Introduction
An estimated 371 million individuals are afflicted
with diabetes worldwide, and the incidence is increasing rapidly,
particularly in developing countries such as China (1,2).
Diabetes doubles the risk of cardiovascular disease (CVD) (3), and CVDs are the leading cause of
mortality in individuals with diabetes. Among the most common
diabetic macrovascular diseases are myocardial infarction, coronary
artery disease and congestive heart failure-diabetic cardiomyopathy
(DCM). Glucose control alone does not benefit macrovascular
diseases (4) as much as diabetic
microvascular complications, suggesting that the pathophysiological
mechanism underlying diabetic macrovascular complications remains
to be elucidated.
DCM is an occult cardiovascular complication
associated with chronic diabetes and is easily overlooked in
clinical practice (5,6). DCM has been known as an independent
cause of congestive heart failure in the absence of coronary artery
disease and hypertension (7). To
date, the potential etiologies have included microangiopathy,
autonomic neuropathy or disrupted cellular metabolism (8–14). The
underlying pathophysiological mechanism of DCM has been associated
with blocked intracellular insulin signaling (insulin resistance in
cardiomyocytes), metabolic substrate shift, enhanced production of
reactive oxygen species, cellular apoptosis and fibrosis in cardiac
muscle (8–14); however, in the absence of adequate
therapeutic options other than blood glucose control, specific
molecular targets or signaling pathways require further exploration
and definition.
Bcl-2/adenovirus E1B 19 kDa-interacting protein 3
(BNIP3), a pro-apoptotic protein with a Bcl-2 homology 3 (BH3)-only
domain, has been suggested to mediate hypoxia or ischemia in
cardiac muscle (15,16). The pro-apoptotic BH3-only proteins
play important roles in the pathogenesis of heart failure, cancer
and inflammatory diseases (17,18).
Compared with other BH3-only proteins, the effect of BNIP3 does not
require the formation of heterodimers with other BH1–4 multidomain
B-cell lymphoma 2 proteins. Instead, BNIP3 homodimers insert onto
the mitochondrial outer membrane by their transmembrane domains.
The increased insertion of BNIP3 homodimers induces mitochondrial
permeablization and impaired mitochondrial membrane potential,
resulting in damaged mitochondrial function and cellular apoptosis
(19,20). Previous studies (15,21,22) have
suggested that BNIP3 is one of the major proteins mediating
ischemia/reperfusion injury of cardiomyocytes, as well as cardiac
remodeling. The ablation of BNIP3 has been shown to significantly
improve post-infarction cardiomyocyte remodeling and cellular
apoptosis (23). Despite this, it is
yet to be elucidated whether hyperglycemia-induced cellular
apoptosis in DCM is also mediated by BNIP3, and if BNIP3 exerts
other effects on cardiac myocytes in response to hyperglycemia.
In our previous studies, BNIP3 expression was shown
to increase in the cardiac muscle of a high-fat diet diabetic rat
model (24,25), suggesting its role in diabetic
cardiovascular complications. These studies have prompted the
present study, which aims to further investigate the role of BNIP3
in the diabetic heart and its underlying molecular mechanism.
Materials and methods
Animal study and maintenance
Animals were purchased from the Shanghai Laboratory
Animal Center (Shanghai, China). The animal protocol was reviewed
and approved by the Animal Care Committee of Shanghai Jiao Tong
University School of Medicine (Shanghai, China). Eight-week-old
Sprague Dawley rats were treated with low-dose streptozotocin (STZ)
by intraperitoneal injection following a high-fat diet for two
months, as described in our previous studies (24,25).
Plasmid construction and
transfection
Whole-length BNIP3 was amplified from rat heart cDNA
using the following primers: Forward, 5′-CTCGAGTTTGCGGAGCCACC-3′;
and reverse, 5′-GGATCCTCAGAAGGCAGATCCAAG-3′. The amplified product
was then cloned into a pIRES-EGFP vector (BD Biosciences, San Jose,
CA, USA) and confirmed by restriction enzyme analysis and DNA
sequencing. Effectene® (Qiagen, Hilden, Germany) was used as a
transfection reagent in the primary cardiac cell culture, while
SuperFect (Qiagen) was used as a transfection reagent both for
plasmid and small interfering RNA (siRNA) in H9c2 cells (American
Type Tissue Collection, Manassas, VA, USA).
siRNA
The sequence of the BNIP3 siRNA was as follows:
Forward, 5′-TCAGCATGAGAAACACAAGCGT-3′; and reverse,
5′-TCCCCAATCCAATGGCTAACAG-3′. The sequence of the control siRNA was
as follows: Forward, 5′-UUCUCCGAACGUGUCACGUTT-3′; and reverse,
5′-ACGUGACACGUUCGGAGAATT-3′. siRNA was synthesized by GenePharma
Co., Ltd. (Shanghai, China).
Primary cardiac cell culture
Hearts of newborn rats (one-to-two days old) were
dissected and washed in icy D-Hank's solution (0.4 g/l KCl; 60 mg/l
KH2PO4; 0.35 g/l NaHCO3; 80 g/ml
NaCl; 48 mg/l Na2HPO4). The isolated hearts
were minced and digested with 0.125% trypsin mixed with 0.01% EDTA,
and Type II collagenase (Thermo Fisher Scientific, Inc., Waltham,
MA, USA) in a 37°C water bath with minor agitation at 50 rpm.
Digestion was terminated by 20% calf serum-Dulbecco's Modified
Eagle's Medium (DMEM; Thermo Fisher Scientific, Inc.) with 5.6 mM
glucose. The digested cells were washed and then preincubated in 5%
CO2 at 37°C for 60–90 min to remove the fibroblasts.
Following preincubation the supernatant was transferred with the
suspended cells into tissue culture with 0.1 mM bromodeoxyuridine
(Sigma-Aldrich, St. Louis, MO, USA). After 12 h, the media were
changed with 1% bovine serum albumin-DMEM (Gibco-BRL, Carlsbad, CA,
USA)/M199 (Sigma-Aldrich) mixed media at a 4:1 ratio, along with
500 ng/ml transferrin, non-essential amino acids (Thermo Fisher
Scientific, Inc.) and 25 mM glucose for 48 h.
Cell apoptosis
Annexin V labeling was performed based on the
manufacturer's instructions (BD Biosciences). The cells were
serum-starved under different glucose concentrations overnight
prior to labeling. Annexin V staining was then detected and
analyzed using flow cytometry (FACSCalibur™; BD Biosciences).
Microarray and quantitative polymerase
chain reaction (qPCR) analysis
RNA was extracted by using of TRIzol® (Life
Technologies) from the cultured cells. cRNA was synthesized and
blotted with an Affymetrix rat GeneChip® 2.0 array. Reverse
transcription was performed using Superscript III Reverse
Transcriptase (Life Technologies), according to manufacturer's
instructions. Data were then confirmed using qPCR analysis with
SYBR® Green (Takara Bio, Inc., Shiga, Japan) on an ABI 7300
Real-Time PCR system (Applied Biosystems, Foster City, CA, USA).
The data were analyzed for target gene expression using the -ΔΔCT
method.
Mitochondrial staining and fractional
extraction
The mitochondria were stained using MitoTracker® Red
CM (ROX) (Thermo Fisher Scientific, Inc.), according to the
manufacturer's instructions with minor modifications. Images were
captured using an LSM510 confocal microscope (Carl Zeiss AG,
Oberkochen, Germany). Mitochondrial isolation was performed in
accordance with the instructions provided by Biovision, Inc.
(Mountain View, CA, USA). Briefly, cells were homogenized using
Dounce tissue grinders (Sigma-Aldrich) to isolate intact
mitochondria, from which the mitochondrial protein was
extracted.
Western blot analysis
Cellular or mitochondrial protein was loaded on
SDS-PAGE gels, separated electrophoretically and transferred to a
nitrocellulose membrane with an MP3 device from Bio-Rad
Laboratories, Inc. (Hercules, CA, USA). Anti-rabbit BNIP3 antibody
was synthesized in our laboratory at a dilution of 1:8,000. In
brief, the whole-length BNIP3 cDNA was cloned into the pGEX-4T2
plasmid (Promega Corp., Madison, WI, USA). The pGEX-4T2-BNIP3
plasmid was subsequently transformed into the E. coli BL21
strain and induced by 1.0 mmol/l
isopropyl-β-D-thiogalactopyranoside, with agitation, at 18°C
overnight. Following transformation, glutathione S-transferase
(GST) fusion protein was purified using a B-PER GST Purification
kit (Thermo Fisher Scientific, Inc.). Following confirmation using
Tandem mass spectrometry (Thermo Fisher Scientific, Inc.), the
purified protein was used to immunize New Zealand white rabbits
(Shanghai Laboratory Animal Center, Shanghai, China) without the
GST tag.
Statistical analysis
All comparisons were analyzed with SPSS 11.0
software (SPSS Inc., Chicago. IL, USA). An independent sample
t-test was used to calculate the statistical significance between
two groups and one way analysis of variance was used to make
comparisons between multiple groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
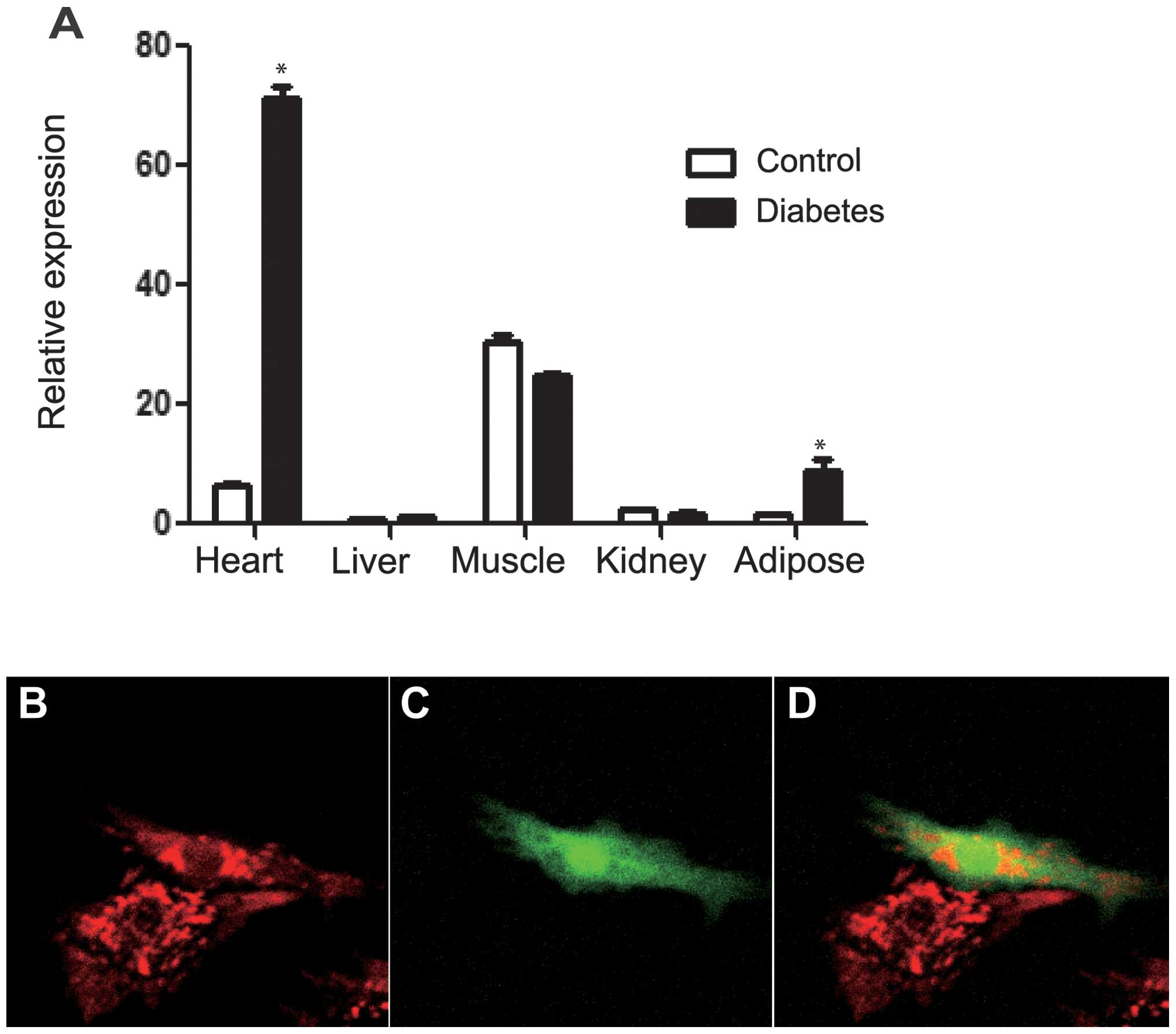
BNIP3 is highly expressed in the heart
and is significantly induced in diabetes
The expression pattern of BNIP3 in different organs
was determined using qPCR at the transcriptional level in
insulin-sensitive tissues, such as the heart, kidney, liver, muscle
and fat. BNIP3 expression was found to be enhanced in the heart,
muscle and adipose tissue compared with that in the liver and
kidney. In the diabetic rat heart, the BNIP3 mRNA level was further
increased to a level 10-fold higher than that in the non-diabetic
control (Fig. 1A). The establishment
of the diabetic rat model is described in our previous studies
(24,25). These results suggest that BNIP3 is
essential for the maintenance of cardiac function in normal and
diabetic conditions.
BNIP3 overexpression induces a loss of
mitochondrial membrane potential
To investigate the effect of BNIP3 on cardiac
muscle, primary cardiac cells were isolated from neonatal rats and
transfected with BNIP3-green fluorescent protein (GFP) plasmid one
day after ex vivo culturing. Transfected positive cells were
shown using GFP fluorescence. Mitochondria were stained red with
MitoTracker Red CM (ROX). Confocal imaging showed that
mitochondrial fusion was increased in BNIP3-GFP positive cells
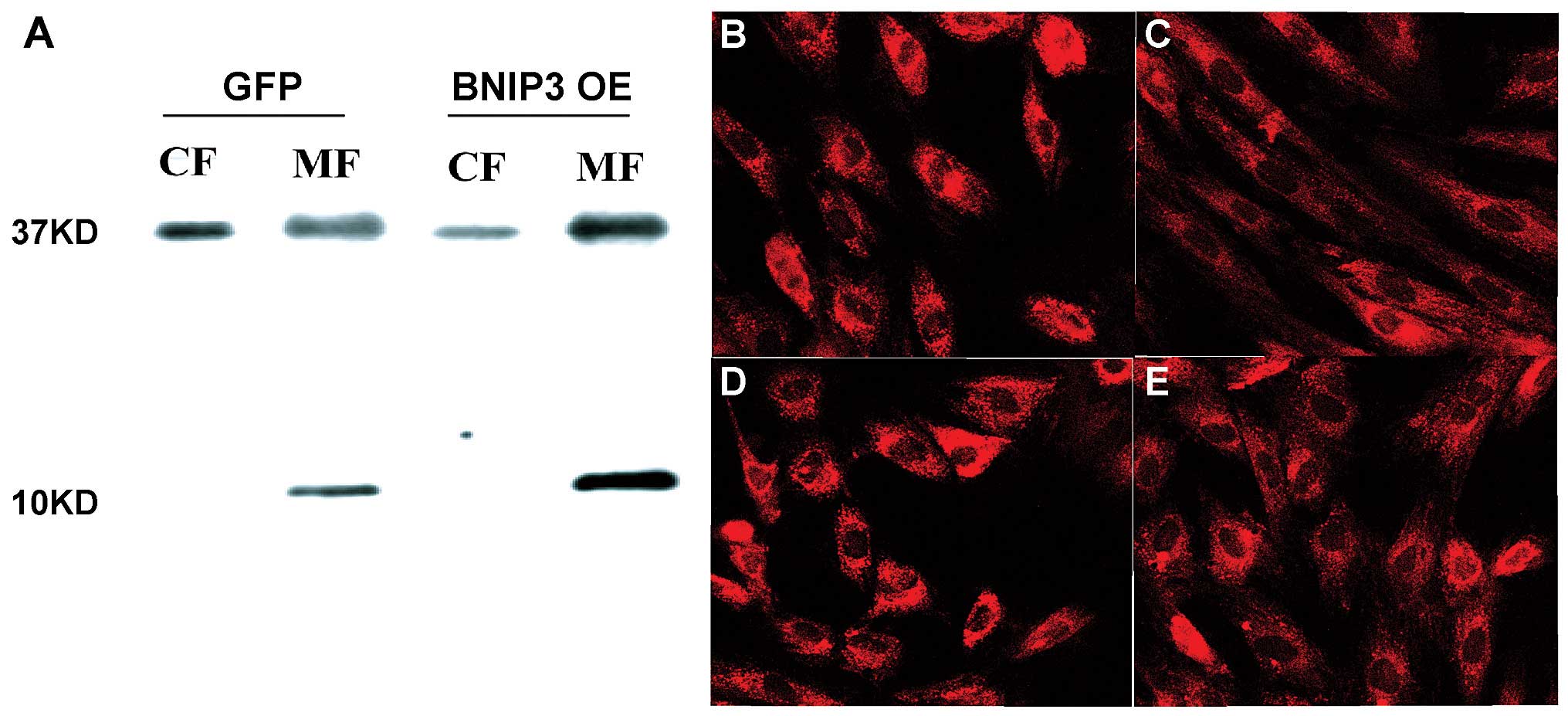
(Fig. 1B–D). Using the BNIP3
antibody produced in our laboratory, it was found that the BNIP3
protein was predominantly localized in the mitochondria instead of
the cytosol (Fig. 2). In the H9c2
cell line, MitoTracker Red CM (ROX) staining showed lower
mitochondrial membrane potential in the cells that overexpressed
BNIP3 compared with the GFP-transfected cells.
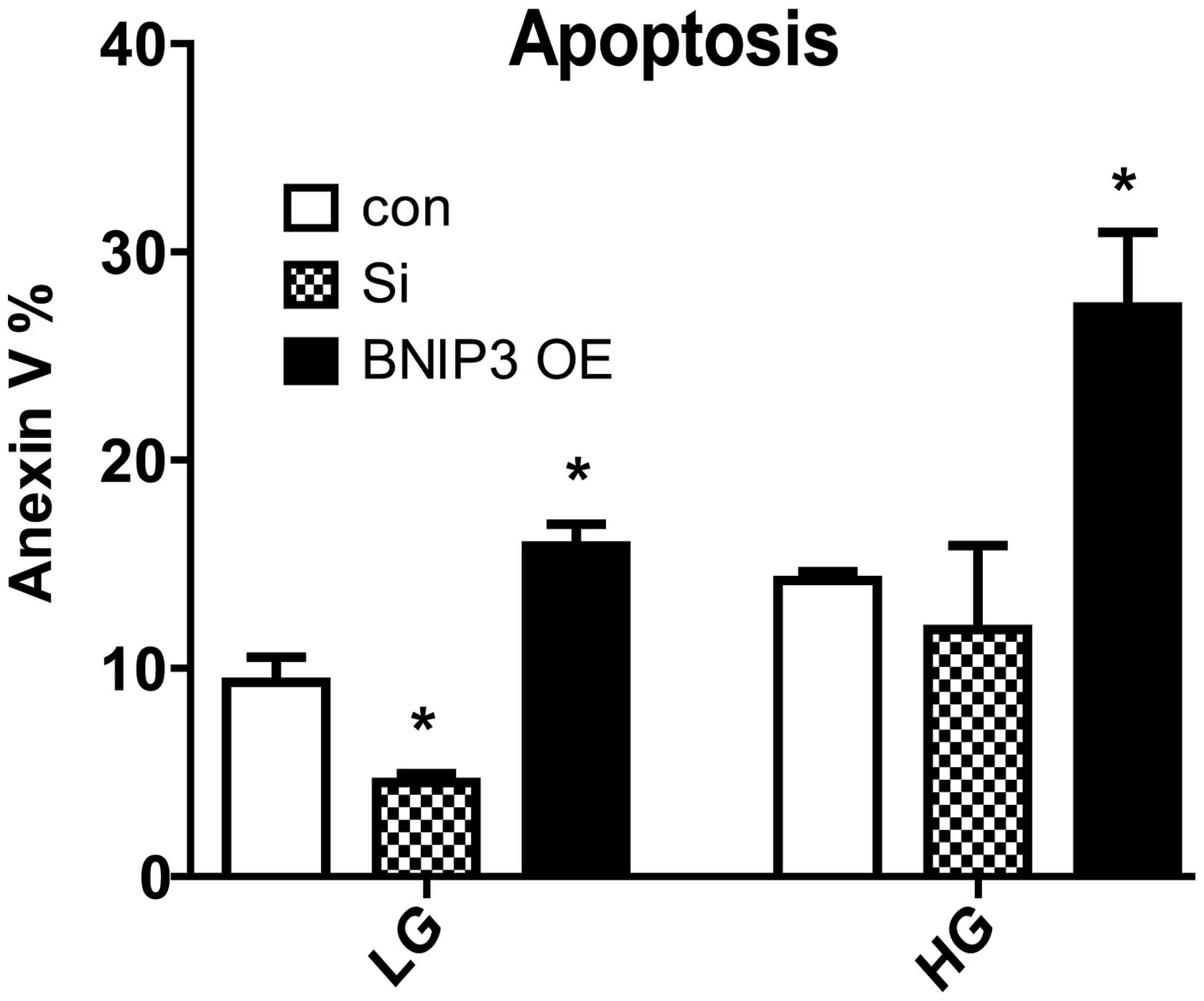
Overexpression of BNIP3 causes
cellular apoptosis and knockdown of BNIP3 fails to significantly
reduce apoptosis in the high-glucose condition
Following the transfection of the H9c2 cells with
BNIP3 plasmids for 72 h, it was found that cellular apoptosis was
significantly induced in the low- (5.6 mM) and high- (25 mM)
glucose conditions. When BNIP3 was knocked down, the number of
Annexin V-positive cells was significantly reduced in the low-, but
not in the high-, glucose culture condition compared with the
control (Fig. 3). BNIP3 is therefore
sufficient but not necessary for apoptosis in the high-glucose
condition.
Comparative transcriptome analysis
reveals new downstream signals of BNIP3 targeting oxidative
phosphorylation, inflammation and heart remodeling
To further explore the downstream targets and
effects of BNIP3, along with its role in the apoptosis of heart
muscle, mRNA microarray analysis was performed to compare the
differential gene expression of GFP and BNIP3-GFP-transfected ex
vivo rat heart cells (Fig. 4A).
Notably, several genes differentially expressed in the two groups
were associated with lipid metabolism, oxidative phosphorylation,
inflammation, fibrosis and apoptosis (Table I). qPCR was employed to confirm the
data from the microarray analysis. The expression of carnitine
palmitoyltransferase 1b (CPT1b), cytochrome c oxidase
subunit VIIIb (COX8b), xanthine dehydrogenase (XDH) and chemokine
(C-X-C motif) ligand 10 was significantly induced by overexpressed
BNIP3, while creatine kinase (brain) (CKB) and matrix
metallopeptidase 9 (MMP9) expression was inhibited. To further
investigate the regulation of these genes by BNIP3, BNIP3 was
knocked down by siRNA in the H9c2 cell line. The genes induced by
BNIP3 were predominantly reduced following treatment with BNIP3
siRNA, with the exception of CPT1b (Fig.
4B). The genes reduced by the overexpression of BNIP3 were all
significantly recovered with BNIP3-knockdown.
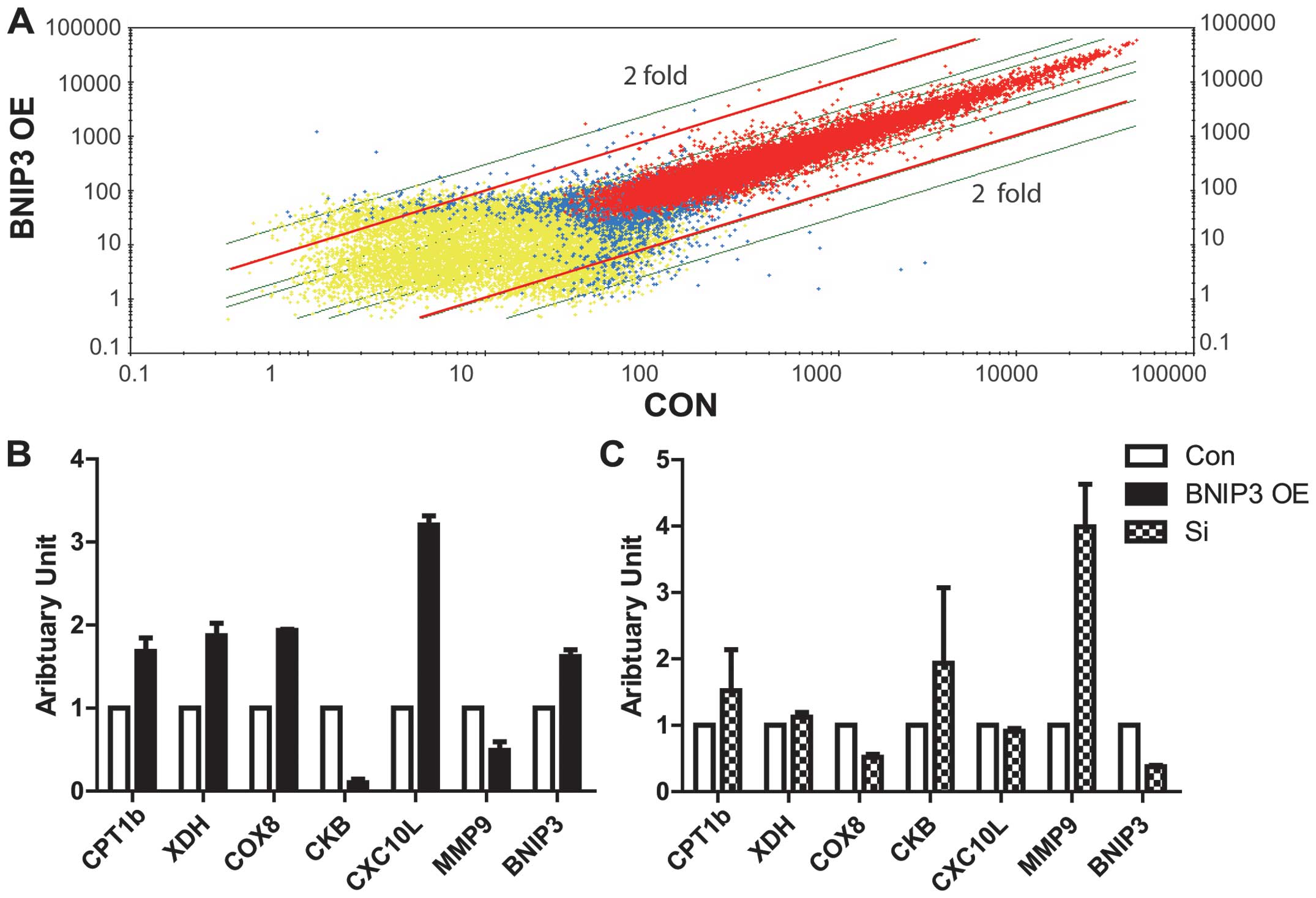 | Figure 4.Differential transcription in cardiac
cells with BNIP3 overexpression. (A) Microarray analysis of H9C2
cells with overexpression of BNIP3 (BNIP3 OE) or GFP (CON). Genes
of interest were selected based on two-fold differences in
expression between the BNIP3 OE and GFP transfected samples. (B)
Confirmation of relative mRNA expression of BNIP3 downstream
targets selected from microarray data with quantitative polymerase
chain reaction data (n=3). (C) Relative expression of BNIP3
downstream target genes in cells transfected with BNIP3 siRNA
(n=3). Con, cells transfected with empty plasmid or non-sense
siRNA; Si, BNIP3 siRNA; BNIP3 OE, cells transfected with
overexpressed BNIP3 plasmid; BNIP3, Bcl-2/adenovirus E1B 19
kDa-interacting protein 3; GFP, green fluorescent protein; siRNA,
small interfering RNA; CPT1b, carnitine palmitoyltransferase 1b;
XDH, xanthine dehydrogenase; COX8, cytochrome c oxidase
subunit VIII; CKB, creatine kinase (brain); CXC10L, chemokine
(C-X-C motif) ligand 10; MMP9, matrix metallopeptidase 9. |
 | Table I.Pathway mining to find downstream
targets of BNIP3 from mRNA microarray. |
Table I.
Pathway mining to find downstream
targets of BNIP3 from mRNA microarray.
| Pathway | Gene | Difference compared
with CON (fold) |
|---|
| Lipid metabolism | Slc2a4 (Glut4) | ↑2.30 |
|
| Acsl1 | ↑2.20 |
|
| Acsl3 | ↓2.00 |
|
| Cd36 | ↑3.00 |
|
| Cpt1b | ↑2.00 |
| Mitochondrial
oxidative phosphorylation | Cox8b | ↑2.50 |
|
| Capn6 | ↑2.50 |
|
| Xdh | ↑2.50 |
|
| CKB | ↓2.30 |
| Cardiac fibrosis | MMP9 | ↓3.03 |
|
| MMP12 | ↓22.67 |
|
| Bdkrb2 | ↓4.00 |
|
| Klk6 | ↓3.20 |
| Calcium
metabolism | P2rx1 | ↓8.00 |
|
| Camk2b | ↓2.82 |
|
| Plcd4 | ↓2.64 |
|
| Cacna1g | ↑2.50 |
| Wnt pathway | Sfrp1 | ↓3.48 |
|
| Wif1 | ↓3.25 |
|
| Axin2 | ↓3.48 |
|
| Ppp2r2b | ↓3.73 |
| Apoptosis and
inflammation factors | Casp12 | ↑2.14 |
|
| Il1a | ↓3.03 |
|
| VCAM1 | ↓2.20 |
|
| Pla2g5 | ↑2.00 |
|
| CXCL10 | ↑4.60 |
| Cell cycle | Cdkn2d | ↓6.96 |
|
| Ccnb2 | ↓2.50 |
|
| Pttg1 | ↓2.00 |
|
|
Espl1_predicted | ↓2.00 |
Discussion
In this study, it was found that BNIP3 mRNA
expression was induced to a level 10-fold higher in diabetes
compared with that in the normal condition. Overexpression and
downregulation of BNIP3 led to changes in the cellular apoptosis.
Microarray data confirmed that, despite the increased expression of
pro-apoptotic genes in diabetes, BNIP3 mediated the effect of
chronic hyperglycemia in cardiac cells via multiple pathways.
Comparable with other studies (26,27),
overexpressed BNIP3 in the present study reduced the mitochondrial
membrane potential, and most overexpressed BNIP3 protein
preferentially bound to the outer membrane. The insertion of BNIP3
homodimers opened the mitochondrial permeability transition pore
and decreased the mitochondrial membrane potential (18,28,29).
Decreased mitochondrial membrane potential indicates mitochondrial
dysfunction and apoptosis. In the present study, overexpression of
BNIP3 was also shown to induce cellular apoptosis in both low- and
high-glucose conditions. The downregulation of BNIP3 using siRNA
interference failed to rescue the apoptosis induced by the high
glucose levels, suggesting that other signaling mechanisms exist in
addition to that involving BNIP3, and that the promotion of
apoptosis may not be a major function of BNIP3 in this
scenario.
Microarray analysis suggested that BNIP3 targeted
genes involved in lipid metabolism and mitochondrial oxidative
phosphorylation, such as CPT-1b, COX8b and XDH. CPT1b is a key
enzyme mediating lipid transport into the mitochondria for fatty
acid oxidation (FAO). COX8H and XDH (30) are crucial regulators of oxidative
stress, which is induced by increased FAO and reduced mitochondrial
efficiency. CKB, inhibited by BNIP3, is a key factor maintaining
the cardiac phosphocreatine level as an energy storage in the
heart. A reduction in the phosphocreatine/adenosine triphosphate
ratio was found to precede the decreasing contractility of cardiac
muscle in diabetic patients (31).
These data suggested that increased BNIP3 could cause mitochondrial
dysfunction in abnormal cardiac muscle energy metabolism, reduce
mitochondrial efficiency, increase oxidative stress and decrease
myocardial energy storage. These changes precede cellular apoptosis
and play an indispensable role in myocardial dysfunction.
The present results also showed that BNIP3 could
negatively regulate the MMP9 expression level. MMP9 denatures
fibronectin and fibrillar collagen (32). Accumulation of these extracellular
proteins promotes intercellular fibrosis and cardiac stiffness;
therefore, elevated BNIP3 expression in high-glucose conditions
induced intercellular fibrosis by inhibiting MMP9, and hence
decreased myocardial compliance.
The observation of increased mitochondrial fusion in
cells overexpressing BNIP3 was unexpected. Mitochondrial fusion has
been shown to be critical for maintaining normal mitochondrial
function in response to metabolic or environmental stress (33). Increased mitochondrial fusion may be
associated with an increased requirement for oxidative
phosphorylation depending on lipid metabolism (33). Furthermore, mitochondrial fission may
contribute to apoptosis induced by hyperglycemia (34). The mechanisms underlying the BNIP3
overexpression-enhanced mitochondrial fusion require further
investigation.
DCM was first recognized in 1955 (35) and defined in 1972 (7) as a disease with heart dilation
dysfunction with or without systolic dysfunction. Histologically,
the ventricular dilation is associated with cardiomyocyte
hypertrophy, as well as intercellular fibrosis. Chronic
hyperglycemia in diabetes alters substrate metabolism to cause
mitochondrial dysfunction, stimulate oxidative stress and calcium
overload, and increase the glycation of interstitial proteins
(8). Notably, it was suggested in
the present study that a number of these pathways, including
altered substrate metabolism, increased oxidative stress and
regulation of interstitial proteins, are potentially regulated by
BNIP3 in response to hyperglycemia. We therefore hypothesized that,
in addition to promoting mitochondria-mediated cellular apoptosis,
BNIP3 could represent an important intracellular signaling
mechanism mediating the effect of hyperglycemia in cardiac muscle
metabolism. BNIP3 may, therefore, provide a potential
pharmaceutical target in DCM. Further studies are required to
confirm the present observations.
Acknowledgements
This study was financially supported by the Science
and Technology Commission of Shanghai Municipality, Experimental
Animal Research Program (no. 11140900500.11140900502), the Pujiang
Program (no. 12PJ1407700) and the National Natural Science
Foundation of China (no. NFSC81200563).
References
|
1
|
Guariguata L: By the numbers: new
estimates from the IDF Diabetes Atlas Update for 2012. Diabetes Res
Clin Pract. 98:524–525. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xu Y, Wang L, He J, et al: 2010 China
Noncommunicable Disease Surveillance Group: Prevalence and control
of diabetes in Chinese adults. JAMA. 310:948–959. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fox CS, Coady S, Sorlie PD, et al: Trends
in cardiovascular complications of diabetes. JAMA. 292:2495–2499.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
No authors listed. Intensive blood-glucose
control with sulphonylureas or insulin compared with conventional
treatment and risk of complications in patients with type 2
diabetes (UKPDS 33). UK Prospective Diabetes Study (UKPDS) Group.
Lancet. 352:837–853. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Poirier P, Bogaty P, Garneau C, Marois L
and Dumesnil JG: Diastolic dysfunction in normotensive men with
well-controlled type 2 diabetes: importance of maneuvers in
echocardiographic screening for preclinical diabetic
cardiomyopathy. Diabetes Care. 24:5–10. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bertoni AG, Tsai A, Kasper EK and Brancati
FL: Diabetes and idiopathic cardiomyopathy: a nationwide
case-control study. Diabetes Care. 26:2791–2795. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Rubler S, Dlugash J, Yuceoglu YZ, Kumral
T, Branwood AW and Grishman A: New type of cardiomyopathy
associated with diabetic glomerulosclerosis. Am J Cardiol.
30:595–602. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fonarow GC and Srikanthan P: Diabetic
cardiomyopathy. Endocrinol Metab Clin North Am. 35:575–599. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Young ME, McNulty P and Taegtmeyer H:
Adaptation and maladaptation of the heart in diabetes: Part II:
potential mechanisms. Circulation. 105:1861–1870. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Boudina S and Abel ED: Diabetic
cardiomyopathy revisited. Circulation. 115:3213–3223. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Crow MT, Mani K, Nam YJ and Kitsis RN: The
mitochondrial death pathway and cardiac myocyte apoptosis. Circ
Res. 95:957–970. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Modrak J: Collagen metabolism in the
myocardium from streptozotocin-diabetic rats. Diabetes. 29:547–550.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shimizu M, Umeda K, Sugihara N, et al:
Collagen remodelling in myocardia of patients with diabetes. J Clin
Pathol. 46:32–36. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Neubauer S: The failing heart - an engine
out of fuel. N Engl J Med. 356:1140–1151. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Crow MT: Hypoxia, BNip3 proteins, and the
mitochondrial death pathway in cardiomyocytes. Circ Res.
91:183–185. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yasuda M, Theodorakis P, Subramanian T and
Chinnadurai G: Adenovirus E1B-19K/BCL-2 interacting protein BNIP3
contains a BH3 domain and a mitochondrial targeting sequence. J
Biol Chem. 273:12415–12421. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lomonosova E and Chinnadurai G: BH3-only
proteins in apoptosis and beyond: an overview. Oncogene. 27:Suppl
1. S2–S19. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sulistijo ES, Jaszewski TM and MacKenzie
KR: Sequence-specific dimerization of the transmembrane domain of
the ‘BH3-only’ protein BNIP3 in membranes and detergent. J Biol
Chem. 278:51950–51956. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bocharov EV, Pustovalova YE, Pavlov KV, et
al: Unique dimeric structure of BNip3 transmembrane domain suggests
membrane permeabilization as a cell death trigger. J Biol Chem.
282:16256–16266. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ray R, Chen G, Vande Velde C, et al: BNIP3
heterodimerizes with Bcl-2/Bcl-X(L) and induces cell death
independent of a Bcl-2 homology 3 (BH3) domain at both
mitochondrial and nonmitochondrial sites. J Biol Chem.
275:1439–1448. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Guo K, Searfoss G, Krolikowski D, et al:
Hypoxia induces the expression of the pro-apoptotic gene BNIP3.
Cell Death Differ. 8:367–376. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Regula KM, Ens K and Kirshenbaum LA:
Inducible expression of BNIP3 provokes mitochondrial defects and
hypoxia-mediated cell death of ventricular myocytes. Circ Res.
91:226–231. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Diwan A, Krenz M, Syed FM, et al:
Inhibition of ischemic cardiomyocyte apoptosis through targeted
ablation of Bnip3 restrains postinfarction remodeling in mice. J
Clin Invest. 117:2825–2833. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang F, Li G, Ding W, et al: Screening
and analysis of early cardiopathology-related gene in type 2
diabetes mellitus. Zhonghua Nei Ke Za Zhi. 41:530–533. 2002.(In
Chinese). PubMed/NCBI
|
|
25
|
Zhang F, Ye C, Li G, et al: The rat model
of type 2 diabetic mellitus and its glycometabolism characters. Exp
Anim. 52:401–407. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Althaus J, Bernaudin M, Petit E, Toutain
J, Touzani O and Rami A: Expression of the gene encoding the
pro-apoptotic BNIP3 protein and stimulation of hypoxia-inducible
factor-1alpha (HIF-1alpha) protein following focal cerebral
ischemia in rats. Neurochem Int. 48:687–695. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Papandreou I, Krishna C, Kaper F, Cai D,
Giaccia AJ and Denko NC: Anoxia is necessary for tumor cell
toxicity caused by a low-oxygen environment. Cancer Res.
65:3171–3178. 2005.PubMed/NCBI
|
|
28
|
Kim JY, Cho JJ, Ha J and Park JH: The
carboxy terminal C-tail of BNip3 is crucial in induction of
mitochondrial permeability transition in isolated mitochondria.
Arch Biochem Biophys. 398:147–152. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Vande Velde C, Cizeau J, Dubik D, et al:
BNIP3 and genetic control of necrosis-like cell death through the
mitochondrial permeability transition pore. Mol Cell Biol.
20:5454–5468. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Minhas KM, Saraiva RM, Schuleri KH, et al:
Xanthine oxidoreductase inhibition causes reverse remodeling in
rats with dilated cardiomyopathy. Circ Res. 98:271–279. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Duncan JG: Mitochondrial dysfunction in
diabetic cardiomyopathy. Biochim Biophys Acta. 1813:1351–1359.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Spinale FG: Myocardial matrix remodeling
and the matrix metalloproteinases: influence on cardiac form and
function. Physiol Rev. 87:1285–1342. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Youle RJ and van der Bliek AM:
Mitochondrial fission, fusion, and stress. Science. 337:1062–1065.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yu T, Sheu SS, Robotham JL and Yoon Y:
Mitochondrial fission mediates high glucose-induced cell death
through elevated production of reactive oxygen species. Cardiovasc
Res. 79:341–351. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ungar I, Gilbert M, Siegel A, Blain JM and
Bing RJ: Studies on myocardial metabolism. IV. Myocardial
metabolism in diabetes. Am J Med. 18:385–396. 1955. View Article : Google Scholar : PubMed/NCBI
|