Introduction
In recent years, the number of patients with
traumatic arthritis, degenerative osteoarthritis and osteoarthritis
has significantly increased, and the articular cartilage defects
caused by these diseases have become a major cause of disability,
seriously affecting the quality of life of the patients. The
articular cartilage is an avascular tissue composed of
chondrocytes, which mainly depend on the articular synovial fluid
and pericellular matrix for their nutrient supply to enable them to
perform anaerobic glycolysis and to supply energy. The self-repair
ability of cartilage is limited (1);
therefore, cartilage tissue engineering has been a focus for
significant development (2). There
remain, however, numerous problems with cartilage tissue
engineering, such as limited resources; insufficient seed cells;
limited passage and proliferation capacities; a lack of
well-performing stent materials; the failure to effectively, stably
and directly induce stem cells to differentiate into cartilage
cells; the susceptibility of tissue-engineered cartilage to
dedifferentiation; and the difficulties with phenotypic maintenance
(3–5).
Bone marrow-derived mesenchymal stem cells (BMSCs)
are a class of stem cells derived from the bone marrow tissue that
exhibit a strong potential for proliferation and differentiation
(6,7). Due to the advantages of BMSCs, such as
being widely sourced, easy to obtain, without medical ethics issues
and suitable for amplification in in vitro culture, these
cells are considered to be the most suitable source of seed cells
for bone tissue engineering (8).
Currently, there is no uniform method of in vitro culture
and directional induction (9). The
aims of the present study, therefore, were as follows: i) To
attempt to prepare an in vitro three-dimensional (3D)
culture to maintain the passage and proliferation capabilities of
BMSCs; ii) to use the transforming growth factor (TGF)-β1 gene to
transfect BMSCs in order to achieve a sustained and stable
expression of TGF-β1 and enable the observation of the effects of
stably expressed TGF-β1 on the directional differentiation of BMSCs
into chondrocytes (10); and to
investigate the conditions and influencing factors that would
affect the differentiation of the stem cells into chondrocytes, in
order to provide the basic theory and methods for the construction
of tissue-engineered cartilage.
Materials and methods
Separation and culture of BMSCs
The whole bone marrow rinsing method was used to
obtain bone marrow blood from the long bone cavity of 4-week-old
New Zealand white rabbits obtained from the animal center of Fuzhou
General Hospital of Nanjing Command (Fuzhou, China), and a
density-gradient centrifugation method was then performed to
isolate the BMSCs. The 24 New Zealand rabbits were sacrificed by an
intravenous air bolus injection into the ear vein. Following
sacrifice, the skin of the four limbs was prepared and locally
disinfected with Anerdian (ShanDong LIRCON Medical Technology
Incor., Co., Ltd., Dezhou, China), and the skin and tissues of the
four limbs were cut to expose the bones. Phosphate-buffered saline
(PBS; Sigma-Aldrich, St. Louis, MO, USA) was then used to rinse out
the bone marrow using a 20-ml syringe at 4°C, and the cells were
centrifuged at 68 × g and resuspended with 20 ml incomplete
Dulbecco's modified Eagle's medium (DMEM; Gibco™ Life Technologies,
Grand Island, NY, USA). The cell suspension was subsequently
injected slowly into a centrifuge tube containing 20 ml Ficoll
separation solution (Pharmacia Corp., Peapack, NJ, USA) and
centrifuged at 27 × g for 30 min. The cell suspension at the
interface was extracted and added into 20 ml incomplete DMEM and
centrifuged at 27 × g for 5 min; the BMSC suspension was then
prepared following the discarding of the supernatant. The BMSC
suspension was seeded in a 75-cm2 culture flask
(5×104 cells/cm2) and cultured at 37°C and 5%
CO2 for 24 h. The medium was changed at 24 and 72 h for
the adherent culture to purify the BMSCs; subsequently, the medium
was changed once every 3 days until the cells grew to 90%
confluence. Trypsin (0.05%)/EDTA (0.02%) (Sigma-Aldrich) was used
for the digestion and passage. The primary cells were recorded as
P0, sequentially followed by P1, P2, P3, etc., until the monolayer
subculture was completed. The cellular morphological changes and
proliferation conditions were observed under an inverted
fluorescence phase contrast microscope (Olympus Corp., Tokyo,
Japan). This study was approved by the ethics committee of Fuzhou
General Hospital of Nanjing Command.
For the 3D culture, P1 BMSCs at 90% confluence were
digested by trypsin and centrifuged. The cell pellets were
suspended in low-viscosity sodium alginate (Sigma-Aldrich) solution
(12.5 g/l) to a concentration of 2×105 cells/ml. The
cell suspension was then added dropwise into 102 mmol/l
CaCl2 solution (5 mmol/l HEPES, pH 7.4; Sigma-Aldrich)
using a 5-ml pipette, allowing the condensed beads to gather in the
CaCl2 solution for ~10 min so that each condensed bead
would contain ~10,000 BMSCs. A five-fold NaCl solution (0.15 mol/l)
was used to wash the suspension three times; the beads were then
inoculated into six 75-cm2 culture flasks and cultured
at 37°C with 5% CO2. The medium [10% fetal bovine serum
(FBS) containing DMEM (Gibco Life Technologies)] was changed every
other day for an 8-week continuous culture. The culture medium was
subsequently discarded, and two-fold 50 mmol/l EDTA (10 mmol/l
HEPES, pH 7.4) was added for the dissolution. Digestion was
performed at 37°C for 15 min, followed by centrifugation at 239 × g
for 10 min; the supernatant was then discarded, and 0.5 ml type II
collagenase was added for the minor digestion. The BMSCs were
subsequently collected for observation under the inverted
fluorescence phase contrast microscope.
Growth curve of the BMSCs
Healthy P1 cells and cells after 1 week of 3D
culture were collected for digestion with 0.05% trypsin/0.02% EDTA
to make the cell suspension. The cell suspension was then seeded in
10 dishes (3.5 cm) at a density of 1×104 cells/dish. Two
dishes were taken for cell counting on days 3, 5, 7, 9 and 11,
respectively, and the remaining cells had their culture medium
changed once every 3 days. The final cell-counting results were
used to establish the cell growth curve.
Construction and identification of
vector
Human spleen tissue mRNA provided by Department of
General Surgery, Fuzhou General Hospital of Nanjing Command, was
extracted and subjected to open reading frame amplification (1,173
bp) according to the GenBank (http://www.ncbi.nlm.nih.gov/genbank/) human TGF-β1
gene sequence (gene sequence no. BC000125). The primers used were
as follows: TGF-β1 forward primer (with the SalI site), CGC
GTC GAC ATG CCG CCC TCC GGG CTG; TGF-β1 reverse primer (with the
HindIII site), CCA AGC TTC AGC TGC ACT TGC AGG AGC. The
TGF-β1 gene was amplified by the reverse transcription-polymerase
chain reaction (RT-PCR) method, and then inserted into the
adenoviral genome-containing cosmid vector pAdEasy1 (Stratagene;
Agilent Technologies, Inc., Santa Clara, CA, USA), and
co-transfected into E. coli BJ5183 (Agilent Stratagene) with
the recombinant plasmid pShuttleCMV.TGF-β1 (Stratagene). The
recombinant cosmid pAd.TGF-β1 was then generated by homologous
recombination. Following the homologous recombination, the
liposomal transfection method was applied to transfect the
recombinant cosmid adenoviral pAd.TGF-β1 into HEK293 cells
(American Type Culture Collection, Manassas, VA, USA) to produce
the recombinant adenovirus. Five clones of HEK293 cells were
selected for second-generation amplification and PCR identification
of whether the recombinant adenovirus carried the target gene.
Transfection and identification
Healthy P1 cells and the BMSCs after 1 week of 3D
culture were obtained and passaged in a 24-well plate. When the
coverage reached 70–80%, trypsin was added for digestion. With
regard to the 3D culture, the condensed beads of sodium alginate
were digested with EDTA using the aforementioned method, and three
wells were randomly selected for cell counting. The recombinant
adenovirus green fluorescent protein (Ad.GFP; Gene Therapy Unit,
Baxter Healthcare Corporation, Deerfield, IL, USA) adenovirus
solution was subsequently added according to the multiplicity of
infection (MOI) values of 0, 5, 20, 50, 100 and 200. Each MOI value
was set in three parallel wells. The total volume was established
as 100 µl with PBS; in addition, 1 ml 5% FBS-containing DMEM (Gibco
Life Technologies) was added to each well. After 1 h, 0.4 ml 5%
FBS-containing DMEM was added for 36–48 h culture at 37°C with 5%
CO2, and the plate was subsequently observed under an
inverted fluorescence microscope. Three vision fields of each well
were observed. The fluorescent cells were counted under a
magnification of x400 to calculate the percentage of fluorescent
cells, and the infection efficiency at each MOI was the average
percentage value of the fluorescent cells in the three vision
fields.
The cells in the experiment were divided into four
groups: i) Ad.TGF-β1-transfected BMSCs from 3D culture (T3D group);
ii) Ad.TGF-β1-transfected monolayer-cultured BMSCs (TMC group);
iii) Ad.Null empty virus (Stratagene)-transfected BMSCs from 3D
culture (ET3D group); and iv) control BMSCs from 3D culture (3D
group). The P1 rabbit BMSCs with good growing conditions were used
to prepare a cell suspension with a density of 1×106
cells/ml. For the monolayer culture, a 10-cm dish was used, while
three 10-cm dishes were used for the 3D culture. When the cells
reached 70–80% confluence, the recombinant adenovirus Ad.TGF-β1 was
transfected into the two cell cultures (MOI, 50:1). The ET3D and 3D
groups were set as the controls. To each dish, 10 ml 10%
FBS-containing DMEM was added, and the dishes were cultured at 37°C
with 5% CO2. The medium was replaced once every 3
days.
RT-PCR
According to the aforementioned GenBank TGF-β1 gene
sequence, the RT-PCR method was performed to detect the expression
levels of the mRNA, with GAPDH as the reference gene. Two weeks
after the transfection, six wells from each experimental group were
taken for the extraction of total RNA by the TRIzol® method (Gibco
Life Technologies). Following washing with pre-cooled PBS, the
NanoDrop™ 2000 ultramicro spectrophotometer (NanoDrop, Wilmington,
DE, USA) was used to detect the optical density values at 260 and
280 nm and calculate the RNA content. Total RNA (1 µg) was
reverse-transcribed into cDNA according to the instructions of the
RT kit (PrimeScript™One Step RT-PCR Kit Ver. 2; Takara
Biotechnology Co., Ltd., Dalian, China), and this cDNA was then
used as the template for the PCR reactions. The PCR reaction
conditions were as follows (Applied Biosystems 7500 Real-Time PCR
System; Applied Biosystems Life Technologies, Foster City, CA,
USA): 95°C pre-denaturation for 3 min; 94°C denaturation for 40
sec, 56°C annealing for 40 sec and 72°C extension for 45 sec (35
cycles); and then final extension at 72°C for 10 min. The primer
sequences (Sangon Biotech Co. Ltd., Shanghai, China) were as
follows: GAPDH upstream primer, 5′-GAA GGT CGG AGT CAA CGG-3′;
GAPDH downstream primer, 5′-GGA AGA TGG TGA TGG GATT-3′; TGF-β1
upstream primer, 5′-CGC GTC GAC ATG CCG CCC GG GCTG-3′; TGF-β1
downstream primer, 5′-CCA AGC TTC AGC TGC ACT TGC AGG AGC-3′. The
PCR products were identified by 1.5% agarose gel electrophoresis;
the scanning and analysis of the results were performed by the gel
imaging system (Bio-Rad Laboratories, Hercules, CA, USA).
Western blot analysis
According to the aforementioned grouping, the
transfected cells were extracted 4 weeks after the transfection;
the medium was removed and the cells were washed twice with
pre-cooled PBS. The cells were then scraped from the dish with a
cell scraper, and the supernatant was discarded following
centrifugation. Proteins were extracted from the lysate according
to the manufacturers instructions. Briefly, 100–200 µl lysis buffer
containing phosphatase inhibitor was added to the collected cells,
and incubated in ice for 0.5 h. Subsequently, the supernatant were
collected by centrifugation at 20,817 × g at 4°C for 20 min, then
stored at −80°C. Next, a bicinchoninic acid assay (Bio-Rad
Laboratories) was performed to determine the protein content.
Following denaturation, the mixture underwent polyacrylamide gel
electrophoresis and transfer to polyvinylidene difluoride
membranes, which were blocked for 2 h with 5% skimmed milk with
Tris-HCl (20 mmol/l), NaCl (13 mmol/l) and 0.1% Tween-20 (TBS-T;
Sigma-Aldrich). Following washing, the polyclonal TGF-β1 primary
antibody (cat. no. sc-146; dilution, 1:1,000; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), with polyclonal GAPDH (cat.
no. sc-25778; dilution, 1:1,000; Santa Cruz Biotechnology, Inc.) as
the control, was added to the membrane for overnight incubation at
4°C; the membrane was then washed three times with 0.25% TBS-T, for
5 min each time. The corresponding horseradish peroxidase
(HRP)-labeled goat anti-rabbit immunoglobulin G (IgG) secondary
antibody (cat. no. sc-2027; dilution, 1:2,500; Santa Cruz
Biotechnology, Inc.) was added for incubation at room temperature
for 1 h. TBS-T was subsequently used to wash the membrane three
times for 10 min each time. An enhanced chemiluminescent solution
(1:1) (Advansta, Menlo Park, CA, USA) was added for the scanning
and data analysis using the gel imaging system (Bio-Rad
Laboratories).
Cartilage differentiation
Cell culture was performed according to the
aforementioned grouping. Dexamethasone (Dex; 100 nmol/l) and
vitamin C (VitC; 50 mg/l; Sigma-Aldrich) were added to the medium
for the continuous 4- to 8-week induction culture. After the 4-week
induction culture, the medium was discarded and the four dishes
were placed on ice (0°C) and rinsed twice with PBS at 0°C. The
cellular changes and the changes in the tissue mass in the 3D
culture dishes were observed under the inverted fluorescence phase
contrast microscope.
ELISA for type II collagen and
aggrecan
On weeks 1, 2, 4 and 8 post-transfection, the
supernatants of the culture media were collected from three wells
from each group. The volume of each well was 2 ml. When the cells
reached 80–90% confluence, the brain-derived neurotrophic factor
standard (Applied Biosystems) and the supernatant were added into a
96-well plate, with three repeated wells for each sample. The
assays were carried out according to the manufacturer's
instructions (R&D Systems, Minneapolis, MN, USA).
Immunohistochemical method
Untransfected P1 BMSCs were set as the control
group, the cell suspension (1×106/ml) was then
inoculated onto the slides of culture dishes for 30-min slide
adherence. Next, DMEM was added into the culture dishes for a 3-h
culture at 37°C and 5% CO2. An inverted fluorescence
phase contrast microscope (Olympus Corporation) was then used to
observe the growth of cells, and the slides were removed when the
cells grew almost fused. Similarly, the cells of the TMC group were
seeded onto the glass slides, type II collagen immunohistochemical
detection was performed on weeks 2, 4 and 6 of induction culture.
The technical steps of detection included slice preparation,
formalin-fixation and overnight culture with the polyclonal
anti-collagen II primary antibody (cat. no. BA1088; dilution, 1:50;
Wuhan Boster Biological Engineering Co., Ltd., Wuhan, China),
followed by incubation with the biotinylated goat anti-rabbit IgG
(1:200) at 37°C for 40 min and streptavidin-HRP (cat. no. BA1003;
dilution, 1:200; Wuhan Boster Biological Engineering Co., Ltd.) at
37°C for 40 min. Following incubation with streptavidin-HRP, the
samples were stained with 0.05% 3,3′-diaminobenzidine plus 0.03%
H2O2, counterstained with hematoxylin, washed
with H2O, differentiated with HCl-EtOH (2 sec),
re-washed with H2O and placed in conventional resin.
Statistical analysis
Data are presented as the mean ± standard deviation
for the indicated number of independently performed experiments.
Statistical significance was determined by an independent samples
t-test. P<0.05 was considered to indicate a statistically
significant difference. The results were analyzed using SPSS
software (version 13.0; SPSS, Inc., Chicago, IL, USA).
Results
Biological characteristics
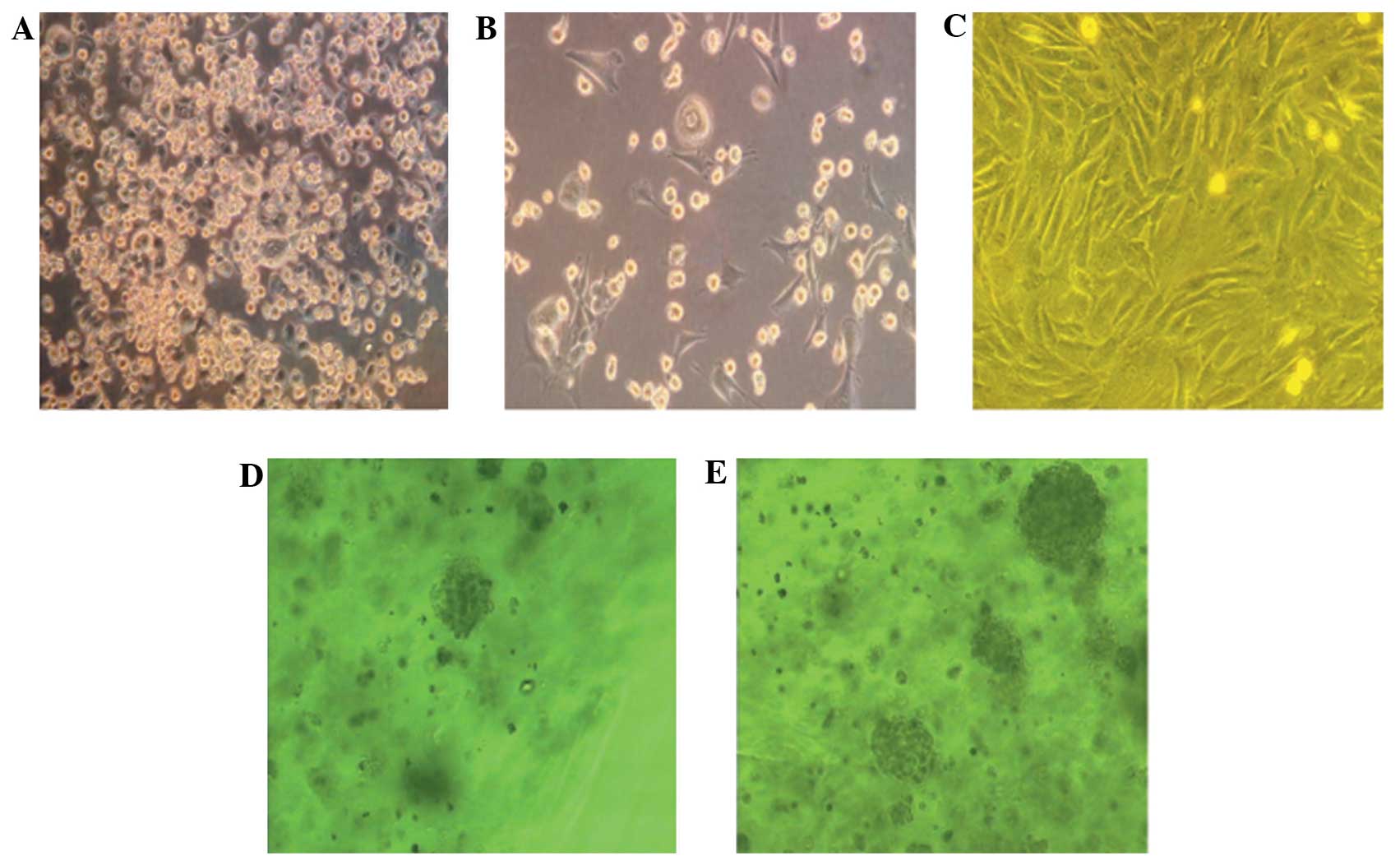
After 48 h of cell culture, the proliferation of the
primary adherent cells appeared dispersed and clonal, and the cells
exhibited a fibroblast-like spindle shape; 4–5 days later, the
adherent cells began to proliferate and transform into triangle,
polygonal and spindle shapes. Partial areas subsequently formed
unequal-sized colonies; the colonies continued expanding and fusing
and the inner cells were morphologically uniform. In the subculture
group, the cells were uniformly long-spindled, fusiform and
polygonal, and proliferation was rapid; 3 weeks later, the cells
began aging and declining. The division and proliferation of the
BMSCs in the sodium alginate 3D gel beads were enhanced, with mass-
and cluster-like growth and the formation of cell microspheres. The
cell microspheres suspended inside the sodium alginate gel remained
round in appearance, more passages were performed and the duration
was considerably longer; after 6–8 weeks, the cells appeared to age
and decline. The cell counts and colonies were significantly higher
in the 3D culture than those in the in vitro monolayer
subculture, indicating that the 3D culture system could generate
more stable BMSCs (Fig. 1).
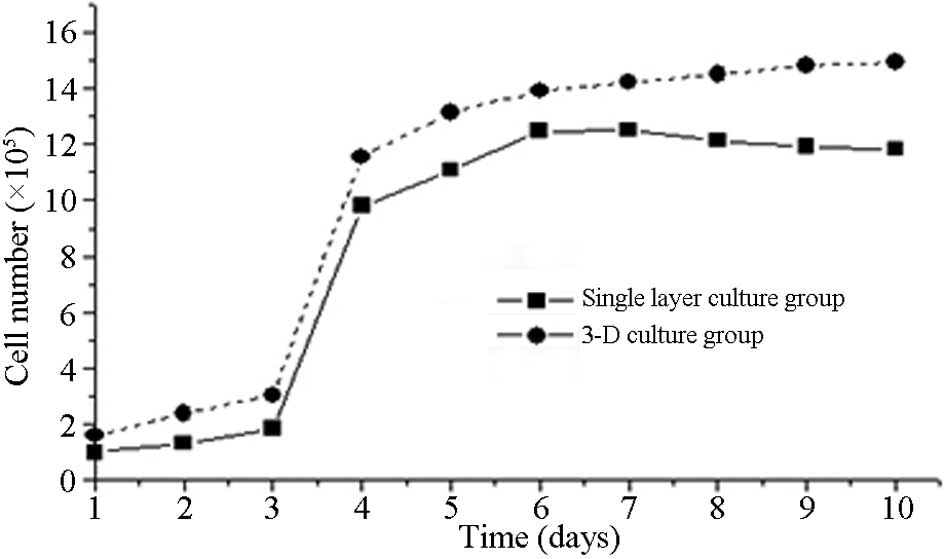
The growth curves of the two BMSC cultures were
similar; however, the 3D culture curve exhibited a more rapid and
early growth period than the monolayer culture, and the plateau
phase was maintained for longer (up to 8 weeks) (Fig. 2).
Transfection and identification
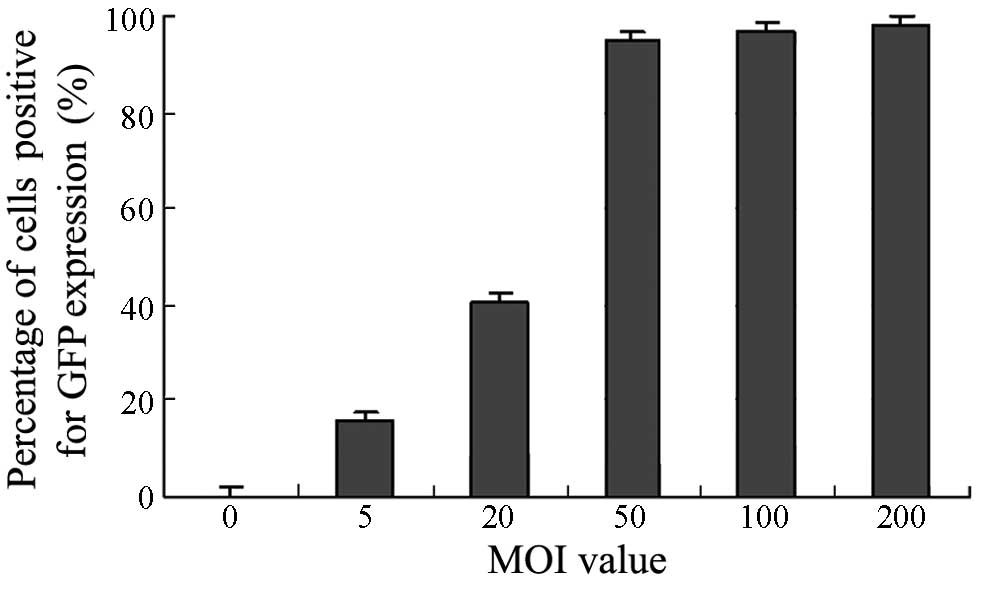
Following transfection, the expression of the green
fluorescent protein (GFP) gene in the Ad.GFP-transfected BMSCs
resulted in the appearance of green fluorescence under the
microscope; by contrast, no fluorescence was observed in the BMSCs
that were not transfected with Ad.GFP. The fluoroscopic observation
results of the Ad.GFP-transfected BMSCs were as follows: 15.8±1.1%
(MOI 5), 40.5±1.7% (MOI 20), 95.0±2.1% (MOI 50), 96.8±2.4% (MOI
100) and 98.1±2.8% (MOI 200). The results showed that with
increasing MOI values, the ratio of GFP-positive cells also
increased. When the MOI was 50, the ratio of green-stained rabbit
BMSCs was >95%, indicating that the in vitro transfection
efficiency of the adenovirus was high, and the target gene could be
effectively expressed. In order to achieve the optimal dose-effect
ratio, an MOI value of 50 was set as the transfection fold for
further experiments (Fig. 3).
The RT-PCR and western blot analyses showed that
strong TGF-β1 expression was present in the cell lysates 48 h after
the transfection; TGF-β1 could also be detected at weeks 2 and 4
post-transfection. The two control groups showed no clear bands.
The expression levels of TGF-β1 mRNA and protein were highest at ~2
weeks. With the extension of culture time and generations, the
expression of TGF-β1 mRNA and protein decreased gradually; among
the groups, the relative expression of the T3D group was notably
higher than that of the TMC group (Figs.
4A and B), indicating that the TGF-β1-carrying recombinant
adenovirus could be used to effectively transfect the rabbit BMSCs
to elicit high levels of mRNA expression. In addition, the protein
expression of the T3D group was evidently stronger than that of the
TMC group.
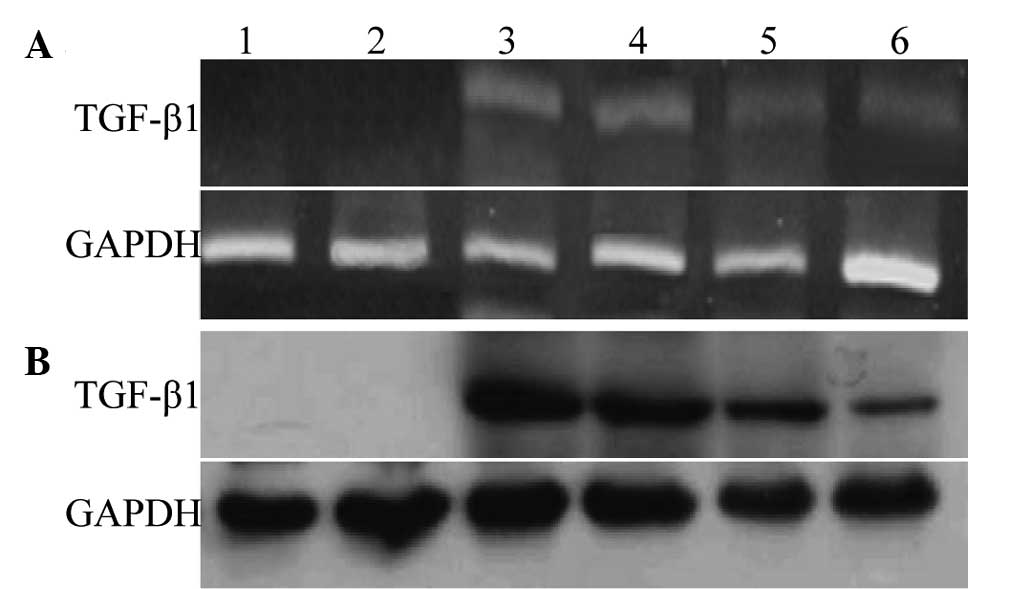 | Figure 4.Detection of TGF-β1 mRNA and protein
expression at different time-points by (A) reverse
transcription-polymerase chain reaction and (B) western blotting,
respectively. Lane 1, blank control BMSCs from 3D culture; lane 2,
ET3D group; lane 3, T3D group (all 2 weeks after transfection);
lane 4, T3D group (4 weeks after transfection); lane 5, TMC group
(2 weeks after transfection); lane 6, TMC group (4 weeks after
transfection). GAPDH was used as the internal control. TGF,
transforming growth factor; T3D, Ad.TGF-β1-transfected BMSCs from
3D culture; ET3D, Ad.Null empty virus-transfected BMSCs from 3D
culture; TMC, Ad.TGF-β1-transfected monolayer-cultured BMSCs; BMSC,
bone-marrow-derived stem cell; 3D, three-dimensional. |
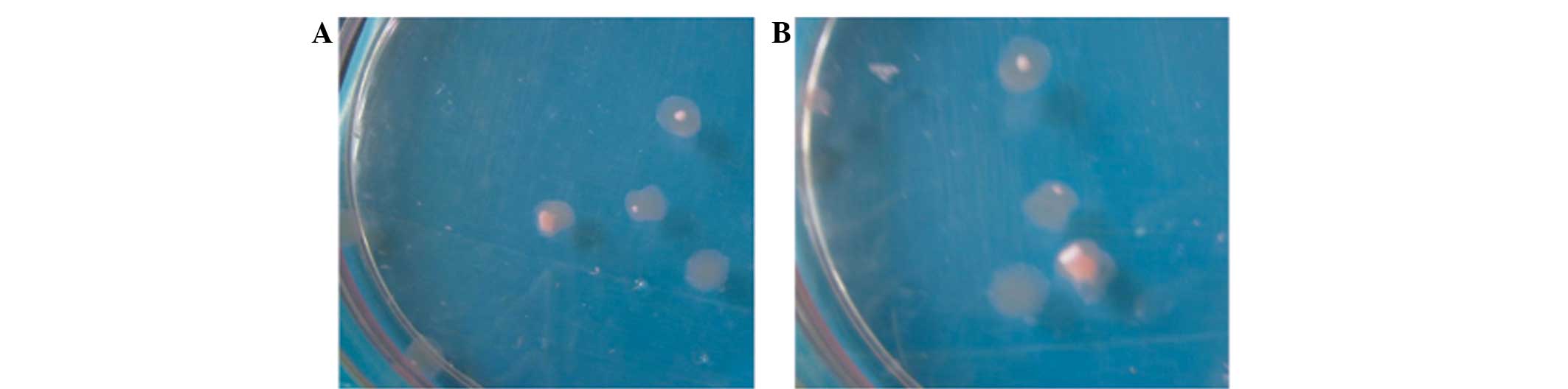
Differentiation promotion
According to the grouping, Dex (100 nmol/l) and VitC
(50 mg/l) were added for the induction culture. An irregular cell
mass could be observed at the bottom of the dish of the T3D group
48 h later; the diameter of this cell mass became progressively
larger, forming small clumps of tissue after 2 weeks. Continued
culturing gradually enlarged the tissue mass, which exhibited a
pink-white color at 4 weeks (Fig.
5). No tissue clumps formed in the TMC and control groups.
ELISA
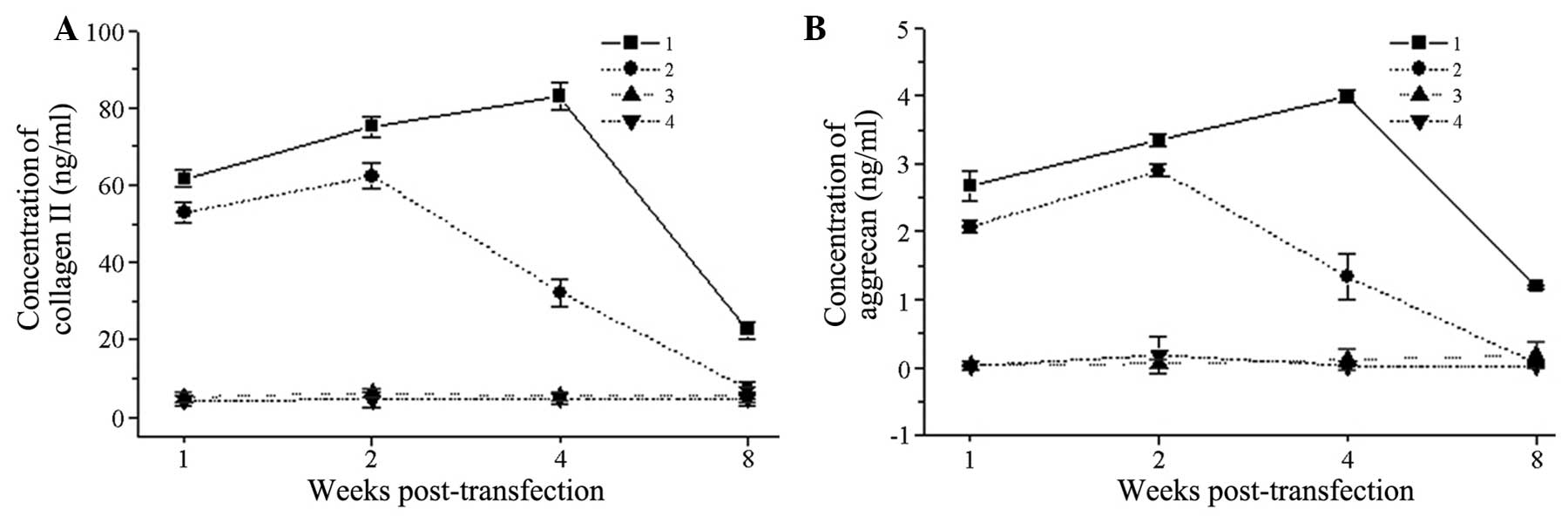
At all time-points following transfection, the type
II collagen expression in the T3D group was significantly increased
compared with that in the TMC group (P<0.05). The expression in
the T3D group reached a peak on day 28 post-transfection
(82.976±3.452 ng/ml) and then decreased gradually, while that in
the TMC group reached its highest point on day 14 post-transfection
(62.331±3.314 ng/ml) and then steadily declined. The two control
groups had almost no expression of type II collagen. On day 56
post-transfection, the type II collagen expression could still be
detected in the T3D group, while almost no type II collagen
expression was observed in the TMC group; the difference between
the groups was statistically significant (P<0.05) (Fig. 6A). The proteoglycan expression was
similar to the type II collagen expression (Fig. 6B).
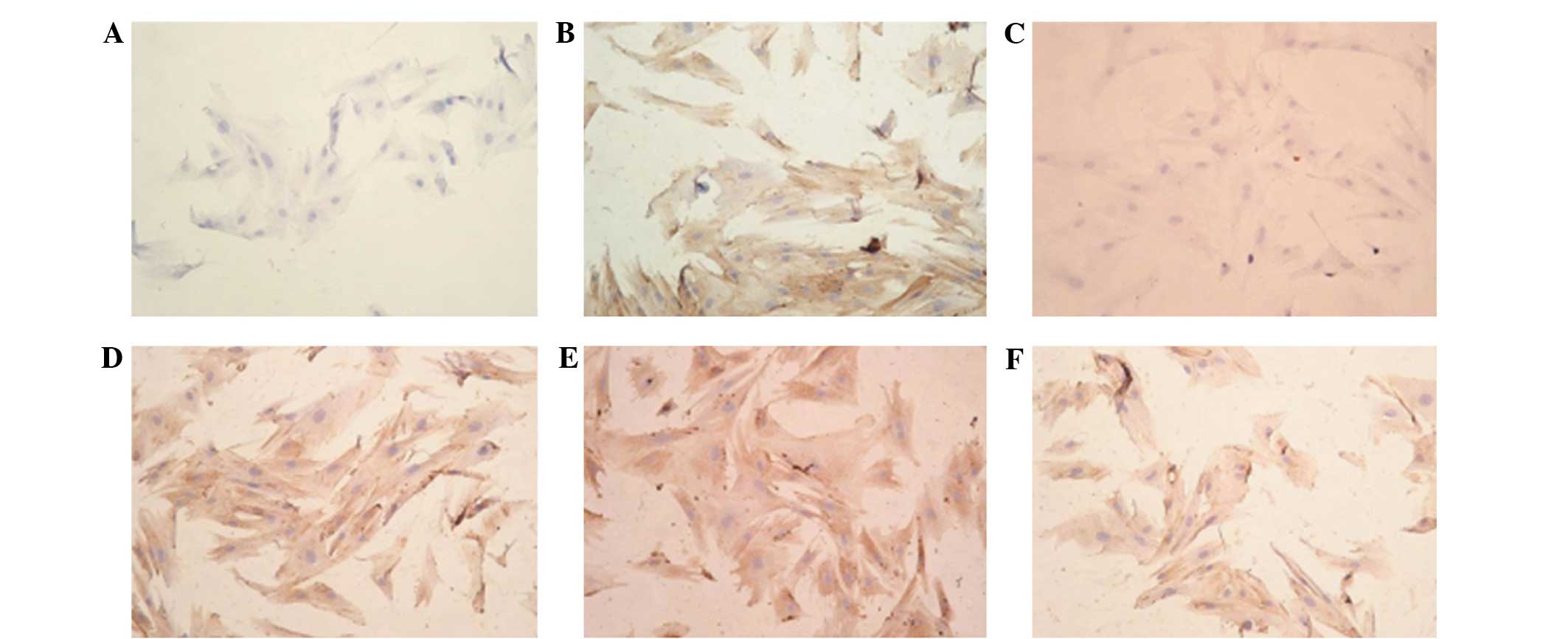
Immunohistochemistry
Following transfection, the cells in the TMC group
at weeks 2 and 4 were seeded onto the slides and subjected to type
II collagen immunohistochemical detection. The tissue clumps of the
T3D group at weeks 2, 4 and 6 were prepared as frozen sections and
then utilized for type II collagen immunohistochemical detection.
The results for the expression of type II collagen were positive in
the TMC group at weeks 2 and 4 (stained brown), while
immunohistochemistry at week 6 did not detect type II collagen
expression. In the T3D group, the type II collagen
immunohistochemistry at weeks 2, 4 and 6 revealed strong, positive
expression (stained yellow-brown); the continued positive result at
the 6th week indicated that the expression of type II collagen
could still be detected. No type II collagen expression was found
in the untransfected BMSC control group (Fig. 7).
Discussion
The principles of cell biology indicate that the
number and density of cells play an important role in the
proliferation and differentiation of BMSCs. During the development
of cartilage, the aggregation of numerous local mesenchymal cells
can be observed, forming a higher cell density; this cell
aggregation has significance toward the differentiation of BMSCs
into cartilage (11). Through the
in vitro culture of BMSCs, Nakahara et al (12) found that, when the cells reached a
certain density, BMSCs could differentiate into chondrocytes. A
study confirmed that the BMSCs passaged with a higher density could
be differentiated into chondrocytes, expressing the chondrocyte
phenotype, and would, therefore, form cartilage nodules when
implanted into nude mice. By contrast those passaged with low
density would fail to differentiate into chondrocytes (13), indicating that certain cell densities
were necessary for the differentiation of BMSCs into chondrocytes.
However, compared with the other types of cells inside the bone
marrow, the abundance of BMSCs inside the bone marrow is not high,
with the ratio being 0.01–1:1×104 among the bone marrow
cells (14).
The construction of a good in vitro culture
system would not only facilitate the proliferation of BMSCs, so
that the seed cells could be obtained at the required scale, but
would also be of use for the differentiation of BMSCs into
cartilage. Currently, the in vitro BMSC culture system has
no consistent standard: The common culture methods of BMSCs are
two-dimensional (2D) culture (including very low-density
subculture, low oxygen tension and gene transfection cultures), 3D
culture, and inactive and active cell cultures (15). At present, the method of 2D culture
is most widely adopted. Although 2D culture techniques are widely
used and gradually improving, they exhibit the following
shortcomings, which are challenging to overcome: i) The surface
area of the adherent cells is limited; therefore the cell yield is
low; ii) the aseptic process is cumbersome, and would be
contaminated easily; iii) the metabolites gradually accumulate, and
untimely exclusion would lead to the poor growth of cells and even
degeneration; iv) the cells remaining in the in vivo 3D
structure with the extracellular matrix would show alterations in
biological behavior, and variation would easily occur. Therefore,
the experimental study of the application of 3D cell-culture
techniques into cartilage tissue engineering has been
initiated.
In 1998, Qiu et al (16) performed a 3D BMSC culture. Glowacki
et al (17) used a collagen
sponge as the carrier for the 3D perfusion culture, which resulted
in a reduction in the accumulation of metabolites; the results
revealed that the content of extracellular matrix was increased and
the activities and functions of the cultured cells were
strengthened. Angele et al (18) applied in vitro-amplified human
BMSCs into a hyaluronic acid-gelatin complex sponge, and a
TGF-β1-exterior chemical limited medium (Dex, VitC, etc.) was
added. After 28 days, cartilage tissue was produced. Ponticiello
et al (19) applied human
BMSCs to a gelatin sponge, with the addition of TGF-β3, and
succeeded in culturing chondrocytes. Compared with the 2D culture,
the 3D culture was able to accommodate high-density cell adhesion
and proliferation, was favorable for intracellular signaling
conduction and could provide a suitable microenvironment to
maintain the metabolic activities of cells. In addition, a 3D
culture facilitates the increased synthesis of extracellular matrix
components, enhances the activities and functions of the cultured
cells and enables the extracellular matrix to be fixed near the
cells, which prevents it from being lost into the medium as easily
as that in the monolayer culture; the 3D culture system therefore
has important regulatory roles in cell proliferation,
differentiation and metabolism. Currently, the biological material
used in 3D culture can take numerous forms, the most common being
the gelatin sponge, hyaluronic acid-gelatin complex sponge,
collagen sponge and alginate beads (20). Since alginate is a linear
polysaccharide with good biocompatibility and can be metabolized
in vivo through hydrolytic and enzymatic pathways, its
metabolites do not generate adverse effects toward the cells
(21). Furthermore, it can undergo
ionic gelation in the presence of Ca2+, exhibiting
mechanical properties similar to those of normal cartilage tissue,
thereby better maintaining the phenotype of the seed cells. The
plasticity of alginate is also strong; thus, it may be an optimum
3D culture material (22). In the
present study, a 3D culture system of sodium alginate gel was
constructed, and the in vitro amplification of BMSC seed
cells was successfully performed. The experimental results showed
that, compared with the 2D culture, the BMSCs obtained from the 3D
culture exhibited a more rapid and early growth, a more evident
increase in total number, a longer maintenance time and a longer
plateau phase (up to 8 weeks).
The sodium alginate 3D culture system constructed in
the present experiment was not only favorable for BMSC
proliferation, generating seed cells at the required scale, but
also aided the differentiation of the BMSCs into cartilage. In the
3D culture conditions, the adenoviral vector was capable of
transfecting BMSCs successfully in vitro (23), and cartilage tissue clumps could be
obtained successfully 4 weeks after the induction of the culture.
Western blot analysis was performed to detect the expression levels
of TGF-β1 protein following the transfection, and RT-PCR analysis
was performed to detect the transcription levels of TGF-β1 mRNA.
The results showed that the expression levels of TGF-β1 protein and
mRNA in the 3D group were significantly higher than those in the
TMC and control groups, indicating that the transfection could
achieve the sustained and effective expression of TGF-β1 and induce
the formation of cartilage tissue in conjunction with other stimuli
(e.g. Dex and VitC) (24). The
detection of cartilage extracellular matrix secretion products
indicated that the TGF-β-transfected BMSCs were capable of
secreting such cartilage-specific matrix components as type II
collagen and proteoglycans. The secretion level in the T3D group
was higher than that in the TMC group, and the duration of the type
II collagen secretion was longer, indicating that the transfected
BMSCs exhibited in vitro differentiation into chondrocytes,
with the T3D group performing in a superior way to the TMC
group.
An explanation for the aforementioned results may be
that the sodium alginate 3D culture system was able to accommodate
high-density cell adhesion and proliferation, which was favorable
for intracellular signal transduction. The use of sodium alginate
may also have reduced the accumulation of metabolic products,
providing a suitable microenvironment that was able to maintain the
metabolic activities of the cells and to form the 3D structure
together with the extracellular matrix. The condensed alginate
beads provided a 3D microenvironment that was similar to the in
vivo conditions, reducing the variation in the biological
behavior of the cells. Furthermore, the content of the
extracellular matrix was improved and could be fixed near the
cells, instead of becoming immersed in the culture solution; thus,
the cells were surrounded by rich extracellular matrix. The 3D
culture system therefore played an important regulatory role in the
cell proliferation, differentiation and metabolism.
The present experimental study completed the
separation and purification of rabbit BMSCs and successfully
established monolayer and 3D cultures in vitro. Compared
with the monolayer culture, the proliferation of the BMSCs in the
3D culture was more active, the number of passages was increased
and the duration of the culture was longer. After 6–8 weeks, the
cells appeared to be aging and declining. Importantly, the 3D
culture system was more comparable with the microenvironment in
vivo, which has laid a foundation for the study of BMSCs in
animal models. The recombinant adenovirus Ad.TGF-β1 was transfected
into the BMSCs, and it was found that the BMSCs in the monolayer
and 3D cultures had a high level of TGF-β1 expression following
transfection, as detected by RT-PCR and western blotting. TGF-β1
expression continued until 6 weeks after the transfection. The
result indicated that the transfection of BMSCs by the recombinant
adenovirus Ad.TGF-β1 was safe and efficient. The protein expression
in the cells in 3D culture was evidently superior to that in the
cells in monolayer culture. Following the RT-PCR and western blot
analyses, ELISA was used to detect the presence of type II collagen
and proteoglycan in the supernatant of the culture after TGF-β1
transfection, as a measure of the BMSC differentiation into
cartilage cells. The results revealed that, following TGF-β1
transfection, the BMSCs could continuously secrete such specific
extracellular matrix components of chondrocytes as type II collagen
and proteoglycans; the levels of the these components were found to
have increased significantly, and the post-gene transfection 3D
culture group exhibited superior results to the monolayer culture
group. In conclusion, the present series of experimental results
has shown that the use of a 3D culture system and gene transfection
in vitro can promote BMSC proliferation and aid the
differentiation of BMSCs into chondrocytes.
References
|
1
|
ODriscoll SW: The healing and regeneration
of articular cartilage. J Bone Joint Surg Am. 80:1795–1812.
1998.PubMed/NCBI
|
|
2
|
Awad HA, Butler DL, Boivin GP, Smith FN,
Malaviya P, Huibregtse B and Caplan AI: Autologous mesenchymal stem
cell-mediated repair of tendon. Tissue Eng. 5:267–277. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ostrander RV, Goomer RS, Tontz WL, Khatod
M, Harwood FL, Maris TM and Amiel D: Donor cell fate in tissue
engineering for articular cartilage repair. Clin Orthop.
389:228–237. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Colther DC, Sekiya I and Prockop DJ:
Identification of a subpopulation of rapidly self-renewing and
multipotential adult stem cells in colonies of human marrow stromal
cells. Proc Natl Acad Sci USA. 98:7841–7845. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huang JI, Zuk PA, Jones NF, Zhu M, Lorenz
HP, Hedrick MH and Benhaim P: Chondrogenic potential of
multipotential cells from human adipose tissue. Plast Reconstr
Surg. 113:585–594. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Meirelles Lda S, Fontes AM, Covas DT and
Caplan AI: Mechanisms involved in the therapeutic properties of
mesenchymal stem cells. Cytokine Growth Factor Rev. 20:419–427.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Boo L, Selvaratnam L, Tai CC, Ahmad TS and
Kamarul T: Expansion and preservation of multipotentiality of
rabbit bone-marrow derived mesenchymal stem cells in dextran-based
microcarrier spin culture. J Mater Sci Mater Med. 22:1343–1356.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Seong JM, Kim BC, Park JH, et al: Stem
cells in bone tissue engineering. Biomed Mater. 5:0620012010.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Augello A, Kurth TB and De Bari C:
Mesenchymal stem cells: A perspective from in vitro cultures to in
vivo migration and niches. Eur Cell Mater. 20:121–133.
2010.PubMed/NCBI
|
|
10
|
Worster AA, Nixon AJ, Brower-Toland BD and
Williams J: Effect of transforming growth factor betal on
chondrogenic differentiation of cultured equine mesenchymal stem
cells. Am J Vet Res. 61:1003–1010. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yoo JU, Barthel TS, Nishimura K, et al:
The chondrogenic potential of human bone-marrow-derived mesenchymal
progenitor cells. J Bone Joint Surg Am. 80:1745–1757.
1998.PubMed/NCBI
|
|
12
|
Nakahara H, Dennis JE, Bruder SP,
Haynesworth SE, Lennon DP and Caplan AI: In vitro differentiation
of bone and hypertrophic cartilage from periosteal-derived cells.
Exp Cell Res. 195:492–503. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bruder SP, Fink DJ and Caplan AI:
Mesenchymal stem cells in bone development, bone repair and
skeletal regeneration therapy. J Cell Biochem. 56:283–294. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mostafa NZ, Uludağ H, Varkey M, Dederich
DN, Doschak MR and El-Bialy TH: In vitro osteogenic induction of
human gingival fibroblasts for bone regeneration. Open Dent J.
5:139–145. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lisignoli G, Remiddi G, Cattinil L, et al:
An elevated number of differentiated osteoblast clonies can be
obtained from rat bone marrow stromal cells using a gradient
isolation procedure. Connect Tissue Res. 42:49–58. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Qiu Q, Ducheyne P, Gao H and Ayyaswamy P:
Formation and differentiation of three-dimensional rat marrow
stromal cell culture on microcarriers in a rotating-wall vessel.
Tissue Eng. 4:19–34. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Glowacki J, Mizuno S and Greenberger JS:
Perfusion enhances functions of bone marrow stromal cells in
three-dimensional culture. Cell Transplant. 7:319–326. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Angele P, Kujat R, Nerlich M, Yoo J,
Goldberg V and Johnstone B: Engineering of osteochondral tissue
with bone marrow mesenchymal progenitor cells in a derivatized
hyaluronan-gelatin composite sponge. Tissue Eng. 5:545–554. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ponticiello MS, Schinagl RM, Kadiyala S
and Barry FP: Gelatin-based resorbable sponge as a carrier matrix
for human mesenchymal stem cells in cartilage regeneration therapy.
J Biomed Mater Res. 52:246–255. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Majumdar MK, Banks V, Peluso DP and Morris
EA: Isolation, characterization and chondrogenic potential of human
bone marrow-derived multipotential stromal cells. J Cell Physiol.
185:98–106. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Diduch DR, Jordan LC, Mierisch CM and
Balian G: Marrow stromal cells embedded in alginate for repair of
osteochondral defects. Arthroscopy. 16:571–577. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ma HL, Hung SC, Lin SY, Chen YL and Lo WH:
Chondrogenesis of human mesenchymal stem cells encapsulated in
alginate beads. J Biomed Mater Res A. 64:273–281. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hwang NS, Im SG, Wu PB, et al:
Chondrogenic priming adipose-mesenchymal stem cells for cartilage
tissue regeneration. Pharm Res. 28:1395–1405. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Taipaleenmäki H, Harkness L, Chen L, et
al: The crosstalk between transforming growth factor-β 1 and delta
like-1 mediates early chondrogenesis during embryonic endochondral
ossification. Stem Cells. 30:304–313. 2012. View Article : Google Scholar : PubMed/NCBI
|