Introduction
Hepatocellular carcinoma (HCC) is the fifth most
prevalent type of cancer and the third most common cause of
cancer-associated mortality worldwide. Each year there are ~630,000
new cases of HCC, more than half of which occur in China (1). Despite recent advances in the
understanding of the molecular basis of HCC and new therapeutic
approaches, the mortality rate has declined only modestly. Drug
resistance remains a major clinical obstacle to successful
treatment in HCC patients (2).
Autophagy (which means self-eating) is a dynamic
process in which subcellular membranes are rearranged to sequester
cytoplasmic constituents, including organelles, for delivery to a
lysosome or vacuole where the sequestered cargo is degraded and
recycled (3). The formation of
autophagosomes, double-membrane vesicles that deliver the
cytoplasmic material, is central to this process. Autophagosome
formation involves the conjugation of cytosolic
microtubule-associated protein light chain 3 (LC3-I) with
phosphatidylethanolamine to form LC3-phosphatidylethanolamine
(LC3-II) as an essential process. Therefore, the ratio of LC3-II
and LC3-I levels can be used as a marker to reflect the activation
of autophagy (4,5).
During periods of nutrient deprivation, autophagy
degrades intracellular proteins to serve as substrates for ATP
generation (6). Autophagy is
essential during starvation, cellular differentiation, cell death
and aging, and also in the prevention of certain types of cancer
and increased tumor cell survival (7,8).
Autophagy induction (autophagic activity above basal levels) may
occur with anticancer drug treatment as an adaptive response and
can lead to chemoresistance (9).
For example, when exposed to low doses of radiation, breast, colon
and prostate carcinoma cells accumulate acidic vesicular organelles
(AVOs) due to autophagy; this is a defense mechanism that increases
the survival of irradiated cells (10). The p53-induced transcription of
proteins involved in the positive regulation of the autophagy
pathway is the common mechanism by which different types of
anticancer drugs such as DNA-damaging agents, microtubule
interfering molecules, and kinase inhibitors trigger autophagy
(11,12).
Defects of autophagy are associated with numerous
diseases and tumors (13,14). However, only a few studies have
examined hepatocellular carcinoma (HCC) in relation to these
processes (15,16). Therefore, in the present study,
inducible lentivirus-mediated RNA interference (RNAi) of LC3 was
used to investigate the association between autophagy and
chemoresistance in HCC. The aim of the study was to improve our
understanding of autophagy in human liver cancers and delineate the
possible role of autophagy as a novel target for anticancer
therapy.
Materials and methods
Cell culture and treatment
conditions
The human hepatoma cell line HepG2 was
purchased from the Cancer Cell Repository (Shanghai Cell Bank,
Shanghai, China) and maintained in Dulbecco’s modified Eagle’s
medium (high glucose; Gibco Life Technologies, Carlsbad, CA, USA)
and supplemented with 10% fetal bovine serum (FBS; Gibco Life
Technologies) in a humidified incubator with 95% air and 5%
CO2 at 37°C. DMEM without serum was also used in certain
experiments. All small hairpin RNA (shRNA)-expressing stable cell
lines were grown in DMEM supplemented with 10% tetracycline
(Tet)-approved FBS (Clontech Laboratories, Inc., Mountain View, CA,
USA) and 1 μg/ml puromycin (Gibco Life Technologies).
Antibodies and agents
The anti-LC3B rabbit polyclonal antibody used in the
western blot assay was from Cell Signaling Technology (#12741S;
Beverly, MA, USA), the anti-p62/sequestosome 1 (SQSTM1) rabbit
polyclonal antibody was obtained from Proteintech (#18420-1-AP;
Chicago, IL, USA) and the anti-GAPDH mouse monoclonal antibody was
from Beijing CoWin Biotech Co., Ltd. (cw0100A; Beijing, China).
Goat anti-rabbit immunoglobulin G-horseradish peroxidase (IgG-HRP)
and goat anti-mouse IgG-HRP (Pierce, Thermo Fisher Scientific,
Rockford, IL, USA) were used as secondary antibodies, and enhanced
chemiluminescence reagent (Pierce, Thermo Fisher Scientific) was
used for developing the blots. The agents epirubicin, doxycycline,
3-methyladenine (3-MA; Sigma-Aldrich, St. Louis, MO, USA) and
purimycin (Invitrogen Life Technologies, Carlsbad, CA, USA) were
dissolved in phosphate-buffered saline (PBS) to form stock
solutions and then added directly into the media to the required
concentration.
Inducible lentiviral shRNA
constructs
To generate shRNA-expressing plasmids, the
double-stranded oligonucleotides encoding the desired shRNA were
cloned into the AgeI and EcoRI restriction sites of
pLKO-Tet-On vector (Addgene, Inc., Cambridge, MA, USA) as described
previously (17). The constructs
containing an LC3-specific shRNA sequence
5′-CTGAGATCGATCAGTTCAT-3′; and scrambled shRNA
5′-GCAAGCTGACCCTGAAGTTCAT-3′ were designated pLKO-Tet-shLC3 and
pLKO-Tet-shCon, respectively.
Virus production and production of stable
cell lines
Lentiviruses were generated by co-transfecting 293T
cells with 1.5 μg shRNA-encoding plasmid and 1 μg pPAX2 and 0.5 μg
pDMG2 (Addgene, Inc.) as helper plasmids, using Lipofectamine 3000
reagent (Invitrogen Life Technologies) according to the
manufacturer’s instructions. Growth media was exchanged after 8–16
h and lentivirus-containing supernatant was harvested 24, 48 and 72
h later.
For target cell transduction, HepG2 cells
were passaged to 40% confluency the following day. Viral medium was
added to the cells with 8 μg/ml Polybrene (Sigma-Aldrich). After 24
h, viral particle-containing medium was removed and replaced with
fresh medium containing 1 μg/ml puromycin. From days 4 to 10, fresh
medium was replaced when necessary and evaluated for cytotoxicity
under a microscope. Finally, the cells, named
HepG2shCon and
HepG2shLC3, were collected for further
experiments.
Immunoblot analysis
Cells were trypsinized, washed with ice-cold PBS and
lysed with RIPA buffer (50 mM Tris, 150 mM NaCl, 1% NP-40, 0.5%
deoxycholic acid and 0.1% SDS), Protein protease inhibitor mixture
(Thermo Fisher Scientific) was added prior to extraction. The
lysates were fractionated on an SDS-PAGE gel and transferred onto a
Trans-Blot polyvinylidene difluoride membrane (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Blots were blocked with 5%
non-fat dry milk followed by probing with the LC3B (1:1,000
dilution, CST) and p62 (1:1,000 dilution, Proteintech) primary
antibodies and GAPDH (1:10,000 dilution, CoWin) overnight at 4°C.
The membranes were then washed thrice with tyrosylprotein
sulfotransferase, incubated with secondary antibodies at room
temperature for 2 h and subsequently washed a further three times
for developing. The corresponding bands were detected using the
Pierce Western HRP protocol.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RNA was isolated with TRIzol reagent (Invitrogen
Life Technologies), treated with DNAse (Promega Corporation,
Madison, WI, USA), and then first strand cDNA was created with
M-MLV reverse transcriptase (Promega Corporation) according to the
manufacturer’s instructions. qPCR was performed in triplicate in
20-μl reactions with iQ SYBR® Premix Ex Taq™ Perfect
Real Time (Bio-Rad Laboratories, Inc.), 50 ng first strand cDNA and
0.2 μg each LC3 primer: Forward, 5′-GAGAAGCAGCTTCCTGTTCTGG-3′ and
reverse, 5′-GTGTCCGTTCACCAACAGGAAG-3′; or 0.2 g each GAPDH primer:
Forward, 5′-GGGTGTGAACCATGAGAAGT-3′ and reverse,
5′-GTAGAGGCAGGGATGATGTT-3′. Samples were cycled once at 95°C for 2
min, then subjected to 40 cycles of 95°C, 58°C and 72°C for 30 sec
each. The relative LC3 mRNA content was calculated using the
2−ΔΔCT method with GAPDH as an endogenous control.
Detection of AVOs with acridine orange
(AO) staining
Autophagy is characterized by the formation of AVOs.
Cells were stained with AO (Sigma-Aldrich) as described previously
(18). Briefly, in AO-stained
cells, the cytoplasm and nucleolus fluoresce bright green and dim
red, respectively, whereas AVOs fluoresce bright red.
HepG2shCon and
HepG2shLC3 cells were cultured with or
without doxycycline (10 μg/ml) for 24 h, then cultured with
serum-free DMEM for 2 h. AO was then added at a final concentration
of 1 μg/ml for a period of 15 min. The cells were washed with PBS 3
times, and the AVOs were visualized using a fluorescence microscope
(Leica DMR, Leica Microsystems GmbH, Wetzlar, Germany) or
quantified by flow cytometry (FACScan system; BD Biosciences, San
Jose, CA, USA.
Determination of the mean red:green
fluorescence ratio in AO-stained cells using flow cytometry
The intensity of the red fluorescence is
proportional to the degree of acidity and/or the volume of the
cellular acidic compartment. Thus, by comparing the mean red:green
fluorescence ratio of different cell populations, the change in the
degree of acidity and/or the fractional volume of their cellular
acidic compartment was measured. Cells were stained with AO for 15
min, removed from the plate with trypsin-EDTA and collected in
phenol red-free growth medium. Green (510–530 nm) and red (>650
nm) fluorescence emissions from 1×104 cells, illuminated
with blue (488 nm) excitation light, were measured using a FACScan
system and CellQuest software (BD Biosciences).
Cytotoxicity assay
Chemotherapy drug cytotoxicity was assessed in
vitro using the Cell Counting kit (CCK)-8 assay (7Sea Biotech,
Shanghai, China) as described previously (19). In brief, 2×103 cells
were seeded in a 96-well flat-bottomed plate, grown at 37°C for 24
h, and then placed in doxycycline (10 μg/ml). Subsequently, cells
were treated with epirubicin at increasing concentrations. After 72
h of culture, 10 μl CCK-8 reagent was added to each well and the
plate was incubated for 1 h. Absorbance was read at 450 nm. All
samples were carried out in sextuplicate. Data are represented as
the percentage reduction in metabolic activity, normalized to
HepG2shCon cells.
Cell cycle analysis
Following treatments, cells were trypsinized, washed
with PBS and fixed in ice cold 75% ethanol. Cells were then washed
with PBS and stained with propidium iodide (Invitrogen Life
Technologies) and analyzed by flow cytometry on a FACScan system.
Cell cycle distribution was analyzed using CellQuest software.
Statistical analysis
Statistical analysis was performed using GraphPad
software (GraphPad Software, Inc., La Jolla, CA, USA). All
quantitative data are expressed as the mean ± standard deviation.
Statistical differences between groups were compared using a
Student’s t-test. P<0.05 was considered to indicate a
statistically significant result.
Results
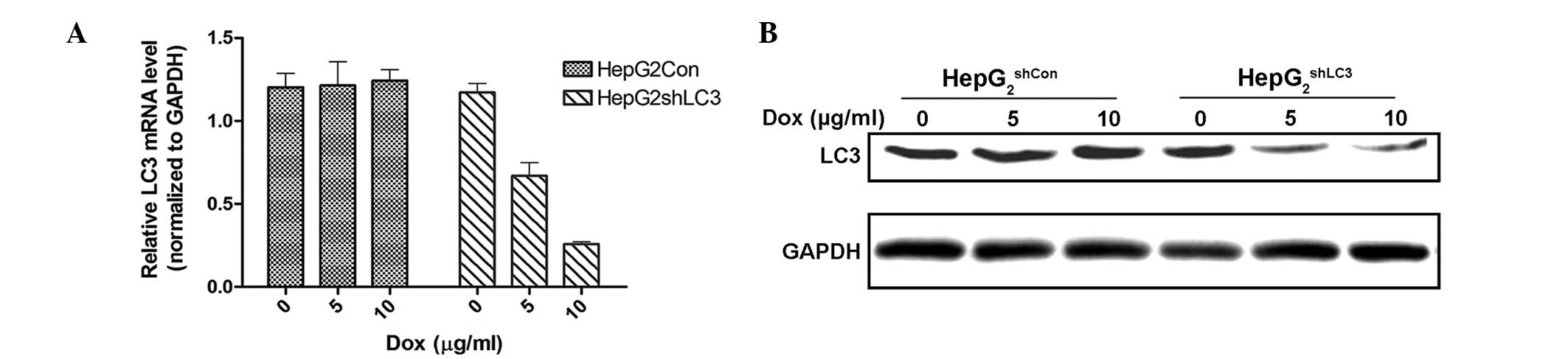
Inducible downregulation of LC3
expression in HepG2 cells
To assess the knockdown of LC3 expression,
HepG2shCon and
HepG2shLC3 cells were cultured in the
presence of increasing amounts of doxycycline (0, 5 and 10 μg/ml)
for 72 h and analyzed by immunoblotting and RT-qPCR. In the absence
of doxycycline, LC3 protein levels did not differ between the two
types of cells. Increasing concentrations of doxycycline, however,
resulted in the effective downregulation of LC3 expression in the
HepG2shLC3 cells (Fig. 1).
Knockdown of LC3 inhibits serum
deprivation-induced autophagy in HepG2 cells
As LC3 has been shown to control the initiation of
autophagy, the effect of LC3 inhibition on serum starvation-induced
autophagy in HepG2 cells was assessed. AO staining was
used to reveal AVOs following serum starvation treatment by
fluorescence microscopy and FACS scanning. The data (Fig. 2A and B) demonstrate that the
inhibition of LC3 resulted in a marked inhibition of the total
number of autophagosomes; the 5 mM 3-MA group served as a positive
control. The p62 protein, also known as SQSTM1, is itself degraded
by autophagy and may serve to link ubiquitinated proteins to the
autophagic machinery to enable their degradation in the lysosome.
Therefore, the induction of autophagy after 2 h serum-starvation
was assessed using immunoblotting to detect the expression level of
p62, with GAPDH as the loading control (Fig. 2C). The degradation of p62 was
repressed in the Dox (10 ug/ml) and 3-MA groups. These results
indicate that the knockdown of LC3 by inducible shRNA may inhibit
starvation-induced autophagy.
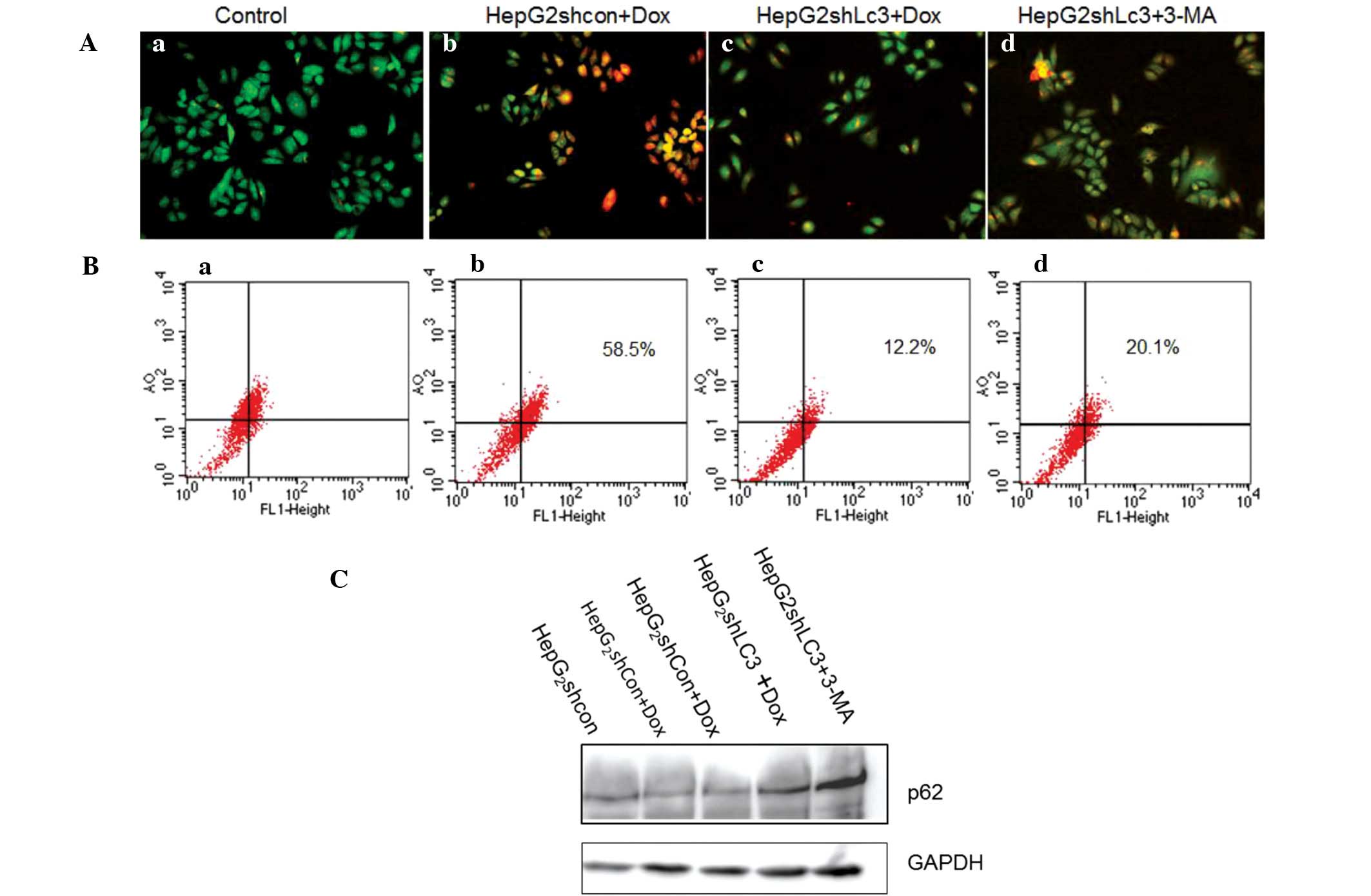 | Figure 2Detection of serum starvation-induced
AVOs by staining with acridine orange. Cells were exposed to the
supravital stain acridine orange 2 h after treatment with serum
starvation. (A) Fluorescent images of AVOs (magnification, ×20):
(a) untreated cells; (b) and (c) HepG2shCon
and HepGshLC3 cell pre-cultured with 10 μg/ml Dox, then
treated with serum-free medium for 2 h. (d)
HepG2shLC3 cells incubated with 5 mM 3-MA for
12 h before serum deprivation (positive control). (B) Determination
of the mean red:green fluorescence ratio in acridine orange-stained
cells by flow cytometry. The mean red:green fluorescence ratio was
determined as described in Materials and methods. (a) Untreated
cells; (b) and (c) HepG2shCon and
HepGshLC3 cell pre-cultured with 10 μg/ml Dox, then
treated with serum-free medium for 2 h. (d)
HepG2shLC3 cells incubated with 5 mM 3-MA for
12 h before serum deprivation (positive control). (C) Detection of
p62 expression. Lane 1, untreated cells; lanes 2–4,
HepG2shCon and HepGshLC3 cells
pre-cultured with 10 μg/ml Dox, then treated with serum-free medium
for 2 h; lane 5, HepG2shLC3 cells incubated
with 5 mM 3-MA for 12 h before serum deprivation (positive
control). AVOs, acidic vesicular organelles; sh, small hairpin;
LC3, microtubule-associated protein 1 light chain 3; Con, control;
Dox, doxycyline; 3-MA, 3-methyladenine. |
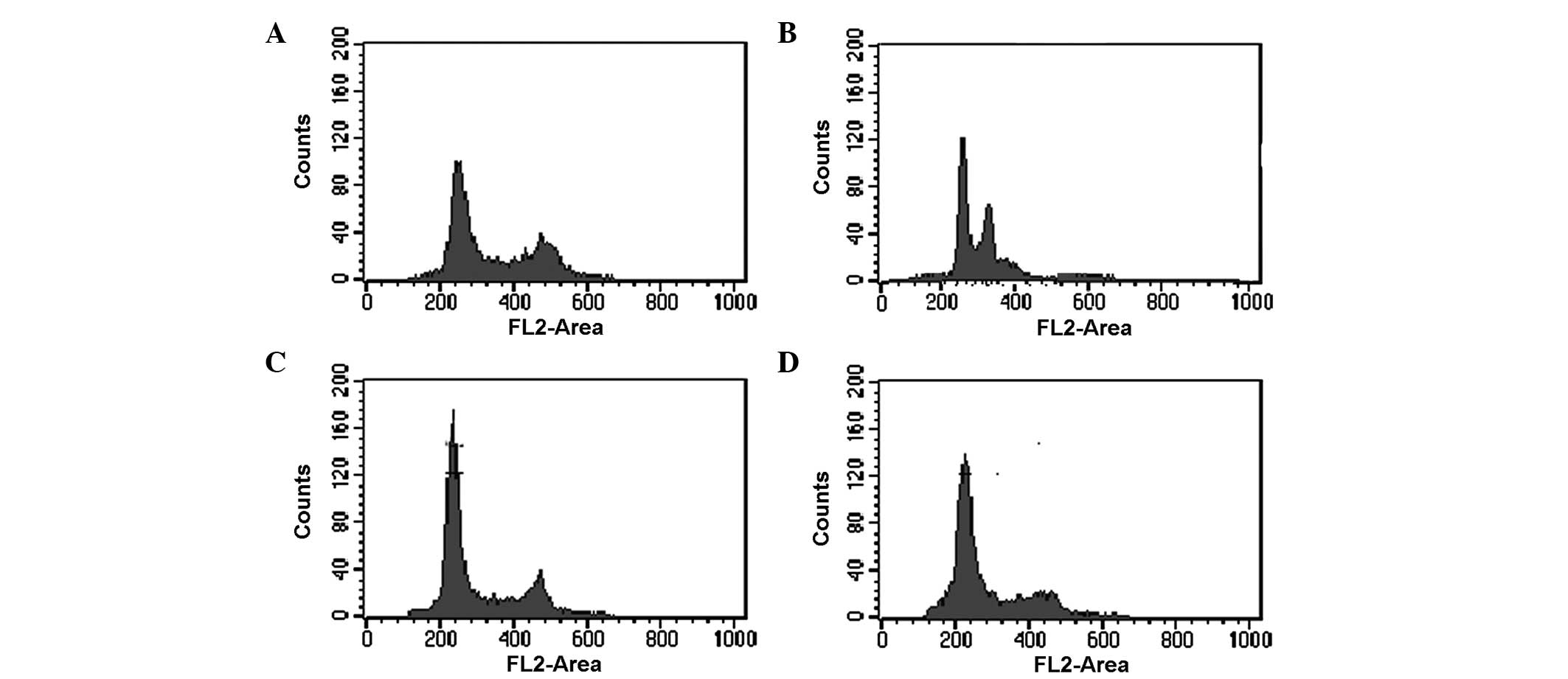
Knockdown of LC3 significantly causes
cell cycle arrest in HepG2 cells
Epirubicin is a widely used anthracycline drug for
chemotherapy that targets DNA by intercalating DNA strands and
inhibits DNA and RNA synthesis. The effect of LC3 inhibition on
epirubicin-induced cell cycle abnormalities in HepG2
cells was investigated using 5 mM 3-MA treated group as the
positive control (Fig. 3). The
results indicated that knockdown of LC3 increased the percentage of
G1 (2N DNA content) phase cells (Fig.
3C) compared with the shCon group (Fig. 3B) following epirubin treatment. The
3-MA group exhibited a similar result (Fig. 3D) to the shLC3 group.
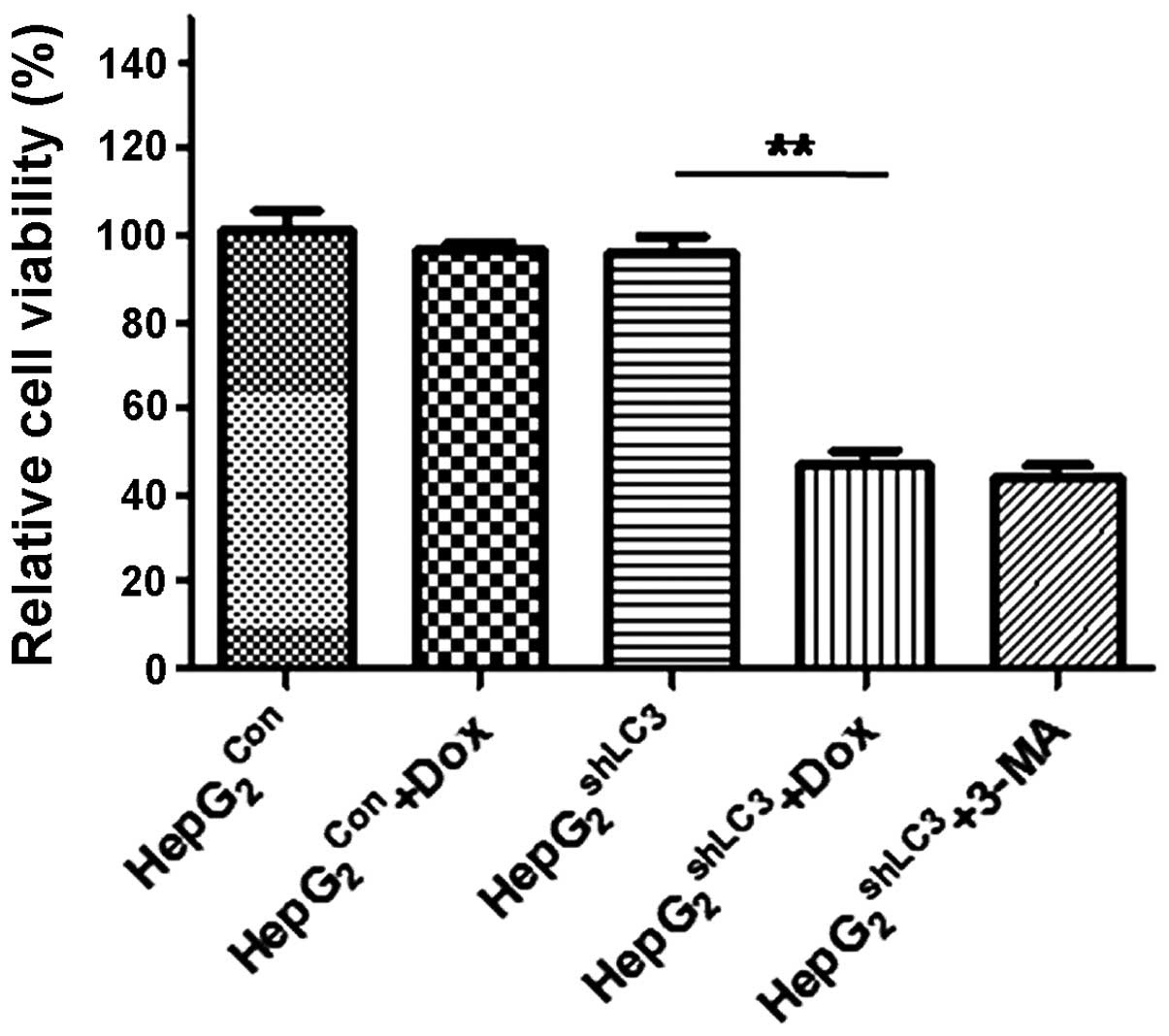
Silencing of LC3 expression sensitizes
HepG2 cells to the therapeutic effect of epirubicin
As autophagy normally promotes the resistance of
tumor cells to chemotherapy, whether knockdown of LC3 increases the
sensitivity of tumor cells to chemotherapy was investigated.
HepG2shCon and
HepG2shLC3 cells were treated with
epirubicin. The effects of LC3 knockdown with and without
epirubicin on cell viability were assessed by CCK-8 assays. LC3
knockdown (induced by 10 μg/ml doxycycline) combined with
epirubicin (4 μM) treatment decrease the viability of
HepG2shLC3 by ~50% (P<0.01) compared with
those of HepG2shLC3 cells treated with
epirubicin and HepG2shCon (Fig. 4).
Discussion
HCC is one of the most prevalent cancers worldwide,
accounting for 85–90% of all primary liver cancers, which
represents ~4% of all newly diagnosed cancer cases (20). Although cytotoxic chemotherapy has
been used for >30 years, definite evidence that it prolongs
survival has been lacking (21).
Various treatment options are available for patients with HCC
according to the degree of background liver damage, tumor diameter
and other factors associated with disease progression (22). Resistance to chemotherapy drugs
remains a significant barrier for cytotoxic agents, often leading
to chemotherapy failure in patients with HCC. Understanding of the
underlying mechanisms is critical if outcomes are to be improved.
There are numerous putative mechanisms for the process by which
chemoresistance is induced (23–25).
Autophagy was first observed in yeast as a survival
mechanism when nutrients were limited, and exists in all eukaryotic
cells from yeast to mammals (26).
Autophagy is a regulated lysosomal pathway for the degradation and
recycling of long-lived proteins and organelles. Nutrient
starvation is the most commonly used method for inducing autophagy
(27). In autophagy, LC3, a
mammalian homolog of yeast ATG8, is activated and relocalizes to
intracellular vesicles when the lipid bilayer structure sequesters
cytoplasm to form autophagosomes. Firstly, LC3 pro-form is cleaved
to form soluble LC3-I. This is then modified to a membrane-bound
form, known as LC3-II, which is recruited onto the autophagosomes
(28,29). Autophagy occurs in response to
various other forms of stress, including oxygen or growth factor
deprivation and chemotherapeutics (30,31).
Recent evidence suggests that autophagy provides a protective
function in tumor cells in response to metabolic stress. A number
of studies have reported that autophagy is activated in cancer
cells in response to various anticancer therapies (32–34).
However, little is known about the role of autophagy in HCC
chemo-resistance. Epirubicin is a structural analog of doxorubicin
that is commonly used to treat HCC. It is usually better tolerated
compared with doxorubicin in the treatment of HCC (35).
In the present study, epirubicin-activated autophagy
was shown to be inhibited by LC3 RNAi. The observed chemoresistance
was found to be associated with the epirubicin-induced activation
of autophagy-associated signaling in HepG2 cells. The
results show that autophagy can be significantly inhibited by
lentivirus-mediated RNAi of LC3 resulting in enhanced cell
sensitivity to chemotherapy. These findings suggest that the
knockdown of LC3 offers a novel approach that increases the
sensitivity of tumor cells to chemotherapeutic agents that target
DNA. In addition, they indicate that epirubicin induced autophagy
as a pro-survival mechanism in HCC, which caused the HCC cells to
be resistant to anthracyclines.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (no. 31100964 and 81372718) and the
Specialized Research Fund for Senior Personnel Program of Jiangsu
University (no. 10JDG45).
References
|
1
|
Ding J and Wang H: Multiple interactive
factors in hepatocarcinogenesis. Cancer Lett. 346:17–23. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li J, Lau GK, Chen L, Dong SS, Lan HY, et
al: Interleukin 17A promotes hepatocellular carcinoma metastasis
via NF-κB induced matrix metalloproteinases 2 and 9 expression.
PloS One. 6:e218162011. View Article : Google Scholar
|
|
3
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yoshimori T: Autophagy: a regulated bulk
degradation process inside cells. Biochem Biophys Res Commun.
313:453–458. 2004. View Article : Google Scholar
|
|
5
|
Tanida I, Ueno T and Kominami E: LC3
conjugation system in mammalian autophagy. Int J Biochem Cell Biol.
36:2503–2518. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Okamoto K, Kondo-Okamoto N and Ohsumi Y:
Mitochondria-anchored receptor Atg32 mediates degradation of
mitochondria via selective autophagy. Dev Cell. 17:87–97. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Reggiori F and Klionsky DJ: Autophagy in
the eukaryotic cell. Eukaryotic Cell. 1:11–21. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Turcotte S and Giaccia AJ: Targeting
cancer cells through autophagy for anticancer therapy. Curr Opin
Cell Biol. 22:246–251. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hollomon MG, Gordon N, Santiago-O’Farrill
JM and Kleinerman ES: Knockdown of autophagy-related protein 5,
ATG5, decreases oxidative stress and has an opposing effect on
camptothecin-induced cytotoxicity in osteosarcoma cells. BMC
Cancer. 13:5002013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Paglin S, Hollister T, Delohery T, Hackett
N, McMahill M, et al: A novel response of cancer cells to radiation
involves autophagy and formation of acidic vesicles. Cancer Res.
61:439–444. 2001.PubMed/NCBI
|
|
11
|
Feng Z: p53 regulation of the
IGF-1/AKT/mTOR pathways and the endosomal compartment. Cold Spring
Harb Perspect Biol. 2:a0010572010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Notte A, Leclere L and Michiels C:
Autophagy as a mediator of chemotherapy-induced cell death in
cancer. Biochem Pharmacol. 82:427–434. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huang G, Jiang QH, Cai C, Qu M and Shen W:
SCD1 negatively regulates autophagy-induced cell death in human
hepatocellular carcinoma through inactivation of the AMPK signaling
pathway. Cancer Lett. Dec 17–2014.(Epub ahead of print).
|
|
14
|
Tian Y, Kuo C, Sir D, et al: Autophagy
inhibits oxidative stress and tumor suppressors to exert its dual
effect on hepatocarcinogenesis. Cell Death Differ. Dec
19–2014.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Pan H, Wang Z, Jiang L, et al: Autophagy
inhibition sensitizes hepatocellular carcinoma to the multikinase
inhibitor linifanib. Sci Rep. 4:66832014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shi YH, Ding ZB, Zhou J, et al: Targeting
autophagy enhances sorafenib lethality for hepatocellular carcinoma
via ER stress-related apoptosis. Autophagy. 7:1159–1172. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wiederschain D, Wee S, Chen L, Loo A, Yang
GZ, et al: Single-vector inducible lentiviral RNAi system for
oncology target validation. Cell Cycle. 8:498–504. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Paglin S, Hollister T, Delohery T, et al:
A novel response of cancer cells to radiation involves autophagy
and formation of acidic vesicles. Cancer Res. 61:439–444.
2001.PubMed/NCBI
|
|
19
|
Yamamoto Y, Mochida J, Sakai D, et al:
Upregulation of the viability of nucleus pulposus cells by bone
marrow-derived stromal cells: significance of direct cell-to-cell
contact in coculture system. Spine (Phila Pa 1976). 29:1508–1514.
2004. View Article : Google Scholar
|
|
20
|
Jemal A, Bray F, Center MM, et al: Global
cancer statistics. CA Cancer J Clin. 61:69–90. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen L, Ye HL, Zhang G, Yao WM, Chen XZ,
et al: Autophagy inhibition contributes to the synergistic
interaction between EGCG and doxorubicin to kill the hepatoma Hep3B
cells. PloS One. 9:e857712014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Alberti A, Clumeck N, Collins S, et al;
ECC Jury. Short statement of the first European Consensus
Conference on the treatment of chronic hepatitis B and C in HIV
co-infected patients. J Hepatol. 42:615–624. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen J, Ding Z, Peng Y, et al: HIF-1α
inhibition reverses multidrug resistance in colon cancer cells via
downregulation of MDR1/P-glycoprotein. PLoS One. 9:e988822014.
View Article : Google Scholar
|
|
24
|
Tao W, Shi JF, Zhang Q, Xue B, Sun YJ and
Li CJ: Egr-1 enhances drug resistance of breast cancer by
modulating MDR1 expression in a GGPPS-independent manner. Biomed
Pharmacother. 67:197–202. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhou Y, Sun K, Ma Y, et al: Autophagy
inhibits chemotherapy-induced apoptosis through downregulating Bad
and Bim in hepatocellular carcinoma cells. Sci Rep.
4:53822014.PubMed/NCBI
|
|
26
|
Meijer AJ and Codogno P: Regulation and
role of autophagy in mammalian cells. Int J Biochem Cell Biol.
36:2445–2462. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li L, Liu GD, Zhang XJ and Li YB:
Autophagy, a novel target for chemotherapeutic intervention of
thyroid cancer. Cancer Chemother Pharmacol. 73:439–449. 2014.
View Article : Google Scholar
|
|
28
|
Tanida I, Minematsu-Ikeguchi N, Ueno T and
Kominami E: Lysosomal turnover, but not a cellular level, of
endogenous LC3 is a marker for autophagy. Autophagy. 1:84–91. 2005.
View Article : Google Scholar
|
|
29
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, et al: LC3, a mammalian homologue of yeast Apg8p, is
localized in autophagosome membranes after processing. EMBO J.
19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mizushima N and Levine B: Autophagy in
mammalian development and differentiation. Nat Cell Biol.
12:823–830. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
White E, Karp C, Strohecker AM, et al:
Role of autophagy in suppression of inflammation and cancer. Curr
Opin Cell Biol. 22:212–217. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sui XB, Kong N, Ye L, Han W, Zhou J, et
al: p38 and JNK MAPK pathways control the balance of apoptosis and
autophagy in response to chemotherapeutic agents. Cancer Lett.
344:174–179. 2014. View Article : Google Scholar
|
|
33
|
Ge YY, Shi Q, Zheng ZY, et al:
MicroRNA-100 promotes the autophagy of hepatocellular carcinoma
cells by inhibiting the expression of mTOR and IGF-1R. Oncotarget.
5:6218–6228. 2014.PubMed/NCBI
|
|
34
|
Yuan H, Li AJ, Ma SL, et al: Inhibition of
autophagy significantly enhances combination therapy with sorafenib
and HDAC inhibitors for human hepatoma cells. World J
Gastroenterol. 20:4953–4962. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cao H, Phan H and Yang LX: Improved
chemotherapy for hepatocellular carcinoma. Anticancer Res.
32:1379–1386. 2012.PubMed/NCBI
|