Introduction
Bile acids have been implicated as causative agents
in cancers of the gastrointestinal tract, including cancers of the
stomach (1), small intestine
(2), biliary tract (3) and colon (4). Bile acids themselves cannot induce
tumors (5); however, their presence
or absence in the colon has been demonstrated to be a strong
determinant of tumor incidence (6).
Bile acids are considered to lack the ability to initiate
tumorigenesis mainly on the basis that they are unable to induce
DNA damage directly; therefore, bile acids are hypothesized to
promote colon tumorigenesis by affecting intracellular signaling
(7). Cholestasis is reported to
cause the intra-hepatic retention of potentially toxic bile acids,
which causes liver injury and biliary fibrosis or cirrhosis
(8). This has been observed in rats
with sustained high levels of bile acids following the intravenous
infusion of bile acids (9) and in
bile duct-ligated rats (10).
Oxidative stress induces DNA damage, which causes chromosomal
aberrations associated with cell transformation (11). Since bile acids induce oxidative
stress, they are thus considered potential carcinogens (12).
Cytochrome P450 (CYP) constitutes a superfamily of
heme-containing enzymes that take part in the metabolism and
elimination of various exogenous and endogenous substances
(13,14). They play critical roles in the
biotransformation of drugs, carcinogens, steroid hormones and
environmental toxicants (15–17).
CYP1A1 and CYP1A2 catalyze the oxygenation of polycyclic aromatic
hydrocarbons (PAHs) and heterocyclic aromatic amines/amides (HAAs)
(18). Changes in the levels of CYPs
may contribute to the development of cancer (13). PAHs induce CYP1A via the aryl
hydrocarbon receptor (AhR), a ligand-activated transcription
factor. When PAHs bind to AhR, sequential signaling events are
initiated that activate the AhR and induce transcription of CYP1A
genes through the xenobiotic response element (XRE) located in the
enhancers of the genes (19).
Humans are exposed to PAHs and HAAs from a wide
range of sources, including tobacco smoke, automobile exhaust,
smoked and cooked food and industrial processes. Such exposure has
been causatively linked to an increased incidence of cancers in
smokers and certain other populations (20). One of the most well-characterized
molecular responses to PAHs is the induction of the CYP1A1 gene,
which encodes the carcinogen-activating enzyme CYP1A1 (21). Moreover, chemicals present in the
diet can activate AhRs; several dietary plant compounds have been
reported to competitively bind to and/or induce AhR-dependent gene
expression. These include 7,8-dehydrorutacarpine (22), indole-[3,2-b]-carbazole (ICZ)
derivatives (23), curcumin
(24) and certain carotinoids
(25). Furthermore, dietary indoles
including indole 3-carbinol (I3C) and tryptophan can be converted
in the mammalian digestive tract into more potent AhR ligands. ICZ,
an acidic condensation product of I3C, the most potent natural AhR
ligand (23), has been detected in
rat feces (26). Certain flavanoids
have also been reported to be AhR ligands, such as quercetin
(27) and tangeritin (28). The metabolic activation of PAHs and
HAAs by CYP1A enzymes is a critical step in the development of
cancer in human populations exposed to PAHs and HAAs (19). CYP1A1/2 enzymes are highly inducible
by a range of chemicals (29). Their
modulation may occur through pre- or post-transcriptional or
pre-translational mechanisms.
A marked activation of CYP1A1 and CYP1A2 genes has
been observed in congenitally jaundiced Gunn rats (30). Despite numerous studies on the
association between bile acids and carcinogenesis, to the best of
our knowledge, the effects of bile acids on CYP1A have not been
fully elucidated. This prompted the investigation of the effects of
bile acids on CYP1A1 induction using one of the most abundant
primary bile acids, chenodeoxycholic acid (CDCA), in the present
study. The effects on CYP1A1 transcription pre- or
post-transcription and protein expression were examined, and the
possible signaling pathways involved in the modulating effects of
bile acids on CYP1A1 expression were elucidated.
Materials and methods
Materials
Fetal bovine serum (FBS), chenodeoxycholic acid
(CDCA) and Sudan III (S.III) were obtained from Sigma-Aldrich (St.
Louis, MO, USA). Polyclonal goat anti-rat CYP1A1 (#299124) was
acquired from Daiichi Pure Chemicals Co., Ltd. (Tokyo, Japan),
horseradish peroxidase-labeled rabbit anti-goat IgG (#A8919) was
obtained from Sigma-Aldrich, and polyclonal goat anti-rat β-actin
(#sc-130657) was from Santa Cruz Biotechnology (Dallas, TX, USA).
Human hepatoma (HepG2) cells were obtained from the American Type
Culture Collection (Manassas, VA, USA). Rat hepatoma (H4IIE) cells
were obtained from the American Type Culture Collection (Rockville,
MD, USA). PD98059, a mitogen-activated protein kinase kinase (MEK)
inhibitor, was from Biomol Research Laboratories, Inc. (Plymouth,
PA, USA) and SB203580, a p38 inhibitor, was from Sigma-Aldrich.
Other chemicals were of analytical grade.
Luciferase activity assay of HepG2
cells
HepG2 cells were grown in Dulbecco's modified
Eagle's medium (DMEM) supplemented with 10% FBS, and antibiotics
(100 U/ml penicillin and 100 µg/ml streptomycin) at 37°C in a
humidified atmosphere of 5% CO2 in air. Cells were
seeded at 70% confluence on 96-well collagen-coated plates. After
24 h, cells were transfected with 50 ng/well pGL3-XRE and 5 ng/well
pRL-SV40 vector (Promega KK, Tokyo, Japan) using
TransIt-pGL3-Transfection Reagent (Mirus Bio LLC, Madison, WI, USA)
according to the manufacturer's instructions for 12 h, after which
the medium was replaced by new medium containing dimethylsulfoxide
(DMSO; control), 0.25 µM S.III and/or 60, 90 or 120 µM CDCA for 24
h. Cells were lysed and firefly and Renilla luciferase
activities were measured from six independent transfections using
the Dual-Luciferase® Reporter assay system (Promega Corporation,
Madison, WI, USA). Transfection data are expressed as fold
induction of firefly to Renilla luciferase activities
relative to empty vector or vehicle control. The experiment was
repeated three times
Treatment of H4IIE cells
Rat H4IIE cells were grown in DMEM supplemented with
10% FBS, and antibiotics (100 IU/ml penicillin and 100 µg/ml
streptomycin) at 37°C in a humidified atmosphere of 5%
CO2 in air. Cells were seeded in 60-mm collagen-coated
dishes and sub-cultured twice a week. H4IIE cells were grown to the
confluent stage, after which they were treated with 0.1% DMSO
(control), 0.25 µM S.III and/or CDCA at concentrations of 60, 90 or
120 µM for a further 24 h, after which cells were harvested for RNA
extraction.
In another experiment, H4IIE cells were grown to the
confluent stage, as described above. They were then treated with 10
µM PD98059 or SB203580 for 40 min prior to the application of 0.1%
DMSO (control), 0.25 µM S.III and/or 90 µM CDCA for a further 24 h.
The cells were then harvested for protein analysis.
RNA extraction
Total RNA was isolated from the H4IIE cell cultures
using TRIzol reagent (Life Technologies Inc., Grand Island, NY,
USA). Briefly cell cultures from the 60-mm dishes were homogenized
in 1 ml TRIzol reagent. Then, 0.3 ml chloroform was added to the
sample. The mixtures were shaken for 30 sec followed by
centrifugation at 4°C and 12,500 × g for 20 min. The supernatant
layers were transferred to a new set of tubes, an equal volume of
isopropanol was added and the samples were shaken for 15 sec and
centrifuged 4°C and 12,500 × g for 15 min. The RNA pellets were
washed with 70% ethanol. RNA was dissolved in diethylpyrocarbonate
(DEPC)-treated water and the prepared RNA was examined by
electrophoresis, which showed that the RNA integrity was
acceptable. The RNA was further checked by measurement of its
optical density using a Nanodrop ND-1000 spectrophotometer (Thermo
Fisher Scientific, Waltham, MA, USA). The ratio of sample
absorbance at 260 and 280 nm for all RNA samples was 1.7–1.9.
Reverse transcription-polymerase chain
reaction (RT-PCR)
A mixture of 5 µg total RNA and 0.5 ng oligo(dT)
primer in a total volume of 24 µl sterilized ultra-pure water, was
incubated at 70°C for 10 min and then made up to 40 µl with a
mixture of 8 µl 5X RT-buffer, 2 µl 10 mM dNTP, 2 µl DEPC water and
2 µl of reverse transcriptase (Toyobo Co., Ltd., Osaka, Japan). The
resultant mixture was incubated in the thermal cycler (Bio-Rad
Laboratories, Inc., Hercules, CA, USA) at 30°C for 10 min, 42°C for
1 h and 90°C for 10 min. For PCR, 1 µl aliquots of the synthesized
cDNA were added to 19 µl of a mixture containing sterilized
ultra-pure water, 2 µl 10X PCR buffer, 2 µl dNTP (2.5 mM), 0.3 µl
10 µM sense and anti-sense primers and 0.1 µl Taq polymerase
(Takara, Kyoto, Japan). The primers were as follows: CYP1A1 sense,
CCATGACCAGGAACTATGGG and anti-sense, TCTGGTGAGCATCCAGGACA [GenBank
accession no., X00469; Parkin (31)]; β-actin sense, ATGTACGTAGCCATCCAGGC
and anti-sense, TCCACACAGAGTACTTGCGC (GenBank accession no.,
V01217). Amplification was initiated by 1 cycle of denaturation at
94°C for 4 min, followed by a 24 cycles, each comprising
denaturation at 94°C for 1 min, annealing at 56°C for 1 min and
extension at 72°C for 1 min. After the last cycle of amplification,
samples were finally incubated for 7 min at 72°C. Amplified PCR
products were separated by electrophoresis through 1.5% agarose
gel. The product size of CYP1A1 was 341 bp and that of β-actin was
628 bp. Bands were stained with ethidium bromide and visualized by
ultraviolet illumination. Photographic images were converted into
computer files with an Epson color-image scanner (Suwa, Japan) in
combination with Adobe Photoshop 6.0 software (Adobe Systems, San
Jose, CA, USA).
Western blot analysis
Following the experimental treatments, H4IIE cells
were washed with ice-cold phosphate-buffered saline (PBS) and
scraped in ice-cold lysis buffer [50 mM HEPES (pH 7.5), 150 mM
NaCl, 5 mM EDTA, 20 mM sodium fluoride, 10 mM sodium pyrophosphate,
2 mM sodium vanadate, 1% Nonidet P-40 and complete protease
inhibitor cocktail]. Harvested cells were incubated on ice for 30
min followed by centrifugation at 12,000 × g for 20 min at 4°C to
obtain cell lysates. Portions of the cell lysate (40 µg cell lysate
supernatant), were resolved by 10–12% SDS-PAGE electrophoresis. The
proteins were transferred electrophoretically to nitrocellulose
membranes, and blocked with 5% skimmed milk in PBS containing 1%
Tween 20 for 2 h at room temperature. The membranes were stained
with Ponceau S to confirm transfer and to ensure equal protein
loading for each sample. The membranes were blocked in 5% skimmed
milk in PBS with 1% Tween 20 for 2 h at room temperature followed
by incubation with goat anti-rat CYP1A1 and β-actin primary
antibodies (1:100 dilution) at room temperature for 3 h and then
exposed to horseradish peroxidase-conjugated rabbit anti-goat
secondary antibodies (1:200 dilution in PBS) for 3 h at room
temperature. Immunoreactive protein bands were detected with the
ECL-Plus chemiluminescence kit (Amersham Life Science, Cleveland,
OH, USA). Intensities of the immunoreactive bands were analyzed
densitometrically using public domain NIH Image software
(http://rsb.info.nih.gov/nih-image/).
CYP1A1 protein bands were normalized against the respective band
intensity of β-actin and normalized to control levels.
Statistical analysis
All data are expressed as means ± standard
deviations. Statistical significances were evaluated using the
Tukey-Kramer honest significant difference test with JMP software
(SAS Institute, Cary, NC, USA). Results were considered to be
statistically significant when P<0.05.
Results
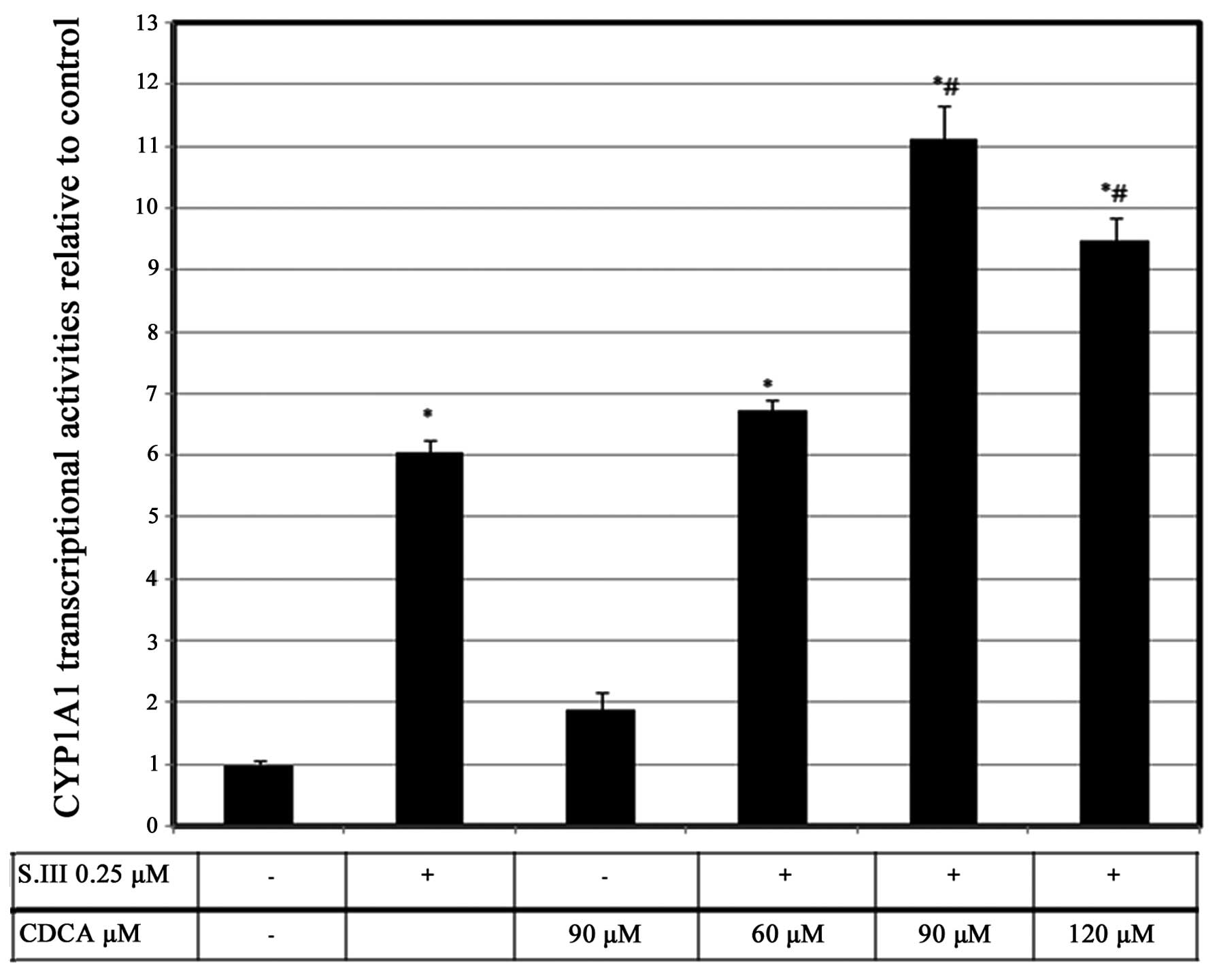
CDCA upregulates S.III-induced CYP1A1
transcriptional activity
The effect of CDCA on CYP1A1 mRNA could potentially
be pre-transcriptional, transcriptional or post-transcriptional. To
confirm the exact level at which CDCA modulates CYP1A1 induction,
CYP1A1 promoter activity was investigated by XRE-luciferase
reporter assays in HepG2 cells. Reporter activity was induced by
0.25 µM S.III to a level of ~6-fold higher than that of the
control. However, 90 µM CDCA induced luciferase activity to only
1.8-fold more than the control levels. Treatment of the cells with
60 µM CDCA plus 0.25 µM S.III slightly increased CYP1A1
transcriptional activity. However, treatment of the cells with 90
µM CDCA plus 0.25 µM S.III induced CYP1A1 transcriptional activity
to a level 11-fold greater than the control. Treatment of the HepG2
cells with 120 µM CDCA plus 0.25 µM S.III did not cause a further
increase of the CYP1A1 transcriptional activity (Fig. 1)
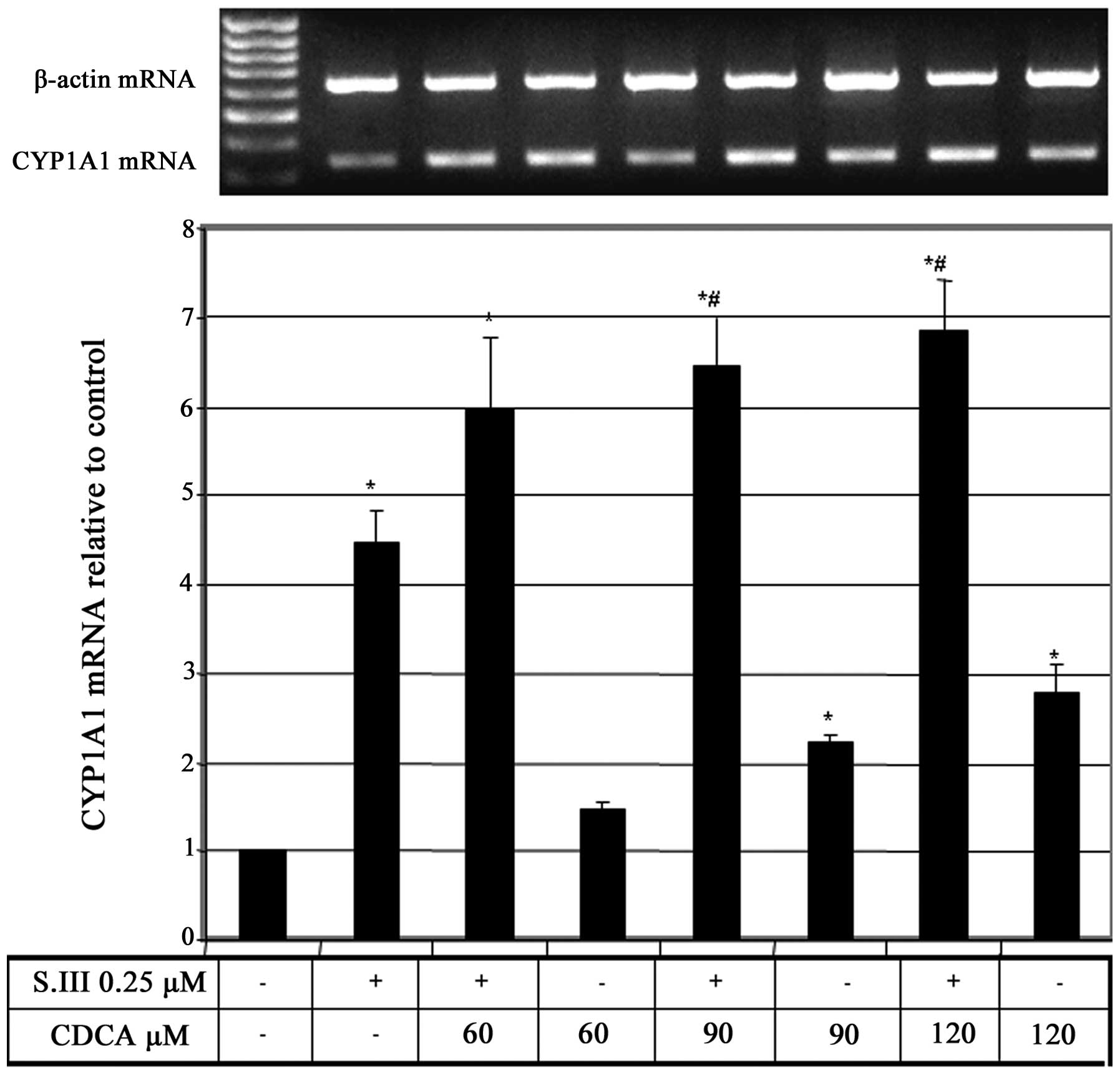
CDCA upregulates S.III-induced CYP1A1
mRNA expression
To further elucidate the effect of CDCA on CYP1A1
transcription, rat H4IIE cells were treated with different
concentrations of CDCA in addition to 0.25 µM of the CYP1A1 inducer
S.III, and the effect of this combination on CYP1A1 mRNA expression
was evaluated. Treatment of H4IIE cells with 0.25 µM S.III plus
increasing doses of CDCA caused upregulation of the S.III-induced
CYP1A1 mRNA in CDCA-dose-dependent manner (Fig. 2).
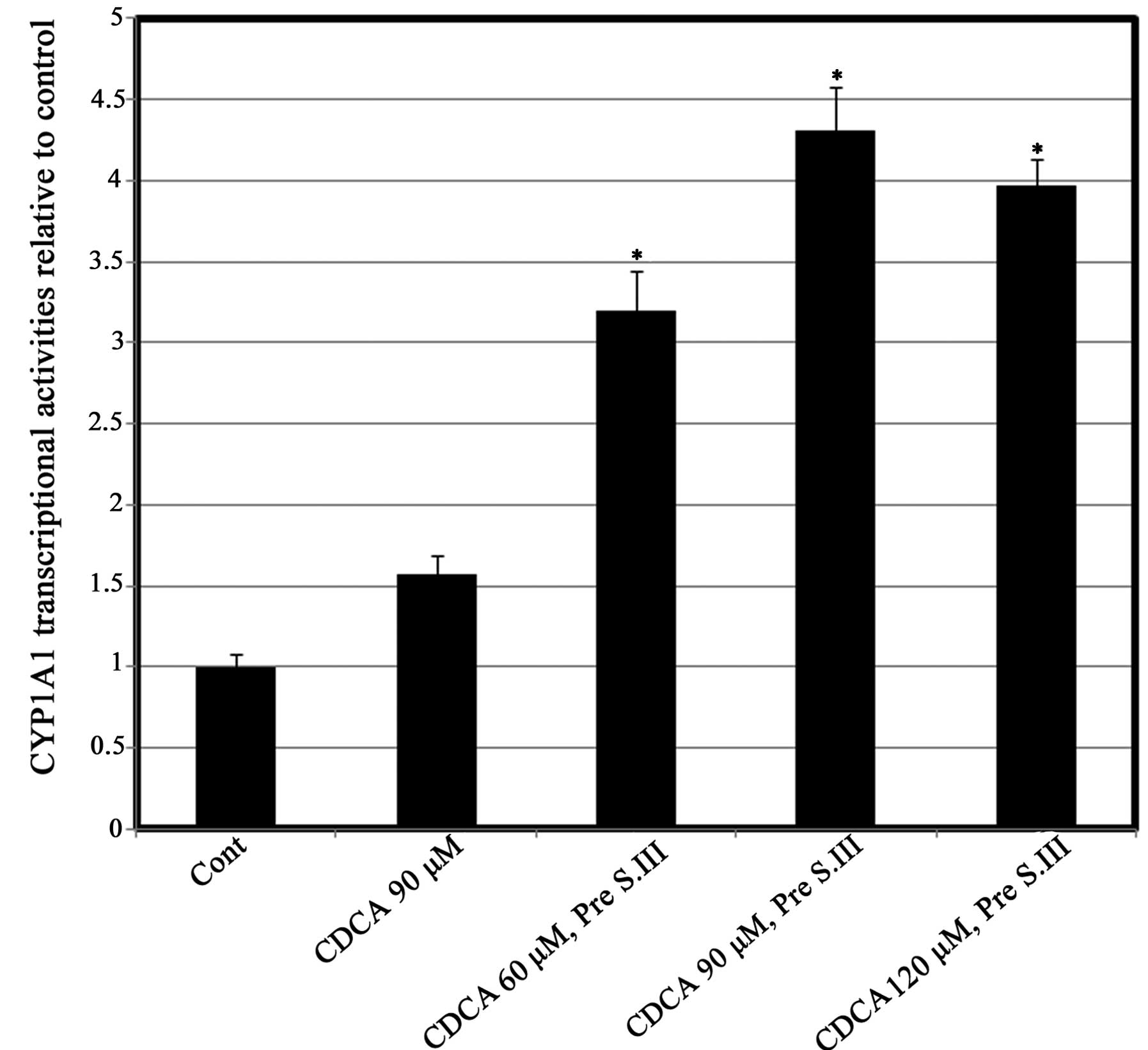
Effect of pre-exposure to CDCA on
CYP1A1 transcriptional activity and mRNA expression
To further determine the exact level of the effect
of CDCA on CYP1A1 transcription, HepG2 cells were treated with
different doses of CDCA for 8 h prior to treatment with 0.25 µM
S.III. The CYP1A1 reporter assay was then conducted. Pre-exposure
of HepG2 cells to different concentrations of CDCA prior to 0.25 µM
S.III resulted in upregulation of the S.III-induced transcriptional
activity of CYP1A1 in a CDAC-dose-dependent manner, with the
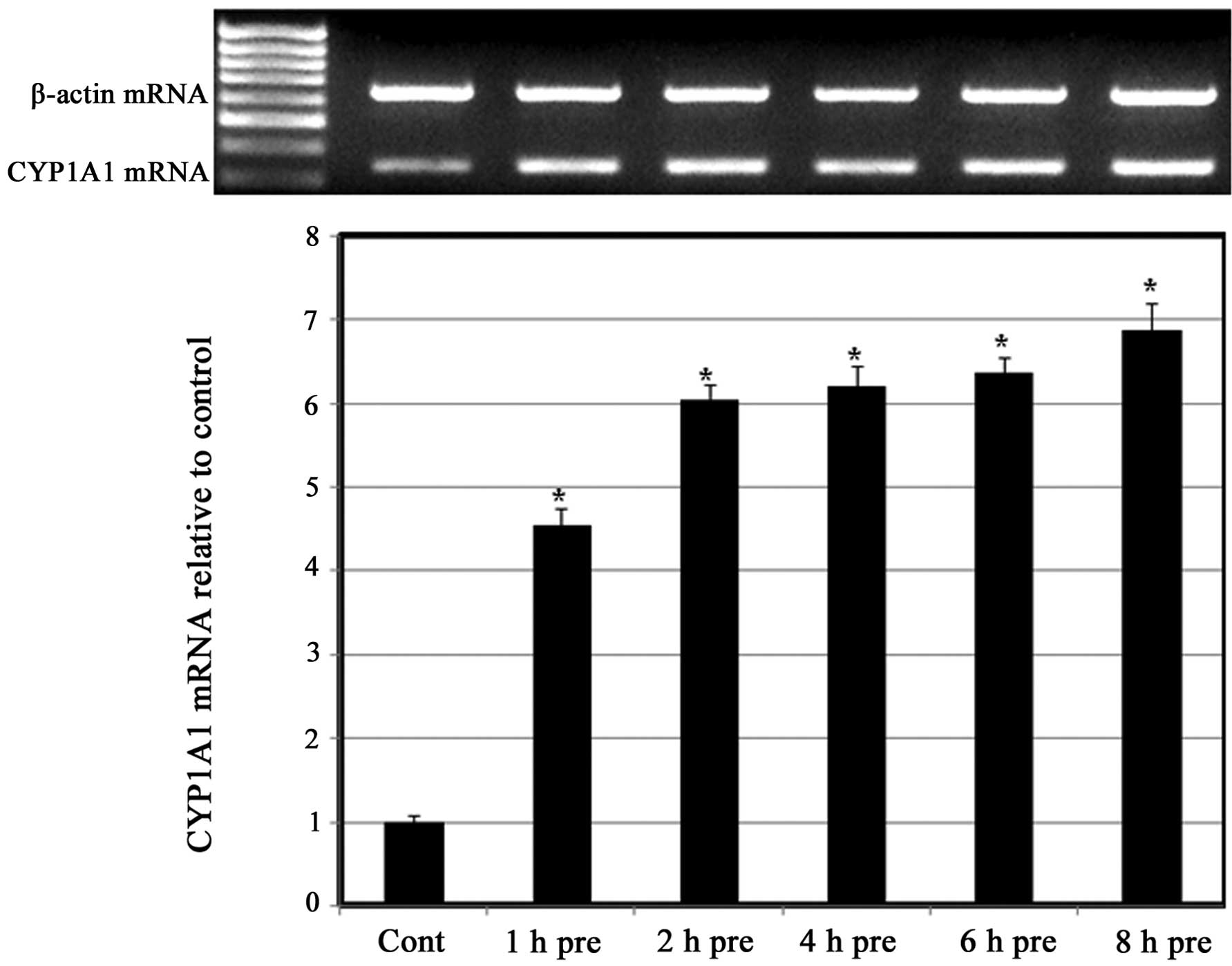
greatest inductive effect at 90 µM CDCA (Fig. 3). To further confirm the predisposing
effect of CDCA on the induction of CYP1A1 by S.III, H4IIE cells
were pre-exposed to CDCA 90 µM for different exposure times prior
to treatment with 0.25 µM S.III and CYP1A1 mRNA expression was
measured. As shown in (Fig. 4),
pre-exposure to 90 µM CDCA for between 1 and 8 h upregulated the
expression of CYP1A1 mRNA in a time-dependent manner.
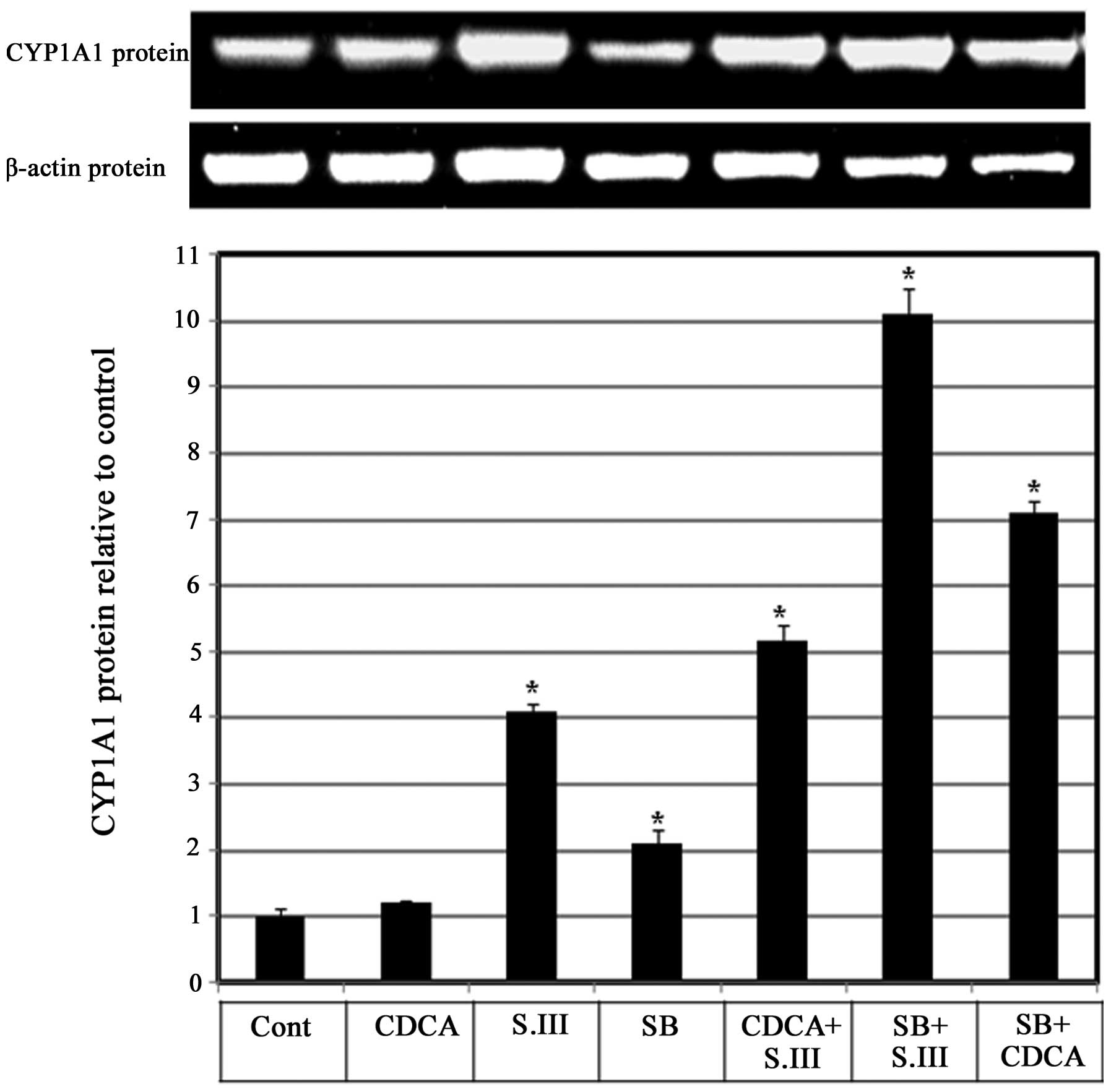
Role of p38 in the CDCA-induced
upregulation of CYP1A1 mRNA expression
Whether p38 is involved in the enhancing effect of
CDCA on CYP1A1 induction by S.III was investigated. H4IIE cells
were treated with the p38 inhibitor SB203580 alone or in
combination with 90 µM CDCA or 0.25 µM S.III, and CYP1A1 protein
expression levels were measured by western blotting. Treatment of
the cells with CDCA in combination with S.III induced a 5-fold
increase in CYP1A1 protein levels compared with the control, and
the levels were higher than those induced by S.III alone. Treatment
with SB203580 alone induced CYP1A1 protein expression to 2-fold the
control levels. When combined with S.III, SB treatment caused a
10-fold induction of CYP1A1 protein expression levels compared with
only 4-fold induction by S.III alone. When combined with CDCA, SB
treatment caused a 7-fold induction of CYP1A1 protein expression
compared with only 1.2-fold induction by CDCA alone (Fig. 5).
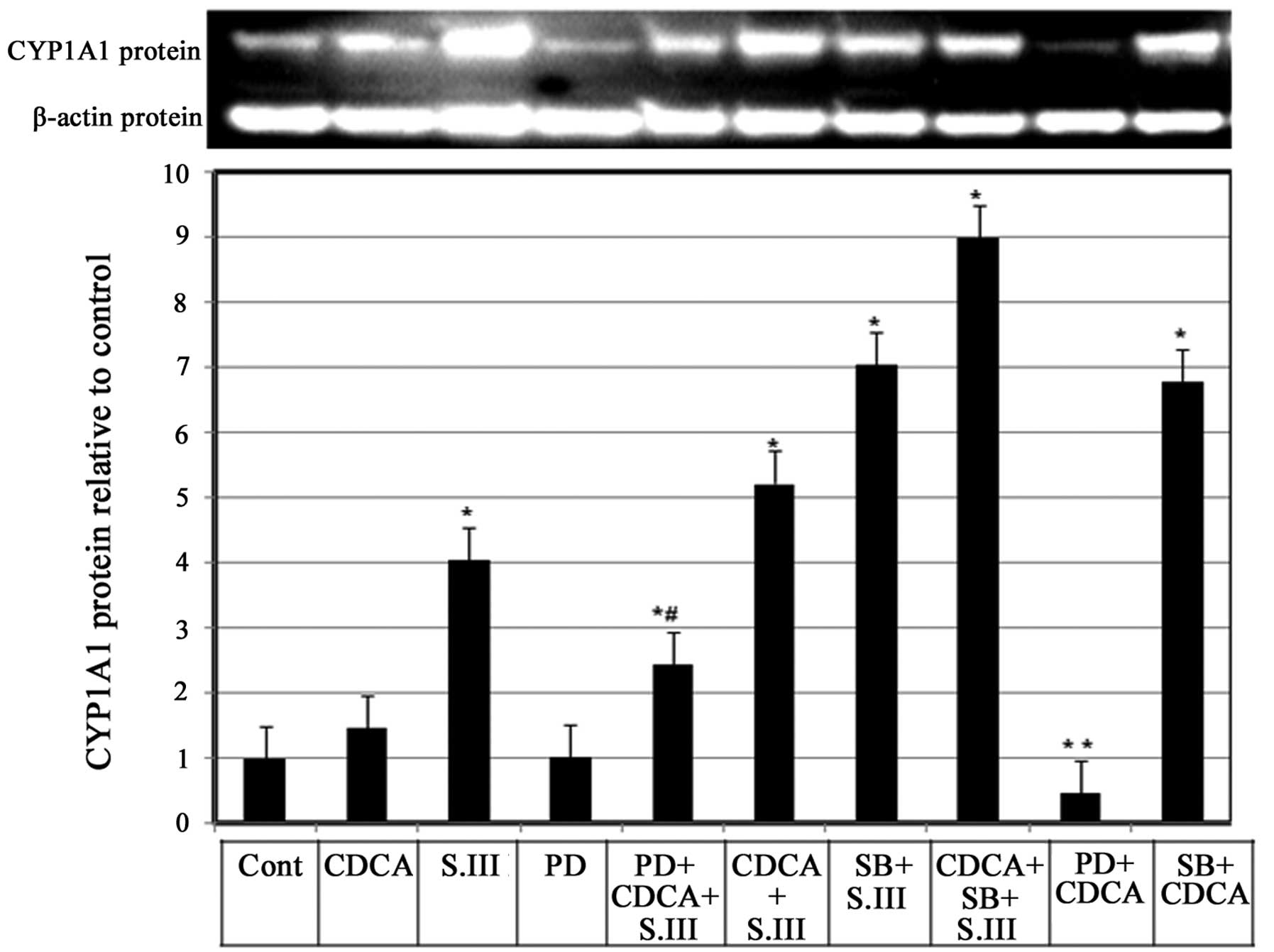
Role of MEK1/2 in the CDCA-induced
upregulation of CYP1A1 protein expression
Whether mitogen-activated protein kinase (MAPK) is
involved in the enhancing effect of CDCA on CYP1A1 induction by
S.III was investigated. H4IIE cells were treated with the MAPK
inhibitor PD98059 alone or in combination with 90 µM CDCA and/or
0.25 µM S.III and CYP1A1 protein expression levels were measured by
western blotting. Treatment of H4IIE cells with PD98059 alone did
not affect the basal CYP1A1 protein expression; however, when
PD98059 was combined with CDCA, a reduction of the CDCA-induced
CYP1A1 protein levels (Fig. 6, bar
9) to only 25% of the levels induced by CDCA alone (Fig. 6, bar 2). When PD98059 was combined
with CDCA and 0.25 µM S.III, CYP1A1 protein expression levels were
2.3-fold greater than control levels, which was lover than the
5.2-fold increase obtained with CDCA combined with 0.25 µM S.III
(Fig. 6, bars 5 and 6). In order to
compare the effects of MEK1/2 inhibitor PD98059 and p38 inhibitor
SB203580, H4IIE cells with were treated with SB203580 and S.III
0.25 µM (Fig. 6, column 7) and
SB203580 combined with S.III and CDCA (Fig. 6, column 8) or in combination with
CDCA only (Fig. 6, column 10). The
results demonstrate that the p38 inhibitor SB203580 upregulates the
induction of CYP1A1 protein by CDCA and/or S.III.
Discussion
The present study has demonstrated the ability of
the bile acid CDCA to upregulate the induction of CYP1A1 in
response to an inducer, a mechanism through which bile acids could
promote liver carcinogenesis. Liver cancer is one of the most
common forms of cancers worldwide, and both genetic and
environmental factors contribute to hepatocarcinogenesis (32). Cholestasis is associated with the
hepatic and systemic accumulation of toxic biliary compounds, such
as bile acids and bilirubin, and subsequent liver damage (33). Bile acids have been shown to promote
liver tumors in a hepatitis B virus transgenic mouse model
(34). The carcinogen-activating
enzyme CYP1A1 (22) is a key
participant in the bioactivation of numerous procarcinogenic
substances such as carcinogenic polycyclic hydrocarbons and
aromatic amines (35). S.III has
been reported to increase the expression levels of human CYP1A1
mRNA in HepG2 cells (36). The
S.III-induced CYP1A1 transcription in HepG2 cells (Fig. 1) and mRNA expression in H4IIE cells
(Fig. 2) were enhanced by the
concurrent administration of CDCA, which indicates a promoting
effect of CDCA on CYP1A1 induction in the liver.
An association between an increased concentration of
bile acids and carcinogenesis has been reported to be associated
with a marked activation of CYP1A1 and CYP1A2 genes in congenitally
jaundiced Gunn rats (30). However,
the mechanism of that association has not been elucidated. In the
present study, the upregulation of S.III-induced CYP1A1
transcription in HepG2 cells (Fig.
3) and S.III-induced CYP1A1 mRNA expression in H4IIE cells
(Fig. 4) following pre-exposure to
CDCA indicates the ability of CDCA to sensitize the cells to the
inductive effect of S.III on CYP1A1. Modulation of CYP enzymes
occurs through pre-transcriptional, post-transcriptional or
pretranslational mechanisms. Mercury has been demonstrated to
downregulate the expression of CYP1A1 at the transcriptional and
posttranslational levels in HepG2 cells (37). In the present study, the upregulation
of S.III-induced CYP1A1 transcription and mRNA due to CDCA
pre-exposure indicates that CDCA modulates CYP1A1 at the
pre-transcriptional level. This implies that CDCA sensitizes the
cells in some way so that their response to CYP1A1 inducers is
increased. Indeed, it has been demonstrated that the bile
acid-mediated induction of adhesion molecule expression occurs by
stimulation of NF-κB and p38 MAPK signaling pathways through an
elevation of reactive oxygen species (38). Therefore, H4IIE cells were exposed to
a selective inhibitor of p38 MAPK in the present study. SB203580
has been reported to suppress induction of the CYP1A1 gene by
2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) through a p38
MAPK-independent pathway in HepG2 human hepatoma cells (39). Pre-exposure of the H4IIE cells to
SB203580 did not suppress the induction of CYP1A1 protein by CDCA
or S.III, instead it upregulated CYP1A1 expression (Fig. 5), indicating the enhancement of
CYP1A1 induction by CDCA did not occur through the p38 MAPK
pathway. Moreover, SB203580 application alone induced CYP1A1
protein expression to increase to double the control level. This
ability of SB203580 to induce CYP1A1 expression is in accordance
with the previously reported ability of SB203580 to directly bind
and activate AhR, and induce CYP1A1 gene expression in an
AhR-dependent manner in human HepG2 cells and Hepa 1c1c7 cells
(40). Previously the bile acids
deoxycholic and chenodeoxycholic acid have been shown to activate
ERK-MEK in mouse hepatocytes (41).
Therefore, this mechanism was further tested using the potent MEK
inhibitor PD98059 (42). The ability
of PD98059 to suppress the CDCA-induced expression of CYP1A1 and
the CDCA-promoted S.III-induced expression of CYP1A1 (Fig. 6), indicates that CDCA may promote
CYP1A1 induction through sensitizing the cells by the modulation of
cell signaling, mainly through the upregulation of MEK1/2.
PAHs induce CYP1A via the AhR. When PAHs bind to the
AhR, they activate the transcription of CYP1A genes through the
dioxin-response element located in the enhancers of the genes
(20). The AhR is an intracellular
mediator of the xenobiotic signaling pathway and is complexed with
HSP90 (43) and hepatitis B virus X
protein associated protein 2 (XAP2) (44) in the cytoplasm. Xenobiotics such as
TCDD and 3-methylcholanthrene (3MC) bind to the AhR with an
extremely high affinity, and the receptor complex subsequently
translocates to the nucleus, where it switches its partner molecule
from HSP90 to the AhR nuclear translocater (Arnt) protein)
(44). In the nucleus, the formed
AhR/Arnt heterodimer binds to the XRE sequences, which are enhancer
DNA elements present in the 5-flanking region of target genes
CYP1A1/1A2, in addition to genes for a series of
xenobiotic-metabolizing enzymes, cell cycle and growth-related
factors (19). The ability of S.III
to induce CYP1A1 in rat liver has previously been demonstrated
(45). Sudan dyes possess a high
affinity for AhRs at the same cavity as other well-known AhR
ligands, such as dioxins and 3-MC (46). S.III, an AhR ligand, has the ability
to stimulate AhR nuclear translocation and binding to the XRE in
the promoter region of CYP1A1 and induce its transcription
(36). The ability of CDCA to
enhance CYP1A1 transcription and expression could be the key
mechanism underlying the reported effect of bile acids on the
promotion of liver tumors in a hepatitis B virus transgenic mouse
model (34).
CYP1A is known to cause the metabolic activation of
promutagens and procarcinogens (47). The enhancing effect of CDCA on the
induction of CYP1A1 by S.III may underlie cholestasis-associated
hepatic liver damage (34).
This study demonstrated the ability of CDCA to
enhance AhR ligand-induced CYP1A1 expression, which may explain the
hepatocarcinogenesis-prompting effect of cholestasis. It also
suggests that the CDCA-induced upregulation of CYP1A1 proceeds
through the CDCA-activated MEK1/2 pathway. Therefore, this pathway
that may be a therapeutic target for the prevention of the
promoting effect of CDCA on pro-carcinogenic activation.
Acknowledgements
The author would like to thank Professors Shoichi
Fujita and Mayumi Ishizuka of the Laboratory of Toxicology,
Department of Environmental Science, Graduate School of Veterinary
Medicine, Hokkaido University (Sapporo, Japan) for their generous
and continuous support. The author would like also to thank Dr
Mohamed Ahmed of the Department of Biotechnology, College of
Science, Taif University (Taif, Saudi Arabia) for his continuous
and kind help in the completion of this study.
Glossary
Abbreviations
Abbreviations:
|
ROS
|
reactive oxygen species
|
|
CYP1A1
|
cytochrome P450 1A1
|
|
PAHs
|
polycyclic aromatic hydrocarbons
|
|
HAAs
|
heterocyclic aromatic
amines/amides
|
|
AhR
|
aryl hydrocarbon receptor
|
|
CDCA
|
chenodeoxycholic acid
|
|
XRE
|
xenobiotic response element
|
|
DMSO
|
dimethylsulfoxide
|
|
HepG2
|
human hepatoma cells
|
|
XRE
|
xenobiotic response element
|
|
H4IIE
|
rat hepatoma cells
|
|
MAPK
|
mitogen-activated protein kinase
|
|
MEK
|
mitogen-activated protein kinase
kinase
|
|
p38
|
p38 mitogen-activated protein
kinase
|
|
XAP2
|
hepatitis B virus X protein associated
protein 2
|
References
|
1
|
Kuwahara A, Saito T and Kobayashi M: Bile
acids promote carcinogenesis in the remnant stomach of rats. J
Cancer Res Clin Oncol. 115:423–428. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ross RK, Hartnett NM, Bernstein L and
Henderson BE: Epidemiology of adenocarcinomas of the small
intestine: Is bile a small bowel carcinogen? Br J Cancer.
63:143–145. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Reveille RM, Van Stiegmann G and Everson
GT: Increased secondary bile acids in a choledochal cyst. Possible
role in biliary metaplasia and carcinoma. Gastroenterology.
99:525–527. 1990.PubMed/NCBI
|
|
4
|
Bayerdörffer E, Mannes GA, Ochsenkühn T,
Dirschedl P, Wiebecke B and Paumgartner G: Unconjugated secondary
bile acids in the serum of patients with colorectal adenomas. Gut.
36:268–273. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Martinez JD, Stratagoules ED, LaRue JM,
Powell AA, Gause PR, Craven MT, Payne CM, Powell MB, Gerner EW and
Earnest DL: Different bile acids exhibit distinct biological
effects: The tumor promoter deoxycholic acid induces apoptosis and
the chemopreventive agent ursodeoxycholic acid inhibits cell
proliferation. Nutr Cancer. 31:111–118. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Morvay K, Szentléleki K, Török G, Pintér
A, Börzsönyi M and Nawroth R: Effect of change of fecal bile acid
excretion achieved by operative procedures on
1,2-dimethylhydrazine-induced colon cancer in rats. Dis Colon
Rectum. 32:860–863. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qiao D, Gaitonde SV, Qi W and Martinez JD:
Deoxycholic acid suppresses p53 by stimulating proteasome-mediated
p53 protein degradation. Carcinogenesis. 22:957–964. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Trauner M, Meier PJ and Boyer JL:
Molecular pathogenesis of cholestasis. N Engl J Med. 339:1217–1227.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Sokol RJ, McKim JM Jr, Goff MC Jr, Ruyle
SZ, Devereaux MW, Han D, Packer L and Everson G: Vitamin E reduces
oxidant injury to mitochondria and the hepatotoxicity of
taurochenodeoxycholic acid in the rat. Gastroenterology.
114:164–174. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dueland S, Reichen J, Everson GT and Davis
RA: Regulation of cholesterol and bile acid homoeostasis in
bile-obstructed rats. Biochem J. 280:373–377. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Choi J and Ou JH: Mechanisms of liver
injury. III. Oxidative stress in the pathogenesis of hepatitis C
virus. Am J Physiol Gastrointest Liver Physiol. 290:G847–G851.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dröge W: Oxidative stress and aging. Adv
Exp Med Biol. 543:191–200. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chang GWM and Kam PCA: The physiological
and pharmacological roles of cytochrome P450 isoenzymes.
Anaesthesia. 54:42–50. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Renton KW: Alteration of drug
biotransformation and elimination during infection and
inflammation. Pharmacol Ther. 92:147–163. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lu AY: The 1996 Bernard B. Brodie lecture:
A journey in cytochrome P450 and drug metabolism research. Drug
Metab Dispos. 26:1168–1173. 1998.PubMed/NCBI
|
|
16
|
Conney AH: Induction of drug-metabolizing
enzymes: A path to the discovery of multiple cytochromes P450. Annu
Rev Pharmacol Toxicol. 43:1–30. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Coon MJ: Cytochrome P450: Nature's most
versatile biological catalyst. Annu Rev Pharmacol Toxicol. 45:1–25.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim D and Guengerich FP: Cytochrome P450
activation of arylamines and heterocyclic amines. Annu Rev
Pharmacol Toxicol. 45:27–49. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ma Q and Lu AY: CYP1A induction and human
risk assessment: An evolving tale of in vitro and in vivo studies.
Drug Metab Dispos. 35:1009–1016. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Proctor RN: Tobacco and the global lung
cancer epidemic. Nat Rev Cancer. 1:82–86. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Whitlock JP Jr, Chichester CH, Bedgood RM,
Okino ST, Ko HP, Ma Q, Dong L, Li H and Clarke-Katzenberg R:
Induction of drug-metabolizing enzymes by dioxin. Drug Metab Rev.
4:1107–1127. 1997. View Article : Google Scholar
|
|
22
|
Gillner M, Bergman J, Cambillau C and
Gustafsson JA: Interactions of rutaecarpine alkaloids with specific
binding sites for 2,3,7,8-tetrachlorodibenzo-p-dioxin in rat liver.
Carcinogenesis. 10:651–654. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Gillner M, Bergman J, Cambillau C and
Alexandersson M: Fernström B and Gustafsson JA: Interactions of
indolo[3,2-b]carbazoles and related polycyclic aromatic
hydrocarbons with specific binding sites for
2,3,7,8-tetrachlorodibenzo-p-dioxin in rat liver. Mol Pharmacol.
44:336–345. 1993.PubMed/NCBI
|
|
24
|
Ciolino HP, Daschner PJ, Wang TT and Yeh
GC: Effect of curcumin on the aryl hydrocarbon receptor and
cytochrome P450 1A1 in MCF-7 human breast carcinoma cells. Biochem
Pharmacol. 56:197–206. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gradelet S, Leclerc J, Siess MH and Astorg
PO: β-Apo-8-carotenal, but not β-carotene, is a strong inducer of
liver cytochromes P4501A1 and 1A2 in rat. Xenobiotica. 26:909–919.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Perdew GH and Babbs CF: Production of Ah
receptor ligands in rat fecal suspensions containing tryptophan or
indole-3-carbinol. Nutr Cancer. 16:209–218. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ciolino HP, Daschner PJ and Yeh GC:
Dietary flavonols quercetin and kaempferol are ligands of the aryl
hydrocarbon receptor that affect CYP1A1 transcription
differentially. Biochem J. 340:715–722. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Canivenc-Lavier MC, Vernevaut MF, Totis M,
Siess MH, Magdalou J and Suschetet M: Comparative effects of
flavonoids and model inducers on drug-metabolizing enzymes in rat
liver. Toxicology. 114:19–27. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Whitlock JP Jr: Induction of cytochrome
P4501A1. Annu Rev Pharmacol Toxicol. 39:103–125. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kapitulnik J and Gonzalez FJ: Marked
endogenous activation of the CYP1A1 and CYP1A2 genes in the
congenitally jaundiced Gunn rat. Mol Pharmacol. 43:722–725.
1993.PubMed/NCBI
|
|
31
|
Parkin DM: Global cancer statistics in the
year 2000. Lancet Oncol. 2:533–543. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zollner G, Marschall HU, Wagner M and
Trauner M: Role of nuclear receptors in the adaptive response to
bile acids and cholestasis: Pathogenetic and therapeutic
considerations. Mol Pharm. 3:231–251. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Barone M, Maiorano E, Ladisa R, Cuomo R,
Pece A, Berloco P, Caruso ML, Valentini AM, Iolascon A, Francavilla
A, et al: Influence of ursodeoxycholate-enriched diet on liver
tumor growth in HBV transgenic mice. Hepatology. 37:880–886. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang CL, Zeng T, Zhao XL and Xie KQ:
Garlic oil attenuated nitrosodiethylamine-induced
hepatocarcinogenesis by modulating the metabolic activation and
detoxification enzymes. Int J Biol Sci. 9:237–245. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ohno M, Ikenaka Y and Ishizuka M: Sudan
III dye strongly induces CYP1A1 mRNA expression in HepG2 cells. J
Biochem Mol Toxicol. 26:16–22. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Amara IE, Anwar-Mohamed A and El-Kadi AO:
Mercury modulates the CYP1A1 at transcriptional and
posttranslational levels in human hepatoma HepG2 cells. Toxicol
Lett. 199:225–233. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Qin P: Tang X, Elloso MM and Harnish DC:
Bile acids induce adhesion molecule expression in endothelial cells
through activation of reactive oxygen species, NF-kappaB, and p38.
Am J Physiol Heart Circ Physiol. 291:741–747. 2006. View Article : Google Scholar
|
|
38
|
Shibazaki M, Takeuchi T, Ahmed S and
Kikuchi H: Suppression by p38 MAP kinase inhibitors (pyridinyl
imidazole compounds) of Ah receptor target gene activation by
2,3,7,8-tetrachlorodibenzo-p-dioxin and the possible mechanism. J
Biol Chem. 279:3869–3876. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Korashy HM, Anwar-Mohamed A, Soshilov AA,
Denison MS and El-Kadi AO: The p38 MAPK inhibitor SB203580 induces
cytochrome P450 1A1 gene expression in murine and human hepatoma
cell lines through ligand-dependent aryl hydrocarbon receptor
activation. Chem Res Toxicol. 24:1540–1548. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Allen K, Kim ND, Moon JO and Copple BL:
Upregulation of early growth response factor-1 by bile acids
requires mitogen-activated protein kinase signaling. Toxicol Appl
Pharmacol. 243:63–67. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Reiners JJ Jr, Lee JY, Clift RE, Dudley DT
and Myrand SP: PD98059 is an equipotent antagonist of the aryl
hydrocarbon receptor and inhibitor of mitogen-activated protein
kinase kinase. Mol Pharmacol. 53:438–445. 1998.PubMed/NCBI
|
|
42
|
Perdew GH: Association of the Ah receptor
with the 90-kDa heat shock protein. J Biol Chem. 263:13802–13805.
1988.PubMed/NCBI
|
|
43
|
Meyer BK and Perdew GH: Characterization
of the AhR-hsp90-XAP2 core complex and the role of the
immunophilin-related protein XAP2 in AhR stabilization.
Biochemistry. 38:8907–8917. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Heid SE, Pollenz RS and Swanson HI: Role
of heat shock protein 90 dissociation in mediating agonist-induced
activation of the aryl hydrocarbon receptor. Mol Pharmacol.
57:82–92. 2000.PubMed/NCBI
|
|
45
|
Refat NA, Ibrahim ZS, Moustafa GG,
Sakamoto KQ, Ishizuka M and Fujita S: The induction of cytochrome
P450 1A1 by sudan dyes. J Biochem Mol Toxicol. 22:77–84. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lubet RA, Connolly G, Kouri RE, Nebert DW
and Bigelow SW: Biological effects of the Sudan dyes. Role of the
Ah cytosolic receptor. Biochem Pharmacol. 32:3053–3058. 1983.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Dewa Y, Nishimura J, Muguruma M, Jin M,
Kawai M, Saegusa Y, Okamura T, Umemura T and Mitsumori K:
Involvement of oxidative stress in hepatocellular tumor-promoting
activity of oxfendazole in rats. Arch Toxicol. 83:503–511. 2009.
View Article : Google Scholar : PubMed/NCBI
|