Introduction
Brugada syndrome (BrS) is a rare, inheritable
arrhythmia syndrome, which is characterized by a coved-type
ST-segment elevation in the right precordial leads (V1 to V3) on
surface electrocardiography (ECG) and a high incidence of sudden
mortality in patients without evident structural heart disease,
electrolyte disturbances or ischemia (1). The condition is considered to be
responsible for 4–12% of all sudden cardiac deaths (SCDs) and 20%
of SCDs in patients with the absence of structural heart disease
secondary to re-entrant polymorphic ventricular tachycardia and
ventricular fibrillation (2–4). The syndrome manifests primarily during
adulthood, with a mean age of SCD of ∼40 years (4). The diagnosis of BrS is based on the
presence of coved-type ST-segment elevations in the right
precordial leads (V1 to V3) on surface electrocardiography (ECG),
which are characteristic of BrS type 1 ECGs, whereas saddle-back
ST-segment elevations correspond to BrS type 2 and 3 ECGs (5). To confirm the diagnosis, the diagnostic
ECGs should be reinforced with either personal symptoms, family
history of premature SCD or at least one additional relative with a
positive type 1 BrS ECG (6).
Since it was first reported by Chen et al in
1998 (7) that loss-of-function
mutations of SCN5A account for the most well-known genetic
substrate for BrS, mutations in 11 other genes that cause BrS have
been reported. These genes include the cardiac L-type calcium
channel subunits encoded by CACNA1C (8), CACNB2B (8) and CACNA2D1 (9), sodium channel subunits encoded by
GPD1L (9), SCN1B
(10), SCN1Bb (11), SCN3B (12) and MOG1 (13), transient outward potassium channel
subunits encoded by KCNE3 (14) and KCND3 (15), the ATP-sensitive potassium channel
encoded by KCNJ8 (16) and
the HCN4 channel encoded by HCN4 (17). However, SCN5A remains the most
frequently reported gene causing BrS to date, accounting for 15–30%
of the clinically diagnosed cases (18–20).
In the present study, a novel heterozygous mutation,
A1428S, was identified in the SCN5A gene in a patient with
BrS. The aim of the study was to characterize the biophysical
properties of this novel mutation, in order to expand the spectrum
of mutations causing BrS and provide evidence for the hypothesis
that the loss of function of the mutant Na+ channel
(Nav1.5) was involved in the pathogenesis of BrS.
Materials and methods
Patients
The study participants were identified and enrolled
at Huazhong University of Science and Technology Union Hospital
(Wuhan, China). Informed consent was obtained from the participants
in accordance with the study protocols approved by the Ethics
Committee of Huazhong University of Science and Technology.
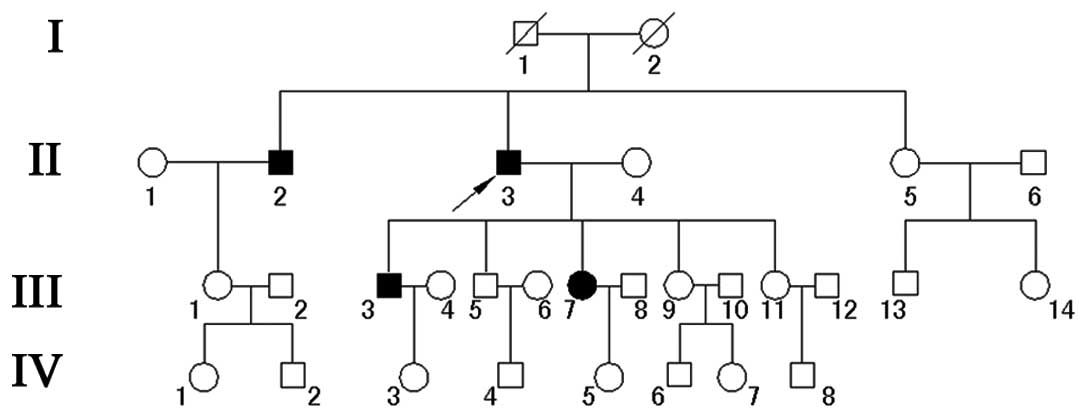
Twenty-eight family members, including 14 males and 14 females were
involved in this study (Fig. 1).
Detailed records on their medical history, physical examinations
and 12-lead ECGs were obtained. The diagnosis of BrS was made on
the basis of symptoms, physical signs and a typical ECG. Among all
the family members, four members exhibited the clinical criteria of
a BrS phenotype.
Direct DNA sequencing analyses
As described previously (21,22),
venous blood (5 ml) was collected from the participants, and total
human genomic DNA was purified with the DNA Isolation kit for
Mammalian Blood (Roche Diagnostics, Indianapolis, IN, USA).
Considering that SCN5A thus far remains the most frequently
reported gene causing BrS, mutation screening of the SCN5A
gene was carried out directly without performing linkage analysis.
The entire coding exons of the SCN5A gene of the proband
were amplified by polymerase chain reaction (PCR). Primers were
designed with intronic flanking sequences according to the gene
sequence described by Wang et al (23). Each amplicon was designed for an
optimal size, with the exon centered within the amplicon. As a
result, a total of 28 amplicons were used to sequence the coding
region of the gene. Briefly, the PCR amplification was performed in
the PTC-200 thermal cycler (MJ Research Inc., Waterdown, MA, USA)
in a 25-µl reaction mixture containing 1.5 mM MgCl2, 0.2
mM of each deoxyribonucleotide triphosphate (Qiagen, Hilden,
Germany), 0.5 µM primers, 1 unit Taq DNA polymerase (Qiagen), and
50 ng genomic DNA. PCR was performed as follows: Initial
denaturation for 5 min at 94°C, followed by nine cycles of 45 sec
at 94°C, 45 sec at 61.5°C and 45 sec at 72°C, followed by 29 cycles
of 45 sec at 94°C, 45 sec at 55°C and 45 sec at 72°C with a
separate annealing temperature at 55°C. Direct bidirectional
resequencing of all PCR-amplified products was performed with the
BigDye Terminator Cycle Sequencing v3.1 kit (Applied Biosystems,
Foster City, CA, USA) and electrophoresed on an ABI PRISM 3730
Genetic Analyzer (Applied Biosystems). Sequencing results from the
subjects and SCN5A gene consensus sequences from GenBank
(GenBank accession no. NM_198056; http://www.ncbi.nlm.nih.gov/genbank/) were compared
using Basic Local Alignment Search Tool analysis. Mutation
description was followed by the nomenclature recommended by the
Human Genomic Variation Society (Carlton South, Australia).
Resequencing of the mutated exon 24 of the SCN5A gene was
performed on the other family members and 100 unrelated controls.
The primers used were as follows: Forward, TGGGGTGGCTTGCTTTTCATAA;
reverse, TGGGGTGCTGGACAAAGAAGAA. The evolutionary conservation of
amino acids in Nav1.5 protein was analyzed among the
following species: Humans (Homo sapiens), orangutans
(Pongo abelii), chickens (Gallus gallus), mice
(Mus musculus), rats (Rattus norvegicus) and dogs
(Canis lupus familiaris). ClustalW was used to align these
protein sequences (http://www.ebi.ac.uk/Tools/msa/clustalw2/).
PCR-restriction fragment length
polymorphism (RFLP) analysis
Mutation c.4282G>T (p.A1428S) disrupts an
Nsi1 restriction site, which allowed us to perform PCR-RFLP
analysis to confirm the mutation and test whether the mutation
co-segregated with the disease in the family as described
previously (24). Briefly, PCR
amplification was performed on exon 24 containing the A1428S
mutation of the SCN5A gene, following the aforementioned
method. The 488-bp PCR product was digested with 10 units
Nsi1 (New England Biolabs, Ipswich, MA, USA) at 37°C
overnight. The resulting digestion products were separated on 1.5%
polyacrylamide gels and the DNA samples were separated by
electrophoresis overnight at 150 V. The DNA bands were visualized
by silver staining.
Cell cultures
Human embryonic kidney 293 (HEK293T) cells were
purchased from the American Type Culture Collection (Manassas, VA,
USA). These cells were cultured in Dulbecco's modified Eagle's
medium (Sigma-Aldrich, St. Louis, MO, USA) containing 10% fetal
bovine serum (Life Technologies, Paisley, UK), 2 mM L-glutamine,
100 U/ml penicillin G and 10 mg/ml streptomycin (Gibco-BRL,
Burlington, ON, Canada) at 37°C in a humidified atmosphere
containing 5% CO2. Cells were plated on
poly-L-lysine-coated glass cover slips (12 mm) (Carl Zeiss, Inc.,
Oberkochen, Germany) and transiently transfected with wild-type
(WT) SCN5A/pEGFP-N2 or mutation type (MT)
SCN5A/pEGFP-N2 (2–5 µg) using Lipofectamine® 2000
(Invitrogen Life Technologies, Carlsbad, CA, USA) as previously
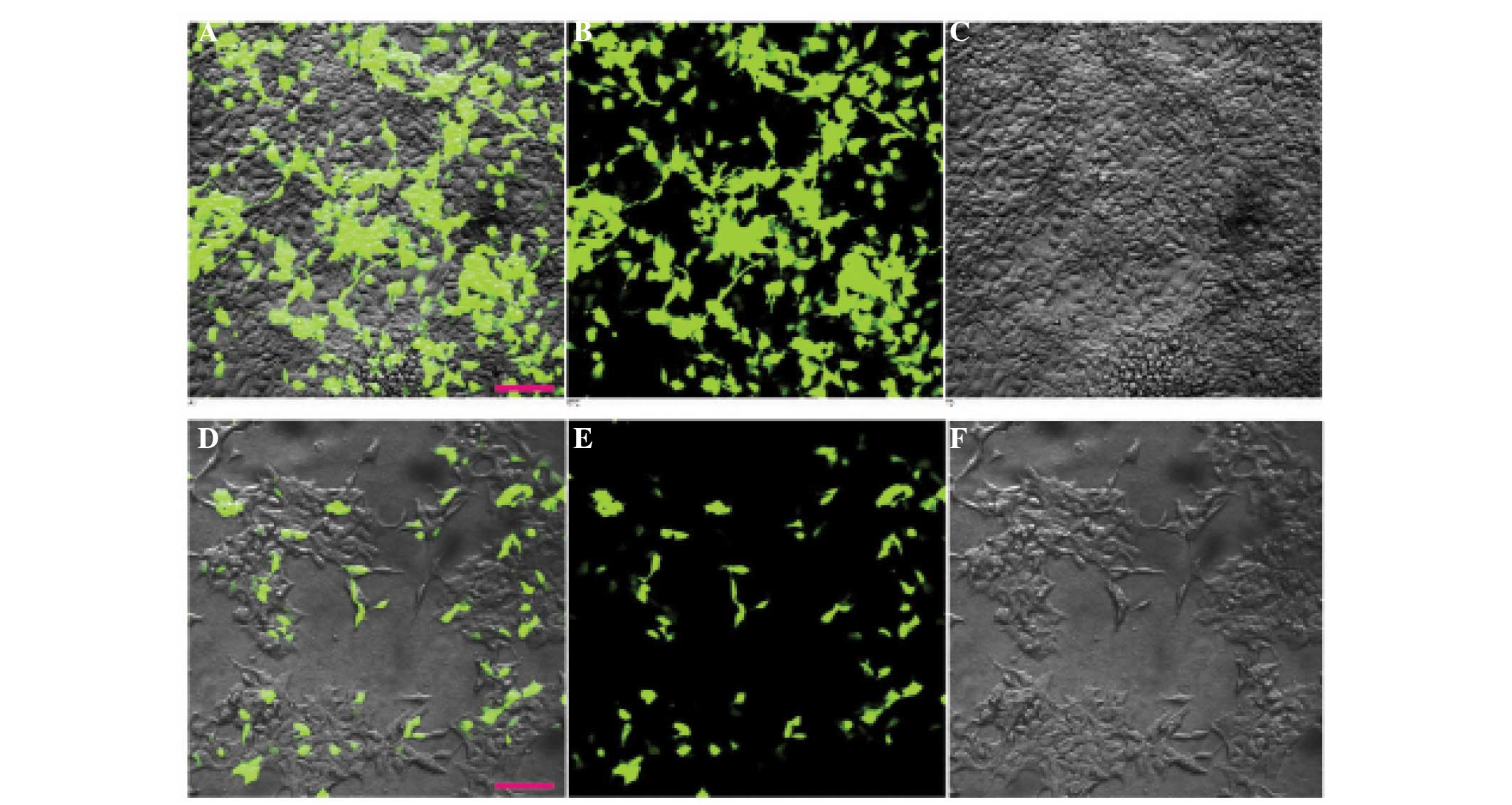
described (20,25). Green fluorescent protein (GFP) was
used as a marker to localize the protein within the cell. Cells
expressing GFP, identified by green fluorescence, were selected for
experiments, as shown in Fig. 2.
Electrophysiological recordings
Macroscopic sodium currents from the transfected
cells were recorded at room temperature (21–24°C) using the
whole-cell patch clamp technique and an Axopatch 200B amplifier
(Axon Instruments, Foster City, CA, USA). The extracellular
solution contained: 70 mM NaCl, 80 mM CsCl, 5.4 mM KCl, 2 mM
CaCl2, 1 mM MgCl2, 10 mM D-glucose and 10 mM
HEPES (adjusted with CsOH to pH 7.3). The patch pipettes were
fabricated from borosilicate glass capillaries (outer diameter, 1.5
mm; inner diameter, 1 mm; VitalSense Scientific Instruments, Wuhan,
China) using a Sutter Model P-97 horizontal puller (Sutter
Instrument Company, Novato, CA, USA). This typically had a
resistance of 1–2 M when filled with the internal solution, which
contained (in mM): 20 mM NaCl, 130 mM CsCl, 10 mM EGTA and 10 mM
HEPES (adjusted to pH 7.2 with CsOH). Currents were amplified and
filtered using an Axopatch 200B amplifier, and digitized with
Digidata 1322A (Molecular Devices, Union City, CA, USA) at 5 kHz
following analog filtering with the four-pole low-pass Bessel (2
kHz) of the amplifier. pCLAMP 9.0 and Origin 8.5 (OriginLab
Corporation, Northampton, MA, USA) software were used for data
acquisition and analysis, respectively. Series resistance
compensation was used to improve the voltage-clamp control
(70–80%). Peak current were normalized by the membrane capacitance
and showed as the current density (pA/pF). Membrane conductance (G)
was defined as I/(V-Erev), where I
was the peak amplitude, V was the test voltage and
Erev was the reversal potential of sodium currents. The
steady-state inactivation curve was studied using a double-pulse
protocol, in which the test voltage was stepped to −10 mV, 200 msec
long, and preceded by 200 msec preconditioning pulses from −140 to
−30 mV in 10-mV steps. The plots of voltage-dependent steady-state
activation and inactivation were fitted by the Boltzmann equation:
1/[1+exp(V-V1/2)/k], where V1/2 was the
voltage at which the sodium current was half-maximally activated,
and k was the slope factor. Recovery from inactivation was examined
by applying a 50-msec conditioning pulse to −20 mV from a holding
potential of −120 mV, followed by a recovery interval of variable
duration (2–10,000 msec) and a test pulse of −20 mV. The recovery
time-course was fitted with the biexponential function:
I/Imax =
Ao+Af[1-exp(-t/τf)]+As[1-exp(-t/τs)],
where τ and A were the time constants and the corresponding
relative amplitudes, respectively.
Statistical analysis
Results are presented as the mean ± standard error
of the mean with sample sizes (n) indicating the number of cells
from which the data were obtained. Statistical significance was
assessed using the Student's t-test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Pedigree and clinical features of the
family
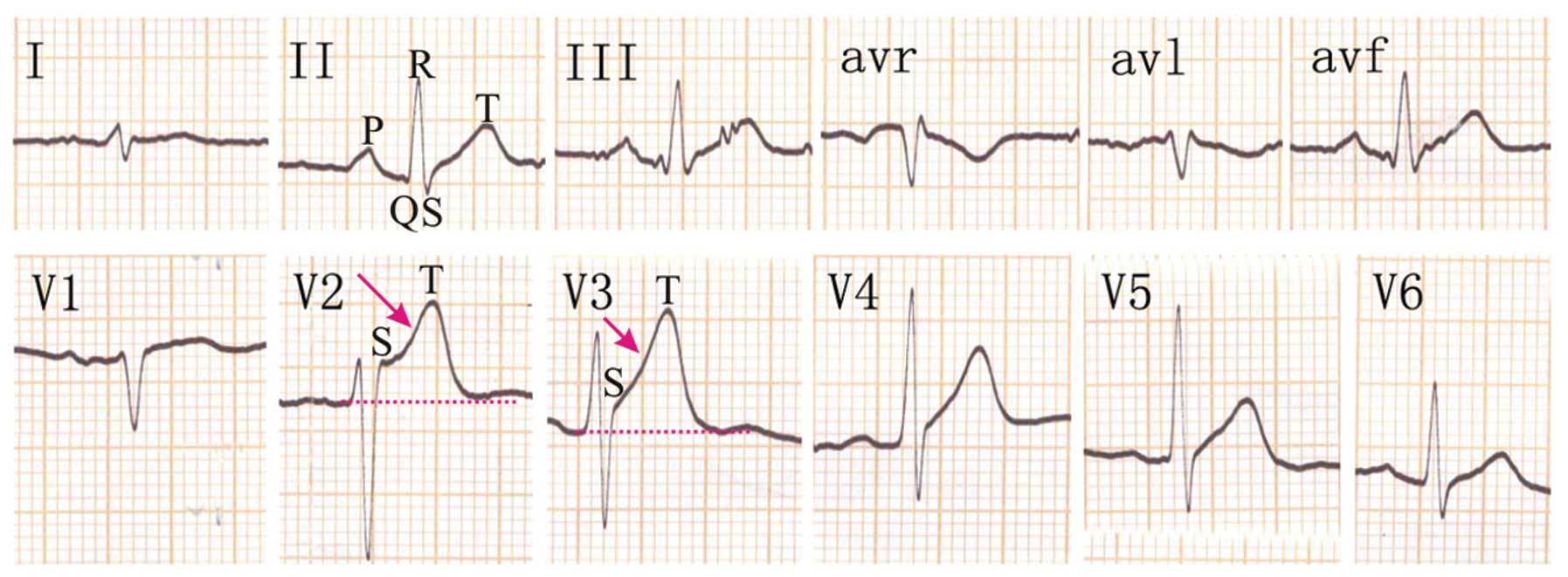
The proband, a 58-year-old patient (II3) was
admitted to the emergency care unit due to syncope during walking
with spontaneous recovery. A 12-lead ECG was performed without a
drug challenge test and showed ST-segment elevation in the right
precordial leads (Fig. 3). In V2 and
V3, the QRS complex showed a typical BrS 1 pattern with a
coved-shape ST-segment. The history of the patient revealed that he
had one episode of syncope in the past, and his familial history
revealed that two children (III3 and III7) and one brother (II2)
had experienced several episodes of dizziness and syncope.
Subsequent to a certain dose of flecainide challenge, all of the
affected individuals showed right bundle branch block and
coved-type ST-segment elevation. However, none of the clinical
features of the proband were identified in the other members of his
family.
Genetic analysis
To identify the molecular basis of BrS in the
proband, exons 1–28 of the dystrophin gene were amplified by PCR.
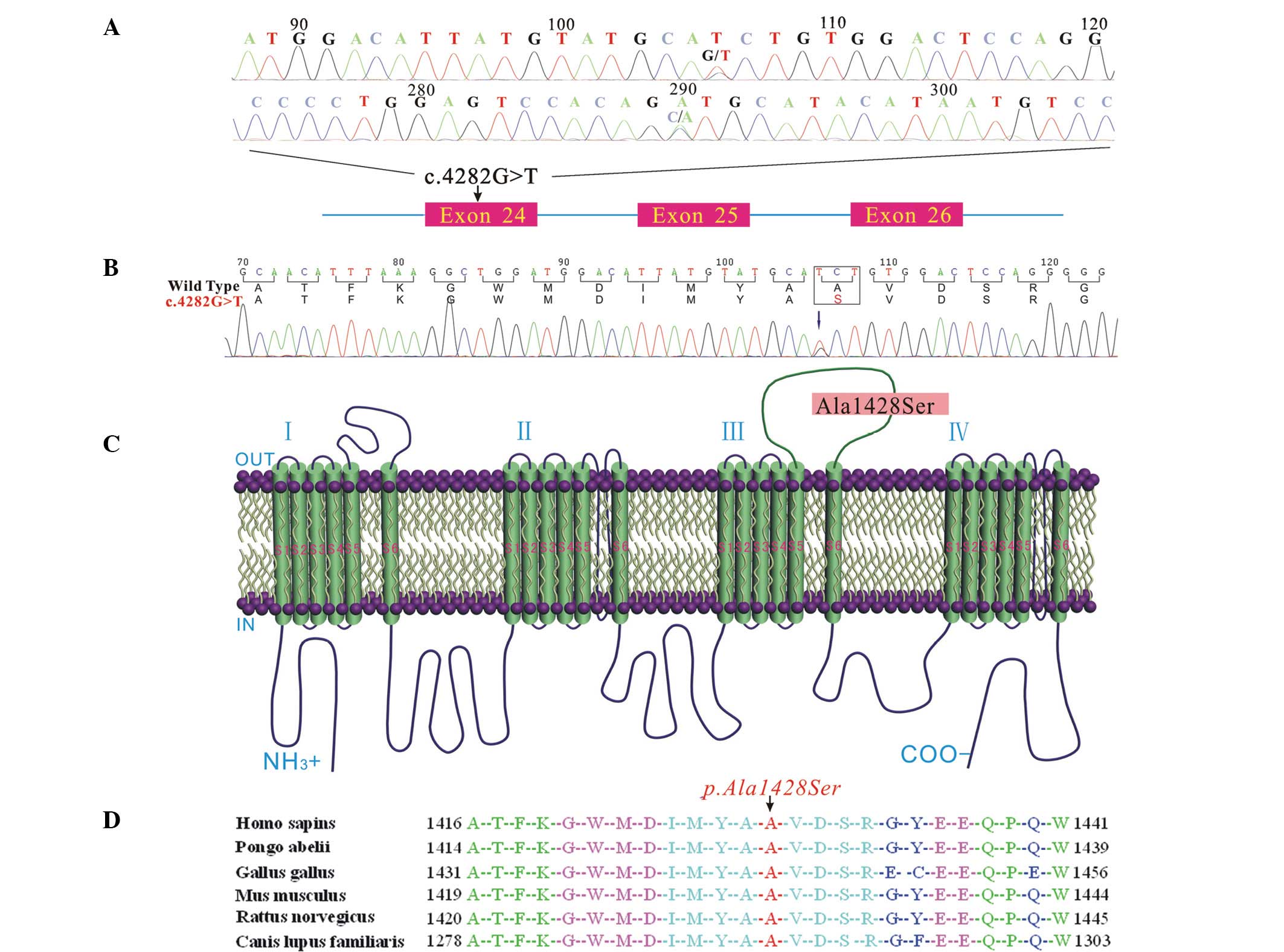
By direct bidirectional sequencing of the PCR products in the
proband, a heterozygous missense single nucleotide at position
4,282 (c.4282G>T) in exon 24 of the SCN5A gene was
revealed (Fig. 4A); this was
confirmed by repeating the experiment. The missense nucleotide
resulted in an amino acid change from alanine to serine at position
1,428 (abbreviated as p.A1428S, Fig.
4B) in the DIII-S5/S6 of the Nav1.5 channel
(Fig. 4C). Ala1428 is highly
conserved among humans (Homo sapiens), orangutans (Pongo
abelii), chickens (Gallus gallus), mice (Mus
musculus), rats (Rattus norvegicus) and dogs (Canis
lupus familiaris) (Fig. 4D).
RFLP analysis
Mutation c.4282G>T (p.A1428S) disrupts an
Nsi1 restriction site. To confirm the mutation and test
whether the mutation co-segregated with the disease in the family,
the novel variation detected in exon 24 of the SCN5A gene
was further evaluated in the other available family members, as
well as in the normal control subjects using RFLP analysis. The PCR
fragment containing mutation Ala1428Ser was able to be cut by the
enzyme; therefore, the RFLP results revealed the presence of the MT
(329 and 159-bp bands) and WT (488-bp bands) alleles (II2, III3,
III7 and proband II3, Fig. 5).
However, the DNA samples from the 100 normal males and the other
unaffected family members were also analyzed by RFLP, and the
results revealed that the unaffected members of the family and the
100 normal controls only had the WT allele (488-bp bands) (Fig. 5). These results further suggest that
this novel mutation (c.4282G>T, p.A1428S) of the SCN5A
gene is not a rare polymorphism, but a causative mutation for BrS
in the proband.
Biophysical properties of the A1428S
mutant
To understand the clinical phenotypes of this
patient, the biophysical properties of the WT and MT channels were
studied. The WT Nav1.5 channels and channels expressed
from the mutated cDNAs of SCN5A were studied in transfected
HEK293 cells. Cells expressing GFP, identified by green
fluorescence (Fig. 2), were selected
for whole-cell patch clamp recordings. The HEK293 cells themselves
did not exhibit a substantial level of INa. By contrast,
cells transfected with the WT SCN5A cDNA had a large
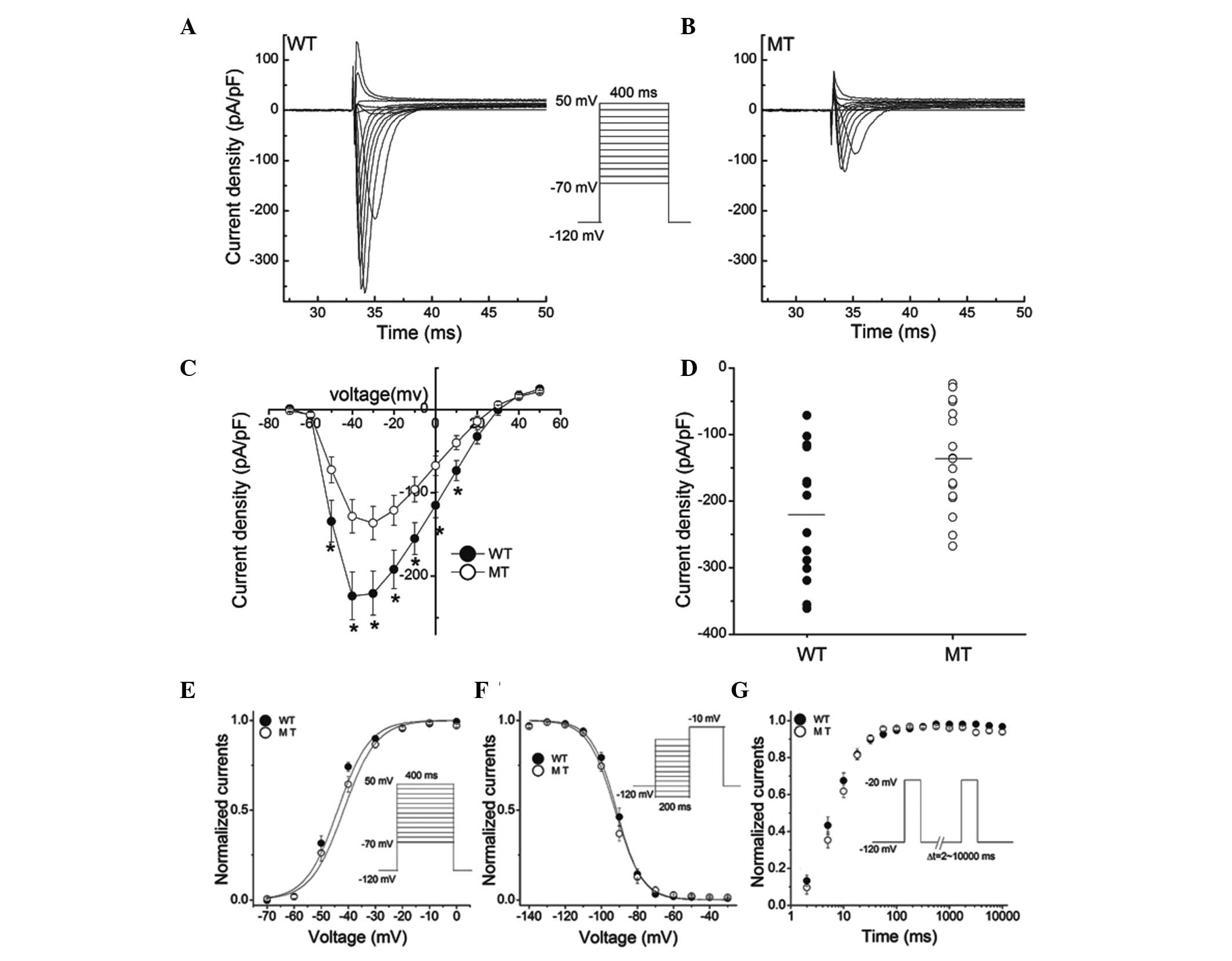
INa (Fig. 6A). A marked
reduction in current amplitudes was observed with the mutation
A1428S (Fig. 6B). Fig. 6C shows the Vm dependence
of the averaged current density. The A1428S mutation did not change
the shape of I-V curve, but significantly reduced the peak current
density without altering the reversal potential and the voltage
dependence of the INa peak. At −30 mV, the mean peak
sodium current density was reduced from −220.82 pA/pF (WT) to
−136.44 pA/pF (A1428S) (Fig. 6D,
P<0.05). The non-significant variation of the reversal
potentials of WT (30.9±1.2 mV, n=10) and A1428S (29.8±2.4 mV, n=10)
indicated that the mutations had no impact on channel selectivity,
as was also evidenced by the I-V curves (Fig. 6C).
The effects of the A1428S mutation on the
Vm dependence of channel activation are shown in
Fig. 6E. The normalized
conductance-Vm association was constructed and it was
revealed that the Vm-dependent activation was the same
in the WT channel (V1/2, −43.86±1.39 mV; k, 5.91±0.76
mV) and the A1428S mutant (V1/2, −41.63±1.36 mV; k,
6.10±0.69 mV). To compare the inactivation kinetics of the WT and
A1428S mutant, whole-cell currents at various potentials were
fitted to a biexponential function. As shown in Fig. 6F, the steady-state inactivation was
not significantly different between the WT channel
(V1/2, −91.50±1.14 mV; k, 7.01±0.48 mV) and the A1428S
mutant (V1/2, −92.33±1.39 mV; k, 7.51±0.73 mV). Recovery
from the inactivation of the WT channel and the A1428S mutant was
then assessed using a double-pulse protocol, as shown in Fig. 6G. The biexponential fit to recovery
curve showed no significant difference between the WT channel
(τf, 5.58±1.36 sec; τs, 40.06±21.32 sec), and
the A1428S mutant (τf, 6.07±1.27 sec; τs,
23.08±6.48 sec) (P>0.05).
Discussion
In this study, a novel heterozygous mutation
c.4282G>T in the SCN5A gene that caused BrS was
presented. It was shown that the mutation caused a significant
reduction in Na+ current, suggesting its involvement in
the pathogenesis of the arrhythmic phenotype seen in the studied
family. Diagnosis of BrS in the proband was based on the ECG, which
showed a coved-type ST-segment elevation in the right precordial
leads, particularly in V2 and V3 (Fig.
3). To identify the disease-causing gene in the proband, all
coding regions (exons 1–28) of the SCN5A gene were
PCR-amplified and sequenced with DNA. A novel missense mutation
(c.4282G>T) in exon 24 of the SCN5A gene was identified,
which resulted in an amino acid change from alanine to serine at
position 1,428 in the DIII-S5/S6 of the Nav1.5 channel
(Fig. 4). This mutation was further
confirmed by PCR-RFLP analysis, which showed that the PCR fragment
containing mutation A1428S could be cut by the Nsi1
restriction enzyme, yielding two shorter DNA fragments of 329 and
159 bp. These fragments were not present in individuals with the WT
allele (Fig. 5). These results
suggested the SCN5A mutation led to the dysfunction of the
Nav1.5 channel, which caused an episode of sudden
collapse during walking.
It is well known that the SCN5A gene encodes
the α subunit of the human cardiac Nav1.5 channel, which
is a transmembrane protein composed of the main pore-forming
α-subunit and two subsidiary β-subunits (β1 and
β2) (26,27). There is considerable evidence that
Nav1.5 forms a section of a macromolecular complex
(28) and that its function is
modulated by cytoskeletal proteins, including tubulin (29), syntrophin and dystrophin (30). Considering these interactions between
Nav1.5 and the cytoskeletal proteins, it is conceivable
that abnormal Nav1.5 proteins affect the function of the
cytoskeleton and the structural integrity of cardiomyocytes. A
report recently showed that patients with BrS with a SCN5A
mutation exhibit enlarged right and left ventricles compared with
individuals without an SCN5A mutation (31). Furthermore, the Nav1.5
channel plays a key role in cardiac excitability and conduction. It
is responsible for the rapid upstroke of the action potential (AP)
caused by the rapid entry of Na+ ions into cardiac
myocytes. In the present study, electrophysiological
characterization of the Nav1.5 mutation, A1428S,
revealed that Na+ current density was significantly
decreased compared with that in the WT, which affected the AP of
cardiomyocytes. Therefore, dysfunctions of this channel can cause
BrS.
The Nav1.5 channel α subunit consists of
four homologous domains, and each domain contains six α-helical
transmembrane repeats. In the present study, the substituted amino
acid p.A1428S was located at the fifth α-helical transmembrane
segment of domain III-S5/S6. Mutations located at this region have
been previously reported, including N1380K (32), although few have been characterized
by functional studies. The present findings indicate that neither
the steady-state activation or inactivation, nor the recovery from
inactivation differs between the A1428S mutation and the WT. These
results revealed that the DIII-S5/S6 of the Nav1.5
channel α subunit was not involved in regulating the recovery from
inactivation, or steady-state activation or inactivation. This
result was consistent with the previous functional studies
reporting that the carboxyl-terminal of the cardiac sodium channel
may play an important role in controlling the gate property of the
channel (33,34). Rivolta et al (35) reported that the mutation Y1795H,
which contributes to BrS, affected steady-state and fast
inactivation, as well as current density. Similar findings were
also observed in the BrS-causing C1859S mutation, as reported by
Petitprez et al (36).
Additionally, change of steady-state activation was observed in the
T1620M and S1710L mutations (37).
These results demonstrated that the carboxyl-terminal of the
cardiac sodium channel plays a critical role in the regulation of
the gating property of the channel. No change in the gating
property of the channel was observed in the A1428S mutation in the
present study.
In conclusion, to the best of our knowledge, this is
the first description of a novel heterozygous missense mutation,
A1428S, in exon 24 of the SCN5A gene in a family with BrS.
This finding expands the mutation spectrum of the SCN5A gene
and may prove useful and valuable for genetic counseling and
prenatal diagnosis in families with BrS. Electrophysiological
characterization of the Nav1.5 mutation, A1428S,
revealed that the fifth α-helical transmembrane segment of
DIII-S5/S6 did not regulate the recovery from inactivation, or
steady-state activation or inactivation. However, the decreased
Nav1.5 activity caused by the A1428S mutation supports
the hypothesis that a reduction in Nav1.5 function is
involved in the pathogenesis of BrS. This structural-functional
study of the Nav1.5 channel enhances the current
understanding of the pathophysiological function of the channel and
provides potential preventive and therapeutic approaches to heart
disease.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (nos. 31301024 and 81400462)
and the Natural Science Foundation of Hubei Province of China (no.
2012FKB02441).
Abbreviations:
|
BrS
|
Brugada syndrome
|
|
ECG
|
electrocardiography
|
|
HEK293T cells
|
human embryonic kidney 293 cells
|
|
MT
|
mutation type
|
|
Nav1.5
|
cardiac Na+ channel
|
|
PCR-RFLP
|
polymerase chain reaction-restriction
fragment length polymorphism
|
|
SCD
|
sudden cardiac death
|
|
WT
|
wild-type
|
References
|
1
|
Moreau A, Keller DI, Huang H, et al:
Mexiletine differentially restores the trafficking defects caused
by two brugada syndrome mutations. Front Pharmacol. 3:622012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brugada P and Brugada J: Right bundle
branch block, persistent ST segment elevation and sudden cardiac
death: a distinct clinical and electrocardiographic syndrome. A
multicenter report. J Am Coll Cardiol. 20:1391–1396. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bhar-Amato J, Nunn L and Lambiase P: A
review of the mechanisms of ventricular arrhythmia in brugada
syndrome. Indian Pacing Electrophysiol J. 10:410–425.
2010.PubMed/NCBI
|
|
4
|
Vohra J: Diagnosis and management of
Brugada Syndrome. Heart Lung Circ. 20:751–756. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wilde AA, Antzelevitch C, Borggrefe M, et
al: Proposed diagnostic criteria for the Brugada syndrome:
consensus report. Circulation. 106:2514–2519. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Antzelevitch C, Brugada P, Borggrefe M, et
al: Brugada syndrome: report of the second consensus conference.
Heart Rhythm. 2:429–440. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chen Q, Kirsch GE, Zhang D, et al: Genetic
basis and molecular mechanism for idiopathic ventricular
fibrillation. Nature. 392:293–296. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Antzelevitch C, Pollevick GD, Cordeiro JM,
et al: Loss-of-function mutations in the cardiac calcium channel
underlie a new clinical entity characterized by ST-segment
elevation, short QT intervals, and sudden cardiac death.
Circulation. 115:442–449. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Burashnikov E, Pfeiffer R,
Barajas-Martinez H, et al: Mutations in the cardiac L-type calcium
channel associated with inherited J-wave syndromes and sudden
cardiac death. Heart Rhythm. 7:1872–1882. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Watanabe H, Koopmann TT, Le Scouarnec S,
et al: Sodium channel β1 subunit mutations associated with Brugada
syndrome and cardiac conduction disease in humans. J Clin Invest.
118:2260–2268. 2008.PubMed/NCBI
|
|
11
|
Hu D, Barajas-Martínez H, Medeiros-Domingo
A, et al: A novel rare variant in SCN1Bb linked to Brugada syndrome
and SIDS by combined modulation of Na(v)1.5 and K(v)4.3 channel
currents. Heart Rhythm. 9:760–769. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hu D, Barajas-Martinez H, Burashnikov E,
et al: A mutation in the beta 3 subunit of the cardiac sodium
channel associated with Brugada ECG phenotype. Circ Cardiovasc
Genet. 2:270–278. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kattygnarath D, Maugenre S, Neyroud N, et
al: MOG1: a new susceptibility gene for Brugada syndrome. Circ
Cardiovasc Genet. 4:261–268. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Delpón E, Cordeiro JM, Núñez L, et al:
Functional effects of KCNE3 mutation and its role in the
development of Brugada syndrome. Circ Arrhythm Electrophysiol.
1:209–218. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Giudicessi JR, Ye D, Tester DJ, et al:
Transient outward current (I(to)) gain-of-function mutations in the
KCND3-encoded Kv4.3 potassium channel and Brugada syndrome. Heart
Rhythm. 8:1024–1032. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Medeiros-Domingo A, Tan BH, Crotti L, et
al: Gain-of-function mutation S422L in the KCNJ8-encoded cardiac
K(ATP) channel Kir6.1 as a pathogenic substrate for J-wave
syndromes. Heart Rhythm. 7:1466–1471. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ueda K, Hirano Y, Higashiuesato Y, et al:
Role of HCN4 channel in preventing ventricular arrhythmia. J Hum
Genet. 54:115–121. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Schulze-Bahr E, Eckardt L, Breithardt G,
et al: Sodium channel gene (SCN5A) mutations in 44 index patients
with Brugada syndrome: different incidences in familial and
sporadic disease. Hum Mutat. 21:651–652. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Priori SG, Napolitano C, Gasparini M, et
al: Clinical and genetic heterogeneity of right bundle branch block
and ST-segment elevation syndrome: A prospective evaluation of 52
families. Circulation. 102:2509–2515. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Priori SG, Napolitano C, Gasparini M, et
al: Natural history of Brugada syndrome: insights for risk
stratification and management. Circulation. 105:1342–1347. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cai F, Zhu J, Chen W, et al: A novel PAX6
mutation in a large Chinese family with aniridia and congenital
cataract. Mol Vis. 16:1141–1145. 2010.PubMed/NCBI
|
|
22
|
Zhu JF, Liu HH, Zhou T and Tian L: Novel
mutation in exon 56 of the dystrophin gene in a child with Duchenne
muscular dystrophy. Int J Mol Med. 32:1166–1170. 2013.PubMed/NCBI
|
|
23
|
Wang Q, Li Z, Shen J and Keating MT:
Genomic organization of the human SCN5A gene encoding the cardiac
sodium channel. Genomics. 34:9–16. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang Q, Liu M, Xu C, et al: Novel CACNA1S
mutation causes autosomal dominant hypokalemic periodic paralysis
in a Chinese family. J Mol Med (Berl). 83:203–208. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tfelt-Hansen J, Winkel BG, Grunnet M and
Jespersen T: Inherited cardiac diseases caused by mutations in the
Nav1.5 sodium channel. J Cardiovasc Electrophysiol. 21:107–115.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Adsit GS, Vaidyanathan R, Galler CM, et
al: Channelopathies from mutations in the cardiac sodium channel
protein complex. J Mol Cell Cardiol. 61:34–43. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Amin AS, Asghari-Roodsari A and Tan HL:
Cardiac sodium channelopathies. Pflugers Arch. 460:223–237. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shy D, Gillet L and Abriel H: Cardiac
sodium channel NaV1.5 distribution in myocytes via interacting
proteins: the multiple pool model. Biochim Biophys Acta.
1833:886–894. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Casini S, Tan HL, Demirayak I, et al:
Tubulin polymerization modifies cardiac sodium channel expression
and gating. Cardiovasc Res. 85:691–700. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gavillet B, Rougier JS, Domenighetti AA,
et al: Cardiac sodium channel Nav1.5 is regulated by a multiprotein
complex composed of syntrophins and dystrophin. Circ Res.
99:407–414. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
van Hoorn F, Campian ME, Spijkerboer A, et
al: SCN5A mutations in Brugada syndrome are associated with
increased cardiac dimensions and reduced contractility. PLoS One.
7:e420372012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Crotti L, Marcou CA, Tester DJ, et al:
Spectrum and prevalence of mutations involving BrS1-through
BrS12-susceptibility genes in a cohort of unrelated patients
referred for Brugada syndrome genetic testing; implications for
genetic testing. J Am Coll Cardiol. 60:1410–1418. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Motoike HK, Liu H, Glaaser IW, et al: The
Na+ channel inactivation gate is a molecular complex: a
novel role of the COOH-terminal domain. J Gen Physiol. 123:155–165.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kass RS: Sodium channel inactivation in
heart: a novel role of the carboxy-terminal domain. J Cardiovasc
Electrophysiol 17 Suppl. 1:S21–S25. 2006. View Article : Google Scholar
|
|
35
|
Rivolta I, Abriel H, Tateyama M, et al:
Inherited Brugada and long QT-3 syndrome mutations of a single
residue of the cardiac sodium channel confer distinct channel and
clinical phenotypes. J Biol Chem. 276:30623–30630. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Petitprez S, Jespersen T, Pruvot E, et al:
Analyses of a novel SCN5A mutation (C1850S): conduction vs.
repolarization disorder hypotheses in the Brugada syndrome.
Cardiovasc Res. 78:494–504. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shirai N, Makita N, Sasaki K, et al: A
mutant cardiac sodium channel with multiple biophysical defects
associated with overlapping clinical features of Brugada syndrome
and cardiac conduction disease. Cardiovasc Res. 53:348–354. 2002.
View Article : Google Scholar : PubMed/NCBI
|