Introduction
Microscopic polyangiitis (MPA) is a systemic
vasculitis involving small- and medium-sized vessels and is
associated with a focal and segmental necrotizing
glomerulonephritis. MPA may be characterized as a pulmonary-renal
syndrome with rapidly progressive glomerulonephritis and alveolar
hemorrhage; however, the manifestations of the disease vary
depending on the organ systems involved (1). The incidence and diagnosis rates of
vasculitis have recently been rising constantly. The detection rate
of anti-neutrophil cytoplasmic antibodies (ANCAs) in cases of
vasculitis can reach as high as 90% (2). The detection of ANCA in the serum has
therefore become an important basis for the diagnosis of vasculitis
(3). ANCA-negative vasculitis is
relatively rare and is thus prone to misdiagnosis. In this study,
we describe a case of microscopic polyangiitis (MPA) and review the
relevant literature with the aim of enhancing the clinical
understanding of ANCA-negative vasculitis and improving the
diagnosis and treatment of the condition.
Case report
The patient was a 69-year-old male who was admitted
to the Department of Respiratory Medicine of the First Affiliated
Hospital of Anhui Medical University (Hefei, China) due to ‘fever
with dyspnea for >40 days’ on March 7, 2014. The highest
measured body temperature of the patient reached up to 40°C,
usually in the afternoon and at night. He was admitted to another
hospital and given an initial diagnosis of upper respiratory tract
infection, which then became pneumonia. The patient was treated
with multiple broad-spectrum antibiotics, to fight the infection,
and hormones; however, the health of the patient did not improve,
and the dyspnea eventually emerged. The outpatient service sent the
patient to our department due to the supposed pneumonia.
Upon physical examination, mild cyanosis was
observed in the patient's lips. The breath sounds from both lungs
were coarse. Velcro-like rales could be heard from the base of both
lungs. The heart rate was 108 beats/min, and the rhythm was
regular. No edema was observed in either lower limb. Laboratory
examination was conducted subsequent to admission. The results from
the routine blood examination showed a white blood cell count of
8.49×109 cells/l, a neutrophil percentage of 79.44%, a
red blood cell count of 3.84×1012 cells/l and a platelet
count of 3.30×1011 cells/l. Routine examination yielded
the following results: Occult blood in urine (++); qualitative
urine protein (+); erythrocyte sedimentation rate (ESR), 55 mm/h;
C-reactive protein, 167.2 mg/l; ANCA, 13 anti-nuclear antibodies,
negative; normal renal function and tumor markers; and no
pathogenic bacterial growth in the sputum culture. The G and
galactomannan tests were negative. The test for 13 antinuclear
antibodies was also negative.
Based on the test results, a diagnosis of
interstitial lung disease (ILD) and pulmonary infection was
reached. Informed consent was obtained from the patient prior to
treatment. The patient was treated with anti-infection and phlegm
elimination drugs with nutritional support; however, the patient's
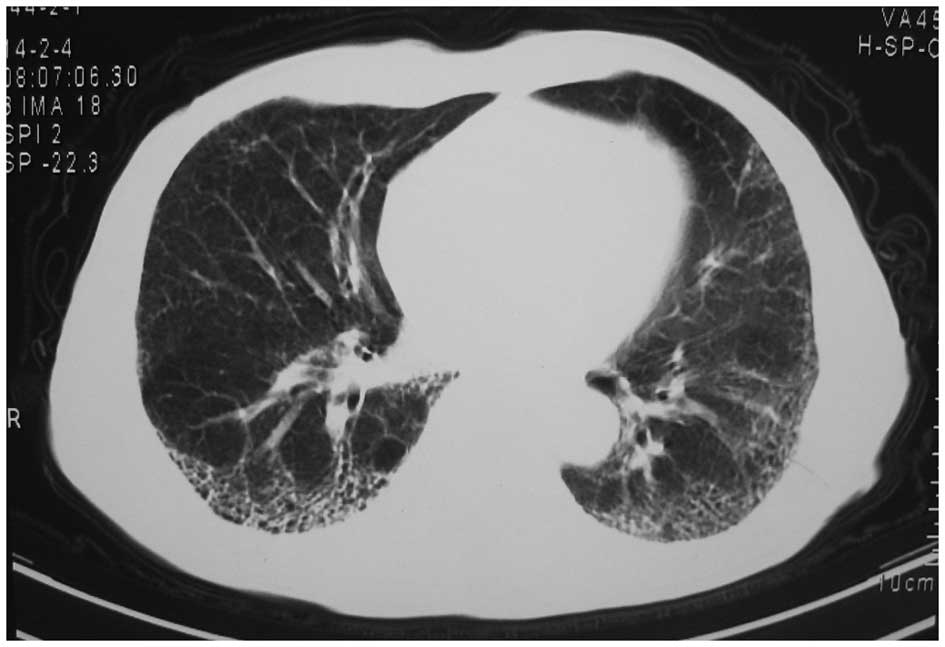
symptoms did not improve. Chest computed tomography (CT) in another
hospital showed interstitial changes in both lungs (Fig. 1). The chest CT was reviewed on March
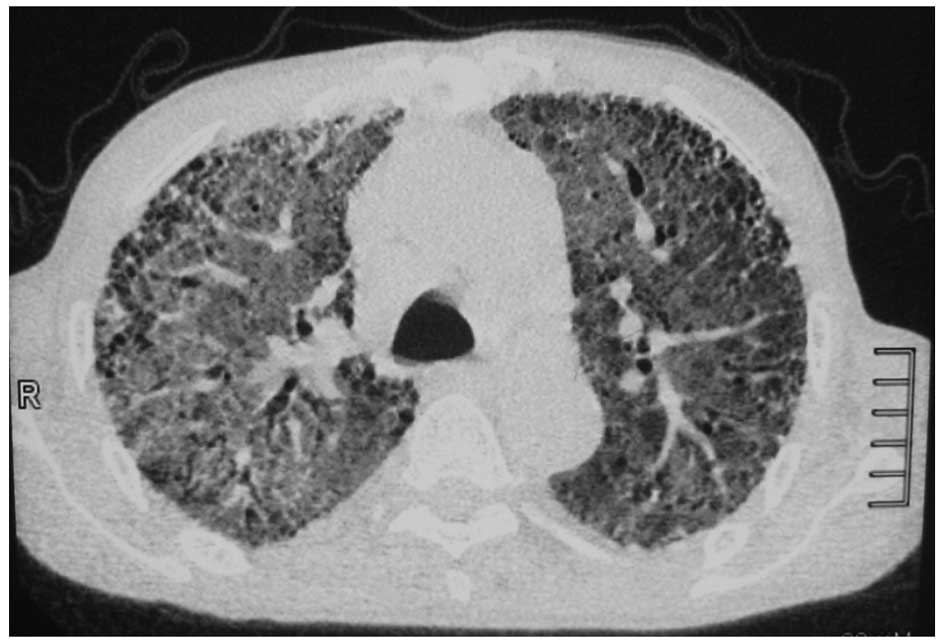
9. The result showed that the transparency in both lungs was
reduced, and patchy, high-density strip shadows were diffusely
distributed and interwoven into a grid shape (Fig. 2). Although the test result for ANCA
was negative, a diagnosis of ANCA-associated vasculitis was
considered. The possibility of MPA was great, but tuberculosis,
fungal infection and lung cancer were excluded.
The patient was treated with methylprednisolone (40
mg every 12 h) and continuous anti-infection drugs; however, the
patient's temperature control was poor and the dyspnea was not
significantly relieved. In addition, the perinuclear type was
reviewed, showing positive results for ANCA (1:80). Positive
results for anti-myeloperoxidase (MPO) antibody were also obtained.
The patient agreed to the addition of cyclophosphamide for
treatment. Through this treatment, the body temperature of the
patient dropped to the normal range, and the dyspnea was relieved.
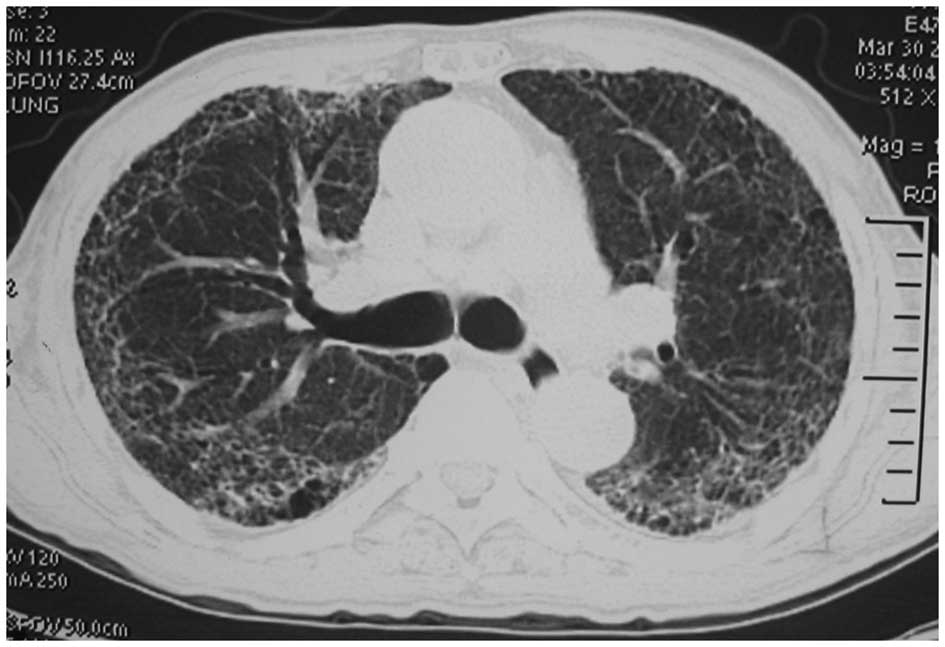
The chest CT on March 30 showed that the diffuse, patchy,
high-density strip shadows in both lungs were significantly
absorbed (Fig. 3). The doses of
administered steroid and cyclophosphamide were gradually reduced.
In the course of treatment, the detected perinuclear types of ANCA
were 1:40 and 1:10, whereas the cytoplasmic ANCA tests were
negative. The routine urine test results, renal function and ESR
were repeatedly reviewed, and all results showed that the patient's
illness was gradually improving. The changes in the condition of
the patient supported the diagnosis of MPA. The patient was again
examined in May 2014, and the tests results for ANCA and ESR were
normal. The patient's general condition was good.
Discussion
Vasculitis refers to a series of heterogeneous
diseases occurring in the vascular wall and its surroundings, which
may present with or without necrosis and can result in different
degrees of stenosis or damage to the vessels and ischemic damage to
the innervated tissues or organs. The majority of vasculitis cases
are primary, often referred to as systemic vasculitis (4). Primary systemic vasculitis affecting
the lungs includes pulmonary capillaritis, ANCA-associated
vasculitis, Wegener's granulomatosis, Churg-Strauss syndrome and
MPA. MPA is a systemic necrotizing vasculitis that affects small
vessels. This condition can invade arterioles, microarteries,
capillaries and venules of the kidneys, skin, lungs and other
organs and is often detected as necrotizing glomerulonephritis and
pulmonary capillaritis. The disease is known as MPA due to it
mainly affecting small vessels, including veins (1). The incidence rate of MPA is (1–3)/100,000
in countries outside of China (5);
in China, the incidence is unclear. With the recent increased
frequency of ANCA examination, however, the number of diagnosed
cases has shown a considerable increase. MPA predominantly occurs
in males, mostly 50–60 years old. The course of MPA varies;
sometimes, the onset is acute, with rapidly progressive
glomerulonephritis and pulmonary hemorrhage (6); sometimes, the onset is insidious with
intermittent purpura, mild renal damage and intermittent
hemoptysis. The disease often involves the kidney, which can cause
necrotizing crescentic glomerulonephritis. Furthermore, MPA can
involve multiple tissues and organs, including the lungs, skin,
joints and digestive tract (6).
According to the literature, lungs infected with
ANCA-associated vasculitis with different etiologies exhibit
different characteristics. MPA mainly manifests as pulmonary
infiltration, diffuse ILD and diffuse alveolar hemorrhage. The
patient in the present report was admitted due to ‘fever and
dyspnea for >40 days’. The patient's renal function was normal,
without cutaneous change, and his chest CT in another hospital
showed interstitial lesions in both lungs. Following the admission
of the patient, he tested negative for ANCA; therefore, the early
diagnosis was unclear. With the progression of the disease, it was
observed that i) the pulmonary lesions progressed rapidly; ii) the
routine urine test results of the patient showed occult blood and
urine protein, which could not be interpreted as being associated
with pulmonary infection; and iii) the symptoms did not improve
significantly following anti-infection treatment. Chest CT showed
that the lesions in both lungs progressed more quickly than
previously. During hospitalization, follow-up tests for ANCA and
consultations with the appropriate departments were conducted. The
final diagnosis was then made.
MPA lacks specific clinical manifestations and is
thus easily misdiagnosed if the pulmonary symptoms are taken as the
main indicators when other clinical symptoms are unspecific.
Pulmonary interstitial lesions can be easily diagnosed but can be
typically mistaken for connective tissue disease or idiopathic
pulmonary fibrosis rather than MPA, thus resulting in the high
incidence rate of early misdiagnosis and poor prognosis.
It has previously been reported that pulmonary
injury in patients with MPA occurs secondary to kidney damage
(7). Chen et al (3) found that, among patients with
ANCA-associated vasculitis, patients aged >65 years exhibited a
significantly higher incidence of pulmonary infection than patients
aged <65 years. The basic pulmonary pathology was pulmonary
capillary inflammation or necrotizing granulomatous vasculitis. A
clinical diagnosis of MPA could be considered in the following
settings: i) Patients showing such symptoms as fever, cough,
expectoration, hemoptysis and dyspnea; interstitial changes in the
lungs shown by chest imaging; detection of fungi and tuberculosis
by sputum culture and smear; exclusion of infectious factors; and
inefficacy of anti-infection and anti-tuberculosis treatments; ii)
involvement of multiple organs from different systems, such as
kidneys, lungs, skin, joints and nerves; and iii) notably increased
ESR. If the patient is suspected to suffer from MPA, then ANCA
examination should be performed to confirm the diagnosis (8).
The diagnosis of MPA lacks a uniform standard. If
multiple system damage, pulmonary and renal infection or palpable
purpura emerge, the diagnosis of MPA should be considered,
particularly for patients testing positive for perinuclear ANCA.
Renal, cutaneous or other visceral biopsies may be of use in the
diagnosis of MPA; however, infective endocarditis has been excluded
for patients. Studies have reported that >80% of patients with
MPA are ANCA-positive (8–11); therefore, a diagnosis of MPA cannot
be excluded for ANCA-negative patients. Renal or other biopsies
should be suggested (12). When the
patient tests negative for ANCA, the result could be a false
negative. A variety of target antigens are aimed at treating ANCA.
These antigens include MPO and proteinase 3; however, ANCA has
other subtypes, which cannot be completely distinguished from serum
ANCA-negative vasculitis (2).
With the progression of the disease in the present
case, the patient tested positive for ANCA. Previous studies have
revealed that the ANCA titer is usually associated with the
activity of the vasculitis and can reflect the curative effect to a
certain extent (13,14). The follow-up examination of the
patient showed that, following treatment with glucocorticoids and
cyclophosphamide, the ANCA titer gradually decreased, which was
consistent with the literature.
Certain scholars believe that a definite diagnosis
for patients with MPA, particularly those who are ANCA-negative,
depends on pathological examination (8). In the present case, respiratory failure
was apparent in the patient during hospitalization, which made
renal and pulmonary biopsies unsuitable. Furthermore, the patient
had no cutaneous lesions, and performing other relatively safe
biopsies was infeasible. The patient fell ill with fever and
expiratory dyspnea. The initial diagnosis was lung infection, and
anti-infection treatment was ineffective; however, a definitive
diagnosis was ultimately made on the basis of medical history,
routine urine and ANCA tests and chest CT. The patient was treated
with glucocorticoids and immunosuppressive therapy, and the
symptoms significantly eased. The ANCA titer and ESR decreased, and
the chest CT improved. The results supported the diagnosis of
MPA.
In summary, MPA is a disease involving multiple
systems. Its clinical manifestation is complicated and changeable,
and the illness rapidly develops. In the present case, the initial
symptoms of the patient pointed to a diagnosis of pulmonary
infection, but pathological examination could not be performed,
which lent difficulty to the clinical diagnosis. By integrating the
clinical symptoms, ANCA detection results and chest CT, a clinical
diagnosis was reached. Combined with a review of the literature, we
conclude that tests for ANCA should be promptly improved and that
changes in the results should be monitored for patients with
suspected MPA. Efforts should be made to improve the early
discovery and diagnosis rates and to achieve timely medication for
MPA to improve the patient prognosis while reducing mortality.
Acknowledgements
This study was supported by a grant from the State
Key Clinical Specialty Construction Project (no. 2012AH001), the
fund for the Academic Backbone of the Excellent Young and
Middle-age People of Anhui Medical University (no. 2013010) and the
fund from the First Affiliated Hospital of Anhui Medical University
for Reserve Talents (no. 2014005).
References
|
1
|
Smyth L, Gaskin G and Pusey CD:
Microscopic polyangiitis. Semin Respir Crit Care Med. 25:523–533.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Collins CE and Quismorio FP Jr: Pulmonary
involvement in microscopic polyangiitis. Current Opin Pulm Med.
11:447–451. 2005. View Article : Google Scholar
|
|
3
|
Chen M, Yu F, Zhang Y and Zhao MH:
Antineutrophil cytoplasmic autoantibody-associated vasculitis in
older patients. Medicine (Baltimore). 87:203–209. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Scott DG and Watts RA: Epidemiology and
clinical features of systemic vasculitis. Clin Exp Nephrol.
17:607–610. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tzelepis GE, Kokosi M, Tzioufas A, Toya
SP, Boki KA, Zormpala A and Moutsopoulos HM: Prevalence and outcome
of pulmonary fibrosis in microscopic polyangiitis. Eur Respir J.
36:116–121. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Villiger PM and Guillevin L: Microscopic
polyangiitis: Clinical presentation. Autoimmun Rev. 9:812–819.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Eschun GM, Mink SN and Sharma S: Pulmonary
interstitial fibrosis as a presenting manifestation in perinuclear
antineutrophilic cytoplasmic antibody microscopic polyangiitis.
Chest. 123:297–301. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kallenberg CG: The diagnosis and
classification of microscopic polyangiitis. J Autoimmunity.
48–49:90–93. 2014. View Article : Google Scholar
|
|
9
|
Wilke L, Prince-Fiocco M and Fiocco GP:
Microscopic polyangiitis: A large single-center series. J Clin
Rheumatol. 20:179–182. 2014.PubMed/NCBI
|
|
10
|
Langford CA: Cyclophosphamide as induction
therapy for Wegener's granulomatosis and microscopic polyangiitis.
Clin Exp Immunol. 164 (Suppl 1):31–34. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Gibelin A, Maldini C and Mahr A:
Epidemiology and etiology of wegener granulomatosis, microscopic
polyangiitis, churg-strauss syndrome and goodpasture syndrome:
Vasculitides with frequent lung involvement. Semin Respir Crit Care
Med. 32:264–273. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chung SA and Seo P: Microscopic
polyangiitis. Rheum Dis Clin North Am. 36:545–558. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kuboshima S, Tsuruoka K, Shirai S, Sasaki
H, Sakurada T, Miura H, Okabayashi J, Konno Y, Shima Y, Yasuda T,
et al: An autopsy case of microscopic polyangiitis complicated with
pulmonary aspergilloma and cytomegalovirus pneumonia. Nihon Jinzo
Gakkai Shi. 49:125–129. 2007.(In Japanese). PubMed/NCBI
|
|
14
|
Zhao MH and Lockwood CM: ANCA defines the
clinical disease manifestations of vasculitis. Sarcoidosis Vasc
Diffuse Lung Dis. 13:221–226. 1996.PubMed/NCBI
|