastases. Postoperative
adjuvant chemotherapy was not administered.
Case 3
In January 2009, a 36-year-old female was admitted
to hospital with abdominal pain and an abdominal mass. On physical
examination, a large and firm mass was evident in the right
abdomen. Laboratory evaluation showed a CA125 level of 57.41 IU/ml
(normal, 35 IU/ml), whereas the CA199 levels were within normal
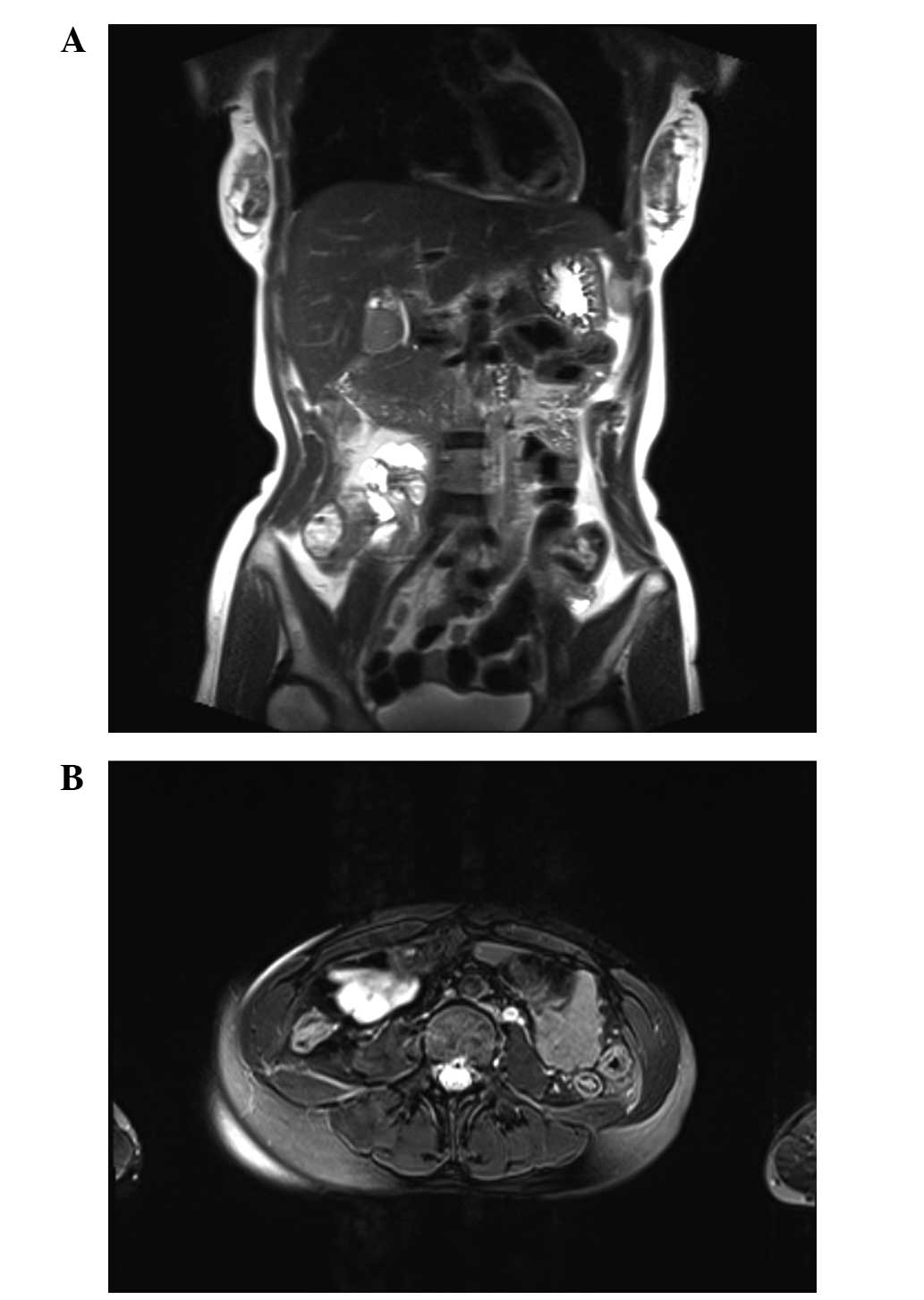
limits. Magnetic resonance imaging confirmed a large mass in the
pelvis (Fig. 1). A provisional
diagnosis of right accessory malignancy was made. Exploratory
laparotomy revealed that the mass was localized in the ileocecal
region and adhered to the sigmoid colon, with no adhesion to the
uterus or fallopian tubes. There was also a large quantity (~500
ml) of bloody, jelly-like liquid within the abdominal cavity. On
gross examination, a huge mass in the ileocecum, measuring 15×15×13
cm, with a cauliflower-like appearance was found. The mass was
grayish-black in color with a hard texture. Histologically, the
tumor consisted of small round cells arranged in diffuse sheets
with uniform rosette formation, large and round nuclei,
hyperchromatic nuclei and scant cytoplasm. Large areas of necrosis
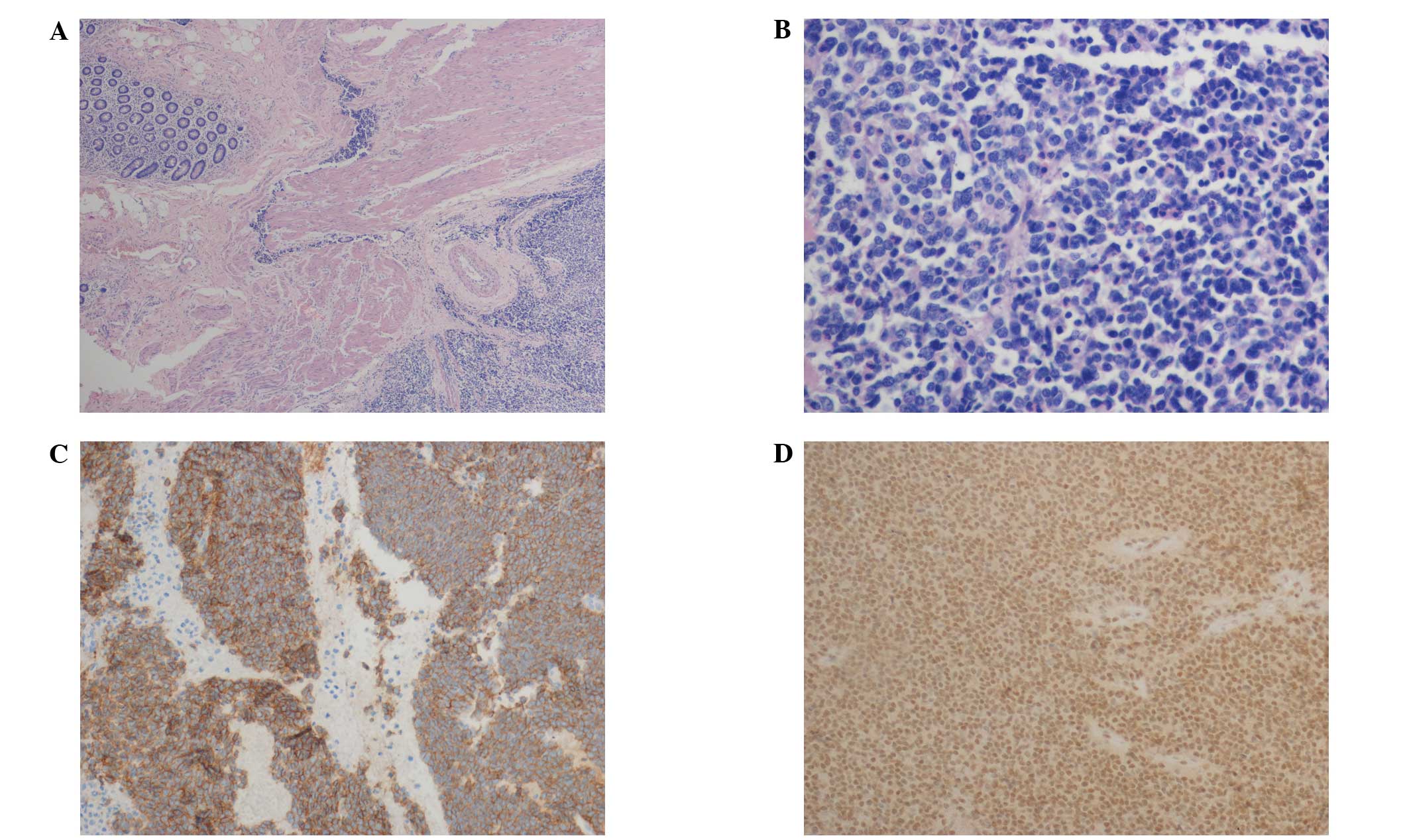
were also present. Immunohistochemistry demonstrated diffuse
membrane positivity of the tumor cells for vimentin, FLI1 and CD99
and negativity for CKpan, CgA, Syn, CD117, HMB45, S100, CD20 and
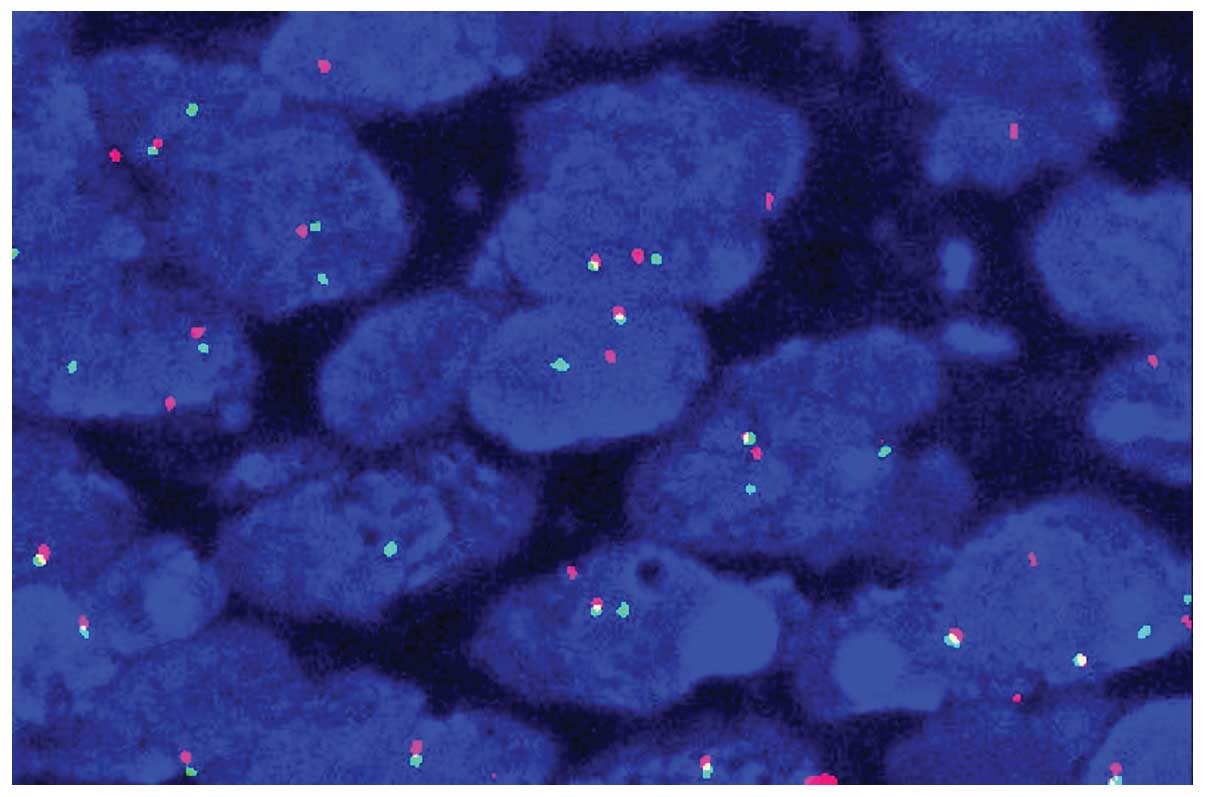
CD3 (Fig. 2). The EWS/FLI1 fusion
transcript of the t(11,22)(q24;q12) translocation was also
detected by fluorescence in situ hybridization (FISH;
Fig. 3). Due to the unusual
location of the tumor in the mesentery, the patient succumbed from
peritoneal recurrence 34 months after the surgery.
Discussion
PNET is a rare aggressive malignant small round cell
tumor most commonly arising in the central nervous system, soft
tissues or bones, which was first recognized by Stout in 1918
(22). The progenitor cells are
possibly neural crest cells (1).
PNET is usually seen along the central axis, particularly in the
soft tissue or bone in children and young adults. This rare and
aggressive tumor has been described in the kidney (23), uterine cervix (24) and pancreas (25). Regardless of the point of origin,
these tumors are highly aggressive, often quickly metastasizing to
the lung and bone. To the best of our knowledge, 18 cases of
intestinal PNET have been reported previously (Table I). The cases were eight males and
10 females, with an average age at presentation of 29 years (range,
9–63 years); 12 cases were aged ≤40 years. Its incidence in older
individuals, as in case 1 in the present study, is quite rare. The
small bowel, and its mesentery, is the most common site (15 cases);
others occurred in the duodenum and mesocolon. The present study
describes another unusual location, namely the ileocecum (case
3).
 | Table IClinical features of previously
reported cases with PNET arising from the intestine and
mesentery. |
Table I
Clinical features of previously
reported cases with PNET arising from the intestine and
mesentery.
| Authors (ref.) | Age
(years)/gender | Location | Tumor size and/or
weight | Positive
immunomarker | Follow-up |
|---|
| 1 Balasubramanian
et al (4) | 53/F | Small bowel
mesentery | 25×26×17.5 cm; 2.6
kg | MIC-2 and PGP9.5
strongly | NM |
| 2 Sethi and Smith
(5) | 44/M | Small bowel | 120 mm diameter | MIC-2 strongly and
NSE weakly | Succumbed with
recurrence, 13 mo |
| 3 Bala et al
(6) | 57/F | Small bowel
mesentery | 12 cm diameter | Vimentin, NSE, O-13,
c-Kit, FLI | NED, 8 mo |
| 4 Horie and Kato
(7) | 40/M | Small bowel
mesentery | 11×8 cm | CD99, NSE, syn and
vimentin. | Succumbed with
recurrence, 5 mo |
| 5 Tokudome et
al (8) | 24/F | Transverse colonic
mesentery | 12×10×7 cm, 590
g | NSE and Mic-2 | NED, 20 mo |
| 6 Maisonnette et
al (9) | 56/F | Mesocolon | 12×14×12 cm | CD99 and FLI1 | Succumbed to acute
respiratory failure, 13 mo |
| 7 Adair et al
(10) | 21/F | Duodenum | NM | CD99 and CK | 10 mo DFS |
| 8 Rodarte-Shade et
al (11) | 32/M | Small bowel | 12×8 cm | CD99 and FLI1 | 6 mo DFS |
| 9 Sarangarajan et
al (12) | 13/M | Jejunum | NM | CD99 and CK | 12 mo DFS |
| 10 Graham et
al (13) | 14/M | Small bowel and
mesentery | 6×3.5×3 cm | CD99 and CK | 10 mo DFS |
| 11 Vignali et
al (14) | 15/F | Ileum | 12×9×8 cm | NM | NM |
| 12 Kim et al
(15) | 63/M | Small bowel | NM | CD99 and CD117 | NM |
| 13 Shek et al
(16) | 9/F | Small bowel and
mesentery | NM | CD99 | 18 mo DFS |
| 14 Boehm et al
(17) | 18/M | Ileum | NM | NM | NM |
| 15 Kie et al
(18) | 20/F | Duodenum | NM | CD99 | 18 mo DFS |
| 16 Prasertvit and
Stoikes (19) | 28/F | Small intestine | NM | NM | NM |
| 17 Turkyilmaz et
al (20) | 15/F | Mesocolon | 10×10×12 cm | Vimentin and
CD99 | NM |
| 18 Kim et al
(21) | 23/M | Mesentery of
jejunum | 12×8×7.5 cm | CD99, CD57 and
NSE | NM |
In the current study, all cases were primary to the
mesentery and ileocecum, with no evidence of the PNET arising
elsewhere. The maximum tumor diameter was 6–15 cm. The histologic
appearance of the tumor typically comprised round-to-ovoid
hyperchromatic cells with minimal cytoplasm, arranged in nests with
variable rosette formation. Immunohistological examinations usually
revealed CD99 and FLI1 positivity. FISH analysis indicated the
presence of EWSR1 gene rearrangement in these three patients.
Making an accurate diagnosis is critical for optimal
patient management and prognostication. Physical examination often
reveals an abdominal or pelvic mass with recurrent abdominal pain.
Imaging examination such as CT scanning is able to provide
important information regarding the size of the mass, the
involvement of adjacent structures and the presence of metastasis.
There are no suggestive blood markers that can be used to diagnose
PNET. Mhawech-Fauceglia et al (26) demonstrated that the most sensitive
and specific test panel for the diagnosis of Ewing’s sarcoma/PNET
is a combination of CD99 and FLI1. Recently, Yoshida et al
(27) reported that the NKX2.2
gene, as an important target of EWS-FLI1, is a valuable marker for
PNET, with a sensitivity of 93% and a specificity of 89%. The
genetic hallmark is the presence of a specific translocation
t(11;22)(q24;q12), which is expressed in 90–95% of patients
(28).
It is difficult to differentiate PNET from other
small round-cell tumors. However, immunohistochemical examinations
with myogenic, neurogenic, and lymphoid cell markers can rule out
many of these tumors. In gastroenteropancreatic neuroendocrine
neoplasms (GEP-NENs), histological examination reveals a trabecular
or solid arrangement and immunohistochemical analysis reveals the
expression of neuroendocrine markers (Syn and CgA) (29). In type II enteropathy-associated
T-cell lymphoma, histological examination of the lymphoma cells
reveals full-thickness infiltration of the intestinal wall. A
notable difference from PNET is the presence of villous atrophy,
cryptal hyperplasia and intraepithelial lymphocytosis. The tumor
cells express CD3, CD43 and CD8 (30). Metastatic carcinoma has a high
incidence in older patients and often has a definite primary
lesion, good adhesion between cells, nested or irregular glandular
structure, cellular atypia and mitotic activity. In addition,
specific immune markers suggestive of tissue origin are positive.
For metastatic malignant melanoma, the majority of cases have an
antecedent history of melanoma (skin and mucous membrane),
morphological diversity is observed, and lipofuscin granules are
visible in the cytoplasm. Malignant melanoma often has diffuse
positivity for S-100 protein as well as possible positivity for
melanocytic markers, including HMB45 and MelanA (31).
There is no established treatment modality for
intestinal PNET. Surgical excision when complete offers the best
chance for survival and adjuvant radiotherapy may reduce local
recurrence (32). Combination
chemotherapy has traditionally included vincristine, doxorubicin,
cyclophosphamide and dactinomycin. The addition of ifosfamide and
etoposide to a standard regimen significantly improves the outcome
for patients with nonmetastatic Ewing’s sarcoma (33). The prognosis of mesenteric PNET is
better compared with that of other sites and is not associated with
the size of the tumor. The 5-year disease-free survival rate of
patients without metastatic disease is >60% compared with 35%
for patients who present with metastatic disease (6). In reviewing the literature, it was
observed that two patients succumbed due to recurrence, one
succumbed to acute respiratory failure, and two survived with no
evidence of disease. The duration of follow-up ranged from 6 to 20
months. The average survival time was 12 months. In the patients of
the present study, case 3 was followed up for 34 months and
succumbed due to peritoneal recurrence. However, cases 1 and 2 were
not followed up.
In conclusion, the present study reviewed 18 known
cases of PNET arising from the intestine and mesentery and reported
three additional cases. Immunohistochemical examination and
molecular characterization are beneficial for differentiating PNET
from other small round-cell tumors.
Acknowledgements
This study was supported in part by the National
Natural Science Foundation of China (81371611, 81171391, 81372743)
and the National Basic Research Priorities Program 973 Project
(2014CB744504) from the Ministry of Science and Technology of
China.
References
|
1
|
Dehner LP: Primitive neuroectodermal tumor
and Ewing’s sarcoma. Am J Surg Pathol. 17:1–13. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Folpe AL, Goldblum JR, Rubin BP, et al:
Morphologic and immunophenotypic diversity in Ewing family tumors:
a study of 66 genetically confirmed cases. Am J Surg Pathol.
29:1025–1033. 2005.PubMed/NCBI
|
|
3
|
Burchill SA: Ewing’s sarcoma: diagnostic,
prognostic, and therapeutic implications of molecular
abnormalities. J Clin Pathol. 56:96–102. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Balasubramanian B, Dinakarababu E and
Molyneux AJ: Primary primitive neuroectodermal tumour of the small
bowel mesentery: case report. Eur J Surg Oncol. 28:197–198. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sethi B and Smith GT: Primary primitive
neuroectodermal tumour arising in the small bowel. Histopathology.
50:665–666. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bala M, Maly A, Remo N, et al: Peripheral
primitive neuroectodermal tumor of bowel mesentery in adults. Isr
Med Assoc J. 8:515–516. 2006.PubMed/NCBI
|
|
7
|
Horie Y and Kato M: Peripheral primitive
neuroectodermal tumor of the small bowel mesentery: a case showing
perforation at onset. Pathol Int. 50:398–403. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tokudome N, Tanaka K, Kai MH, et al:
Primitive neuroectodermal tumor of the transverse colonic mesentery
defined by the presence of EWS-FLI1 chimeric mRNA in a Japanese
woman. J Gastroenterol. 37:543–549. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Maisonnette F, Roux ET, Abita T, et al:
Ewing sarcoma of the mesocolon. Gastroenterol Clin Biol.
31:552–554. 2007.(In French). View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Adair A, Harris SA, Coppen MJ and Hurley
PR: Extraskeletal Ewings sarcoma of the small bowel: case report
and literature review. J R Coll Surg Edinb. 46:372–374. 2001.
|
|
11
|
Rodarte-Shade M, Palomo-Hoil R, Vazquez J,
et al: Primitive neuroectodermal tumor (PNET) of the small bowel in
a young adult with lower gastrointestinal bleeding. J Gastrointest
Cancer. 4:2012.
|
|
12
|
Sarangarajan R, Hill DA, Humphrey PA, et
al: Primitive neuroectodermal tumors of the biliary and
gastrointestinal tracts: clinicopathologic and molecular diagnostic
study of two cases. Pediatr Dev Pathol. 4:185–191. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Graham DK, Stork LC, Wei Q, et al:
Molecular genetic analysis of a small bowel primitive
neuroectodermal tumor. Pediatr Dev Pathol. 5:86–90. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Vignali M, Zacchè MM, Messori P, et al:
Ewing’s sarcoma of the small intestine misdiagnosed as a voluminous
pedunculated uterine leiomyoma. Eur J Obstet Gynecol Reprod Biol.
162:234–235. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim DW, Chang HJ, Jeong JY, et al: Ewing’s
sarcoma/primitive neuroectodermal tumor (ES/PNET) of the small
bowel: a rare cause of intestinal obstruction. Int J Colorectal
Dis. 22:1137–1138. 2007. View Article : Google Scholar
|
|
16
|
Shek TW, Chan GC, Khong PL, et al: Ewing
sarcoma of the small intestine. J Pediatr Hematol Oncol.
23:530–532. 2001. View Article : Google Scholar
|
|
17
|
Boehm R, Till H, Landes J, et al:
Ileoileal intussusception caused by a Ewing sarcoma tumour. An
unusual case report. Eur J Pediatr Surg. 13:272–275. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kie JH, Lee MK, Kim CJ, et al: Primary
Ewing’s sarcoma of the duodenum: a case report. Int J Surg Pathol.
11:331–337. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Prasertvit S and Stoikes N: A rare case of
ewing’s sarcoma of the small intestine. Am Surg. 79:E78–E79.
2013.PubMed/NCBI
|
|
20
|
Turkyilmaz Z, Sonmez K, Karabulut R, et
al: Extraskeletal Ewing sarcoma of the mesocolon in a child. J
Pediatr Surg. 47:E1–E3. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kim JM, Chu YC, Choi CH, et al: Peripheral
primitive neuroectodermal tumor with osseous component of the small
bowel mesentery: a case study. Korean J Pathol. 47:77–81. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Stout AP: A tumor of the ulnar nerve. Proc
NY Pathol Soc. 18:2–12. 1914.
|
|
23
|
Risi E, Iacovelli R, Altavilla A, et al:
Clinical and pathological features of primary neuroectodermal
tumor/ewing sarcoma of the kidney. Urology. 82:382–386. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Malpica A and Moran CA: Primitive
neuroectodermal tumor of the cervix: a clinicopathologic and
immunohistochemical study of two cases. Ann Diagn Pathol.
6:281–287. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shi L, Guo Z and Wu X: Primary pulmonary
primitive neuroectodermal tumor metastasis to the pancreas: a rare
case with seven-year follow-up. Diagn Pathol. 8:512013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mhawech-Fauceglia P, Herrmann F,
Penetrante R, et al: Diagnostic utility of FLI-1 monoclonal
antibody and dual-colour, break-apart probe fluorescence in situ
(FISH) analysis in Ewing’s sarcoma/primitive neuroectodermal tumour
(EWS/PNET). A comparative study with CD99 and FLI-1 polyclonal
antibodies. Histopathology. 49:569–575. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yoshida A, Sekine S, Tsuta K, et al:
NKX2.2 is a useful immunohistochemical marker for Ewing sarcoma. Am
J Surg Pathol. 36:993–999. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Saxena R, Sait S and Mhawech-Fauceglia P:
Ewing sarcoma/primitive neuroectodermal tumor of the kidney: a case
report. Diagnosed by immunohistochemistry and molecular analysis.
Ann Diagn Pathol. 10:363–366. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lindholm DP and Oberg K: Biomarkers and
molecular imaging in gastroenteropancreatic neuroendocrine tumors.
Horm Metab Res. 43:832–837. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chan JK, Chan AC, Cheuk W, et al: Type II
enteropathy-associated T-cell lymphoma: a distinct aggressive
lymphoma with frequent γδ T-cell receptor expression. Am J Surg
Pathol. 35:1557–1569. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
McCluggage WG: Ovarian neoplasms composed
of small round cells: a review. Adv Anat Pathol. 11:288–296. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Marec-Bérard P, Chotel F and Claude L:
PNET/Ewing tumours: current treatments and future perspectives.
Bull Cancer. 97:707–713. 2010.(In French).
|
|
33
|
Grier HE, Krailo MD, Tarbell NJ, et al:
Addition of ifosfamide and etoposide to standard chemotherapy for
Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N Engl
J Med. 348:694–701. 2003. View Article : Google Scholar : PubMed/NCBI
|