Introduction
Cerebral ischemia-reperfusion injury often occurs
following the restoration of blood flow in cerebral stroke
patients, and causes neurological deficits (1,2). Despite
the fact that great progress has been made over the years in
studies of cerebral ischemia-reperfusion injury, numerous unsolved
clinical problems remain. It is, therefore, of great importance to
explore novel drugs that could contribute to the prevention and/or
treatment of this condition.
Cerebral ischemia-reperfusion injury is closely
associated with increases in the extracellular glutamate
concentration, glial cell swelling and neuronal necrosis (3–5).
Glutamate transporter 1 (GLT-1) and glutamate aspartate transporter
(GLAST) are cation-dependent glutamate transporters, which not only
transfer glutamate into glial cells, but also critically maintain
appropriate glutamate gradients across intra- and extracellular
environments. Ischemia- and hypoxia-induced astrocyte swelling may
therefore be associated with GLT-1 and GLAST dysfunction, occurring
due to the loss of glutamate balance between the inside and outside
of the cell (6–8). Erythropoietin (EPO) reduces the
extracellular glutamate concentration, which is increased by
ischemia and hypoxia, and the glutamate-induced neural cell death
(9). Furthermore, EPO has been shown
to protect against cerebral ischemia-reperfusion injury in both
experimental and clinical research (10,11);
however, little is known about the involvement of GLT-1 and GLAST
in the protective effect of EPO against cerebral
ischemia-reperfusion injury.
We hypothesized that EPO would upregulate the GLT-1
and GLAST expression to promote the transport of glutamate into
astrocytes, thereby reducing the extracellular glutamate
concentration and excitatory glutamate neurotoxicity induced by
cerebral ischemia-reperfusion injury. The aim of the present study
was to explore the protective effect of EPO against rat cerebral
ischemia-reperfusion injury and its effect on the GLT-1 and GLAST
expression.
Materials and methods
Animals
This study was approved by the Ethics Review Board
of Tangdu Hospital (Xi'an, China; no. 2011036). A total of 140 male
Sprague Dawley rats with an average body weight of 320–350 g were
included in the present study and were randomly and evenly
allocated into the following four groups: Sham (control group;
neither occlusion nor reperfusion was performed), EPO-sham [neither
occlusion nor reperfusion was performed but the rats received an
intravenous injection of EPO (Sigma-Aldrich, St. Louis, MO, USA) at
a dosage of 5,000 U/kg body weight], middle cerebral artery
occlusion (MCAO; blood perfusion was restored 2 h after the MCAO)
and EPO-MCAO (the rats received an intravenous injection of EPO 15
min prior to the MCAO at a dosage of 5,000 U/kg body weight and
blood perfusion was restored 2 h later).
MCAO model
Rats were anesthetized by an intraperitoneal
injection of 10% chloral hydrate (350 mg/kg body weight) and fixed
on the operating table in a supine position. A 2-cm incision was
made in the middle of the neck and the right common carotid artery
(CCA) was isolated so that the proximal portion could be ligated.
The right external carotid artery (ECA) and internal carotid artery
(ICA) were then isolated and the pterygopalatine artery (PPA) was
isolated along the ICA without ligation. The ECA was ligated near
the branching point of the CCA. The distal part of the CCA was
nipped with a bulldog clamp. A loosely knotted silk thread was
placed at the distal part of the CCA and a tiny incision was made
at the bottom of the thread where a plug thread was inserted. The
silk thread was then tightened to prevent the plug thread from
slipping out and causing subsequent hemorrhage. The bulldog clamp
was loosened and the plug was threaded into the calvarium, along
the CCA and through the ICA. When a drag force was encountered, the
plug thread was slightly retracted. The insertion depth of the plug
thread was 17.5–40.5 mm away from the branching point so that it
was located at the start of the MCA and blocked blood flow. The
incision was then sewn up. Animals that woke up with the following
four signs were used for further study: i) Adduction of the right
forelimb with flexion when the tail was lifted up; ii) ipsilateral
Horner's sign; iii) crawling to the right in circles and iv)
falling down to the right upon standing. Animals in the control
group underwent isolations of the CCA, ICA and PPA without
ligation. Filament lamps were used to keep the animals at a normal
body temperature.
Assessment of functional neurological
deficit
Functional neurological deficit scores were obtained
from animals 24, 36 and 72 h after reperfusion (12) based on the following scoring system:
Score 0, no functional neurological defect; score 1, failure to
fully extend the right forepaw; score 2, crawling in circles to the
right; score 3, falling down to the right; score 4, failure to walk
spontaneously with loss of consciousness.
Measurement of rat cerebral infarct
volume
Infarct volume was evaluated 72 h after obtaining
the neurological scores. Following the sacrifice of the rats
through decapitation, the rat brains were rapidly removed, frozen
for 20 min at −20°C and then sectioned at 2-mm intervals from the
frontal to the occipital pole in the ischemic hemisphere,
generating four coronal slices. 2,3,5-Triphenyltetrazolium chloride
(TTC) (2%) was subsequently used to stain the slices for 30 min at
37°C, and the slices were fixed in 4% paraformaldehyde overnight.
The infarcted lesion area appeared white, in contrast to the
red-stained normal areas. A digital scanner was used to capture
images of the TTC-stained sections, which were then analyzed using
the Motic Images Advanced 3.2 image analysis system (Motic, Hong
Kong SAR, China). The degree of brain damage was assessed by
measuring brain edema/swelling (13). In order to calculate the infarct zone
(IZ) in each brain slice, the area of normal tissue of the
ipsilateral hemisphere was subtracted from the area of the
contralateral hemisphere; the total IZ, expressed as the percentage
of the damaged area relative to the total area of the contralateral
hemisphere, was obtained by adding the damaged area of the four
slices.
Detection of apoptotic neurons in rat
brains
Rat apoptotic neurons in the same infarct area of
the ipsilateral cortex were detected 72 h after obtaining the
neurological scores using terminal deoxynucleotidyl
transferase-mediated dUTP nick end labeling (TUNEL) fluorescence
double staining with a TUNEL kit from Sigma-Aldrich.
Measurement of the GLT-1 and GLAST
mRNA levels by reverse transcription-quantitative polymerase chain
reaction (RT-qPCR)
Rat brains from the control and experimental groups
were removed 72 h after obtaining the neurological scores for the
total RNA isolation. RNA was then reverse-transcribed into cDNA.
With the synthesized cDNA as a template, primer pairs
5′-AGTATGTGGCGGGCTGCTTCG-3 (upstream) and
5′-GGAAATAATGAGAGGGAGGAT-3 (downstream) for GLT-1;
5′-AACTTTGCCTGTTACCTTC-3′ (upstream) and 5′-CAGTCACAATCTGACCTCC-3
(downstream) for GLAST; and 5′-CGGCAGTCGCCTTGGACGTT-3 (upstream)
and 5′-GCCCTTTCCCATCTCAGCAGCC-3′ (downstream) for β-actin (Primer
Express, Applied Biosystems; Life Technologies, Carlsbad, CA, USA)]
were used for the qPCR using the ABI Prism® 7500 qPCR system
(Applied Biosystems). Amplification of β-actin with SYBR® Green
II-labeled primers was used as an internal control. The data were
analyzed using the ABI Prism 7500 SDS software, version 2.0.6
(Applied Biosystems). Quantification was performed using the
2−ΔΔct method.
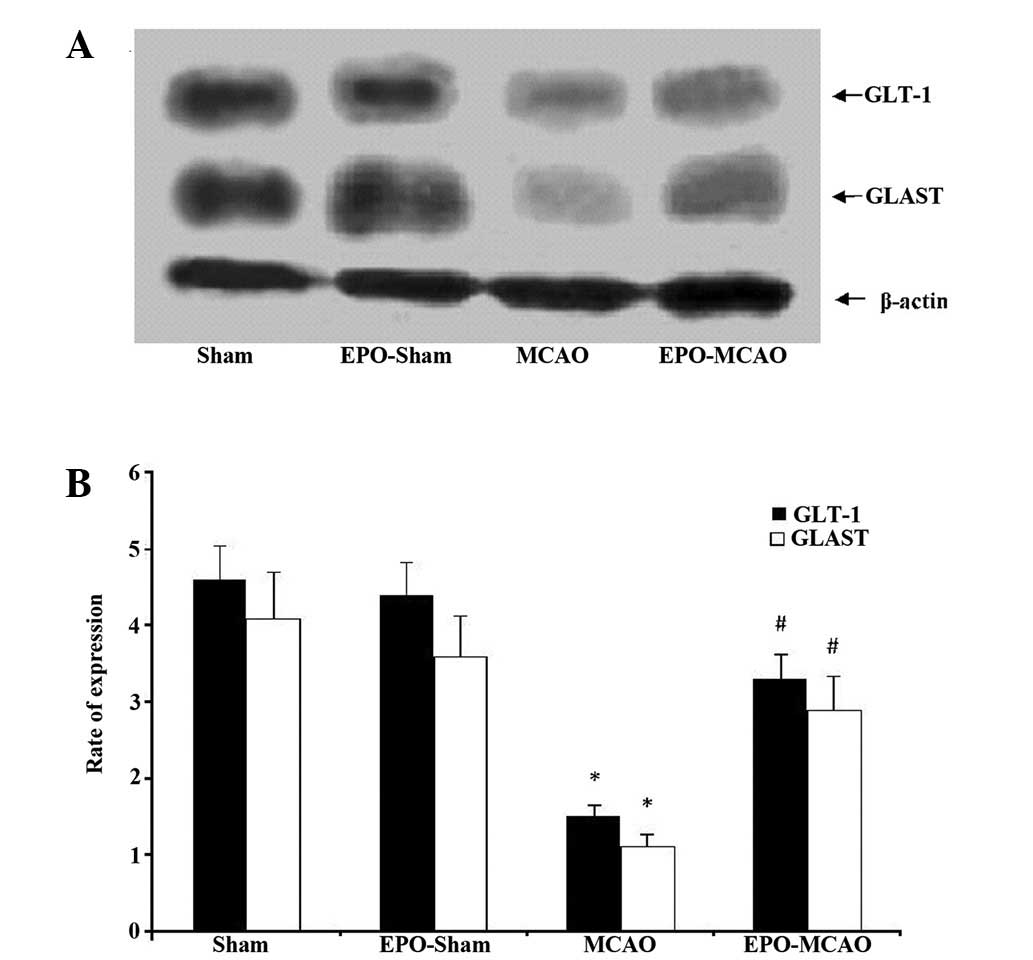
Detection of GLT-1 and GLAST protein
expression using western blotting
Rat brains were extracted 72 h after obtaining the
neurological scores, and then homogenized and lysed in
radioimmunoprecipitation assay buffer containing protease
inhibitors (Sigma-Aldrich). The total amount of protein in the
brain homogenate was quantified using Biuret colorimetry (Bio-Rad,
Hercules, CA, USA). Sodium dodecyl sulfate-polyacrylamide gel (10%)
was used to separate equal amounts (20 µg) of protein and
electrotransfer them onto the polyvinylidene fluoride membrane
(Hybond-C; GE Healthcare, Freiburg, Germany). The membrane was then
blocked with 5% bovine serum albumin for 1 h and incubated with
polyclonal anti-GLT-1 (#SEE380Mu) and anti-GLAST antibodies
(#SEE806Mu; dilution, 1:1,000; Sigma-Aldrich) for another 2 h at
room temperature. The membrane was washed three times in 1X
phosphate-buffered saline containing 0.5% Tween 20 (PBST) and then
incubated with horseradish peroxidase-labeled anti-mouse secondary
antibody (dilution, 1:1,000) for 1 h. The membrane was further
washed in PBST three times, for 10 min each time. Proteins were
visualized using an enhanced chemiluminescence protein detection
kit (Pierce Biotechnology, Inc., Rockford, IL, USA). The intensity
of the bands was analyzed using Scion Image software, version 4.0.3
(Scion Corp., Frederick, MD, USA). β-actin was used as an internal
control for quantifying the protein expression of GLT-1 and
GLAST.
Statistical analysis
The number of apoptotic neurons in the corresponding
ischemic and non-ischemic sites was counted using a microscope
(magnification, ×400) by an investigator blinded to the experiment
design. The data are expressed as the mean ± standard deviation and
were analyzed using analysis of variance followed by post hoc
analysis using the least significant difference test (SPSS 13.0;
SPSS Inc., Chicago, IL, USA). P<0.05 was considered to indicate
a statistically significant difference.
Results
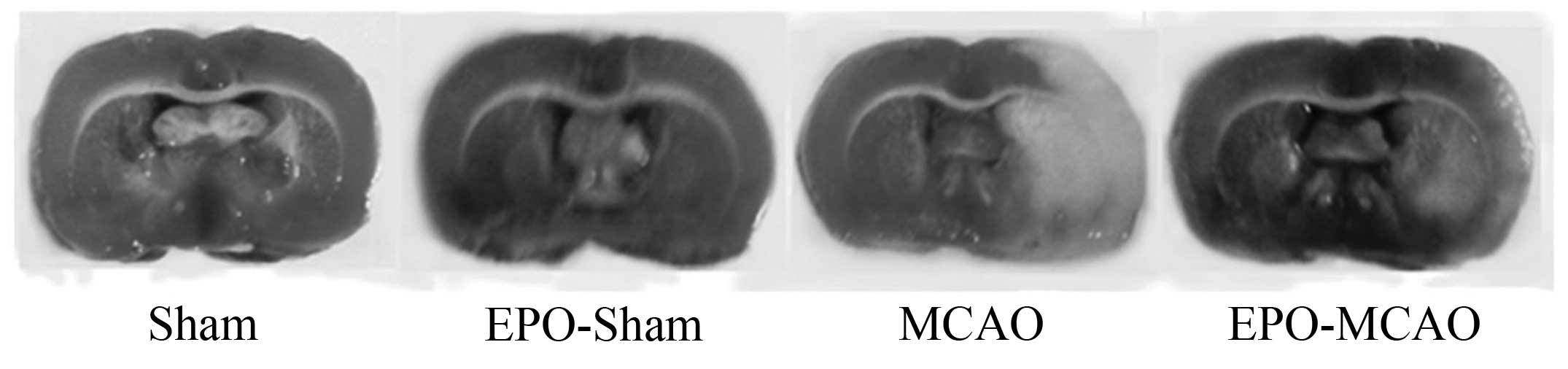
EPO preconditioning reduces the
infarct volume and improves the neurological function 24 h after
ischemia-reperfusion
The neurological deficit score and infarct volume in
the model groups 24, 36 and 72 h after the ischemia-reperfusion
injury were significantly higher than those in the control group
(P<0.01). Pretreatment with EPO significantly reduced the
neurological deficit score and infarct volume compared with the
MCAO group at the same time-point (P<0.01) (Fig. 1 and Table
I).
 | Table I.Neurological deficit score and infarct
volume of rats 24, 36 and 72 h after the ischemia-reperfusion
injury. |
Table I.
Neurological deficit score and infarct
volume of rats 24, 36 and 72 h after the ischemia-reperfusion
injury.
|
| Neurological
deficit |
|
|---|
|
|
|
|
|---|
| Group | n | 24 h | 36 h | 72 h | Infarct volume,
mm3 (n=5/group) |
|---|
| Sham | 36 | 0 | 0 | 0 | 0 |
| EPO-sham | 31 | 0 | 0 | 0 | 0 |
| MCAO | 39 | 3.5±0.4a | 3.1±0.5a | 2.6±0.8a | 166±21a |
| EPO-MCAO | 34 |
1.5±0.3a,b |
1.4±0.4a,b |
1.2±0.6a,b |
52±15a,b |
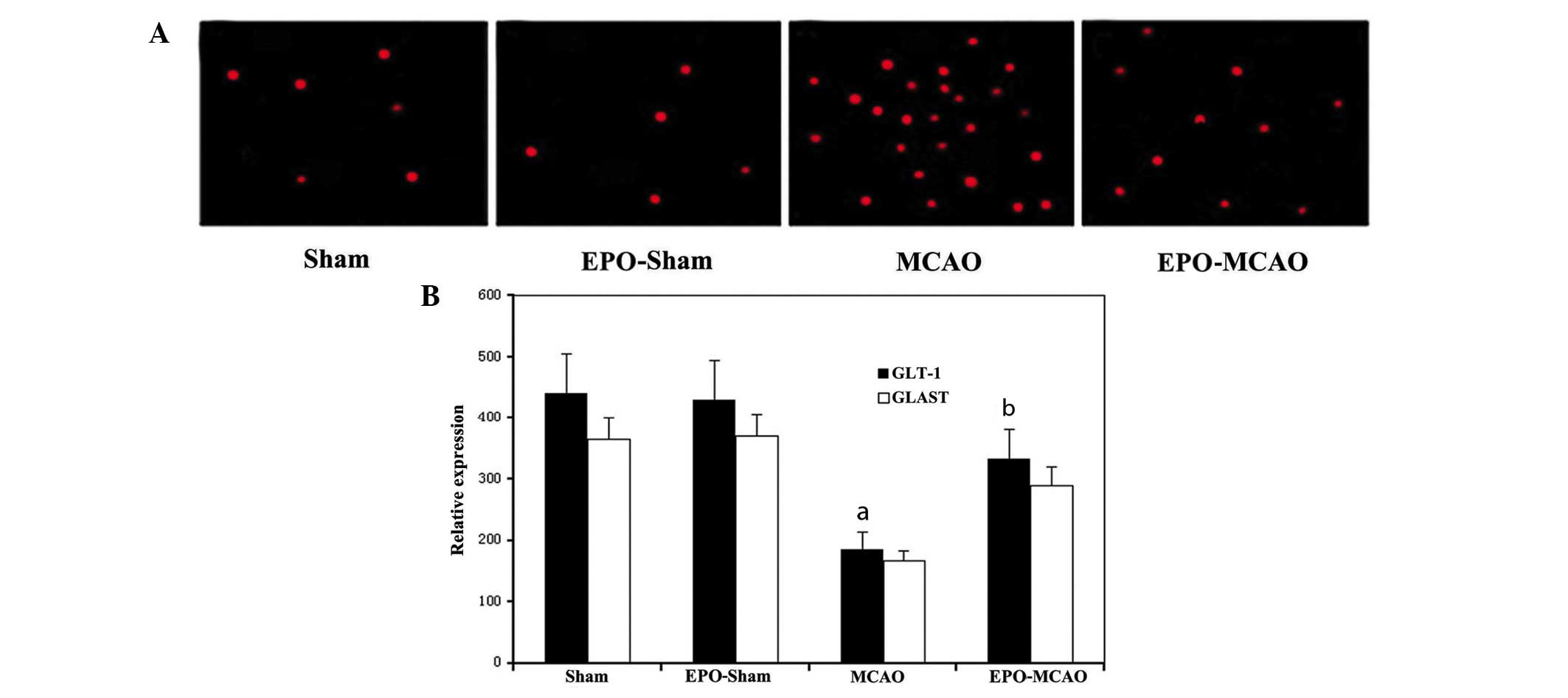
EPO preconditioning reduces the number
of apoptotic cells 72 h after ischemia-reperfusion
More TUNEL-positive apoptotic cells were observed in
the IZ of rats in the MCAO group than in the IZ of the control
group rats (P<0.05). The number of TUNEL-positive cells in the
EPO-MCAO group was significantly lower than that in the MCAO group
(P<0.05) (Fig. 2).
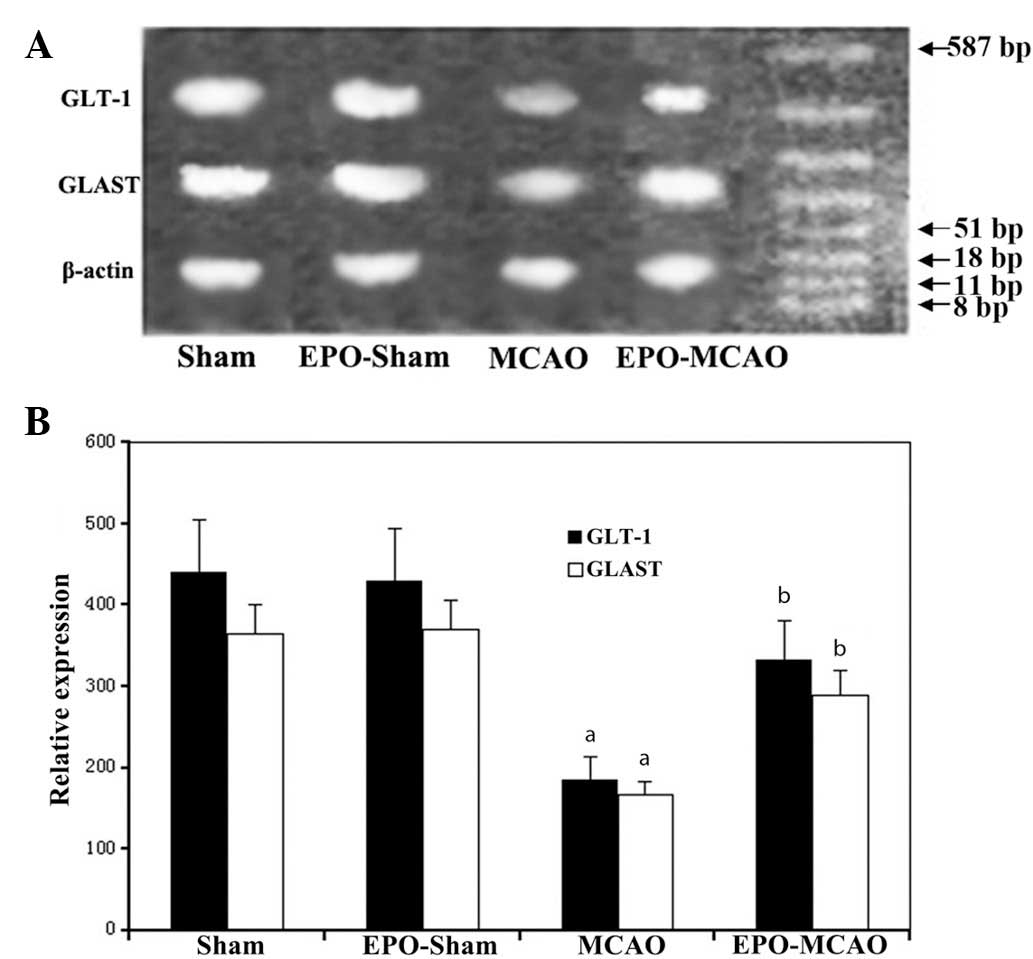
EPO-preconditioning increases the
expression levels of GLT-1 and GLAST mRNA and protein 24 h after
the ischemia
The expression levels of GLT-1 and GLAST mRNA and
protein in the MCAO group were significantly reduced 72 h after the
local ischemia compared with those in the control group
(P<0.01). By contrast, EPO-preconditioning significantly
increased the GLT-1 and GLAST mRNA and protein expression levels
above the levels of the MCAO group (P<0.01) (Figs. 3 and 4).
Discussion
Despite the restoration of blood flow following a
period of cerebral ischemia, full cerebral function is not restored
in certain stroke patients; instead, reperfusion exacerbates the
injury initially caused by the ischemia (14). At present, the most promising
approach for the prevention and treatment of ischemic cerebral
injury is to address the injury caused by reperfusion. Curative
approaches using medical, physical and biological methods have been
successful in animal experiments (15–17);
however, not one of these methods has been successfully
demonstrated in clinical practice. It is, therefore, of the utmost
importance to explore novel therapeutic strategies against
ischemia-reperfusion injury.
EPO has been shown to exert a protective effect on
cerebral ischemia-reperfusion injury. The protection mediated by
EPO may not be due to an increased erythropoietic activity; rather,
EPO is believed to inhibit the neuronal apoptosis induced by drugs
and traumatic brain injury (11). A
study based on liposome-mediated directional transport of EPO to
the central nervous system revealed that EPO could effectively
protect the central nervous system from ischemia-reperfusion injury
(18–20). Furthermore, it was found in the
present study that EPO pretreatment could significantly reduce the
neurological deficit score, infarct volume and number of apoptotic
cells following cerebral ischemia-reperfusion. The present results,
in combination with previous reports, demonstrated that EPO could
be a promising therapeutic drug against cerebral
ischemia-reperfusion injury.
Cerebral ischemia-reperfusion injury may involve
multiple pathological processes. The release of glutamate, an
excitatory neurotransmitter, during cerebral ischemia and hypoxia
leads to cerebral injury. Extensive evidence from animal and
clinical experiments suggest that high concentrations of
extracellular glutamate are closely associated with neural injury
(21). Excessive neurotoxicity
induced by extracellular glutamate can lead to neuronal death.
Under physiological conditions, glutamate is stored in presynaptic
vesicles and is released into the synaptic cleft when action
potentials arrive, allowing signals to be transmitted to
postsynaptic neurons. Generally, glutamate released into the
synaptic cleft is transported out of the extracellular space by
glutamate transporters (i.e. GLT-1 and GLAST) expressed on
astrocytes to reduce the noise-signal ratio and excitatory
neurotoxicity. Subsequent to entering the astrocytes, glutamate is
converted into glutamine and then transported back into neurons.
Under pathological conditions, the transporting activity or
expression of the GLT-1 and GLAST decreases, leading to an
increased extracellular glutamate concentration that can cause
excitatory neurotoxicity (21).
GLT-1 and GLAST are also expressed in other types of cells besides
astrocytes, including neurons, but only a small quantity of
glutamate is imported into these cells via these transporters
(22). Furthermore, glutamate can be
transported in a bidirectional manner under pathological
conditions, which can result in a net transport of glutamate from
the inside to the outside of the cell (23). Overexpression of GLT-1 can markedly
reduce hypoxia-induced cerebral injury (9). Specific overexpression of GLT-1 in
astrocytes under the control of a glial fibrillary acidic protein
promoter showed that GLT-1 protected the neurons in the brain
slices that were under oxygen and glucose deprivation conditions
(24). EPO has been found to exert
its neuroprotection by stimulating Janus kinase 2 phosphorylation,
which leads to the activation of signal transducer and activator of
transcription 5, Akt and nuclear factor-κB pathways and initiates
the expression of neuroprotective proteins, such as glutamate
transporters (e.g. GLT-1 and GLAST) (25–28). Of
note, the present results showed that cerebral ischemia-reperfusion
reduced the mRNA and the protein levels of GLT-1 and GLAST, whereas
EPO preconditioning significantly increased these levels following
ischemia-reperfusion. This suggests that the protection against
cerebral ischemia-reperfusion injury, as a result of EPO
preconditioning, may be partly due to the upregulation of GLT-1
and/or GLAST expression; however, since GLT-1 and GLAST are
expressed in both astrocytes and other cells, the cell type that is
involved in the protective role of EPO against cerebral
ischemia-reperfusion injury remains to be elucidated. In addition,
the occlusion in the right side of the brain in the MCAO model
could affect the blood brain barrier and allow EPO to also enter
the left side of the brain and upregulate the GLT-1 and GLAST
expression in general. Thus, the exact effects and mechanisms of
EPO on MCAO-induced alterations in GLT-1 and GLAST require further
study using the contralateral side of the brain as a control.
In conclusion, EPO pretreatment effectively relieved
acute cerebral ischemia-reperfusion injury by reducing the
neurological deficit score, infarct volume and number of apoptotic
cells. This finding may be associated with the increased expression
and transport activity of GLT-1 and GLAST. Further studies are
necessary in order to fully comprehend the detailed molecular
mechanisms underlying the protective effects of EPO preconditioning
against cerebral ischemia-reperfusion injury.
Glossary
Abbreviations
Abbreviations:
|
EPO
|
erythropoietin
|
|
GLT-1
|
glutamate transporter 1
|
|
GLAST
|
glutamate aspartate transporter
|
|
MCAO
|
middle cerebral artery occlusion
|
|
CCA
|
right common carotid artery
|
|
ECA
|
right external carotid artery
|
|
ICA
|
internal carotid artery
|
|
PPA
|
pterygopalatine artery
|
|
IZ
|
infarct zone
|
References
|
1
|
Koudstaal PJ, Stibbe J and Vermeulen M:
Fatal ischaemic brain oedema after early thrombolysis with tissue
plasminogen activator in acute stroke. BMJ. 297:1571–1574. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Clark RK, Lee EV, White RF, Jonak ZL,
Feuerstein GZ and Barone FC: Reperfusion following focal stroke
hastens inflammation and resolution of ischemic injured tissue.
Brain Res Bull. 35:387–392. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mitani A and Kataoka K: Critical levels of
extracellular glutamate mediating gerbil hippocampal delayed
neuronal death during hypothermia: Brain microdialysis study.
Neuroscience. 42:661–670. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Keelan J, Bates TE and Clark JB:
Differences in the amount of glutamate released by neonatal and
adult synaptosomes under conditions of in vitro
ischaemia/reperfusion. Biochem Soc Trans. 24:425S1996. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Semkova I, Schilling M, Henrich-Noack P,
Rami A and Krieglstein J: Clenbuterol protects mouse cerebral
cortex and rat hippocampus from ischemic damage and attenuates
glutamate neurotoxicity in cultured hippocampal neurons by
induction of NGF. Brain Res. 717:44–54. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Perego C, Vanoni C, Bossi M, Massari S,
Basudev H, Longhi R and Pietrini G: The GLT-1 and GLAST glutamate
transporters are expressed on morphologically distinct astrocytes
and regulated by neuronal activity in primary hippocampal
cocultures. J Neurochem. 75:1076–1084. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fukamachi S, Furuta A, Ikeda T, Ikenoue T,
Kaneoka T, Rothstein JD and Iwaki T: Altered expressions of
glutamate transporter subtypes in rat model of neonatal cerebral
hypoxia-ischemia. Brain Res Dev Brain Res. 132:131–139. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Han BC, Koh SB, Lee EY and Seong YH:
Regional difference of glutamate-induced swelling in cultured rat
brain astrocytes. Life Sci. 76:573–583. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Harvey BK, Airavaara M, Hinzman J, Wires
EM, Chiocco MJ, Howard DB, Shen H, Gerhardt G, Hoffer BJ and Wang
Y: Targeted over-expression of glutamate transporter 1 (GLT-1)
reduces ischemic brain injury in a rat model of stroke. PLoS One.
6:e221352011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Calapai G, Marciano MC, Corica F, Allegra
A, Parisi A, Frisina N, Caputi AP and Buemi M: Erythropoietin
protects against brain ischemic injury by inhibition of nitric
oxide formation. Eur J Pharmacol. 401:349–356. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tang Z, Sun X, Shi Q, Wang X, Xie Y, Huo
G, Zhou S and Liao Z: Beneficial effects of carbamylated
erythropoietin against oxygen-glucose
deprivation/reperfusion-induced astrocyte swelling, Proposed
molecular mechanisms of action. Neurosci Lett. 530:23–28. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Longa EZ, Weinstein PR, Carlson S and
Cummins R: Reversible middle cerebral artery occlusion without
craniectomy in rats. Stroke. 20:84–91. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shimakura A, Kamanaka Y, Ikeda Y, Kondo K,
Suzuki Y and Umemura K: Neutrophil elastase inhibition reduces
cerebral ischemic damage in the middle cerebral artery occlusion.
Brain Res. 858:55–60. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Patt A, Rutherford RB, Pearce WH and
Repine JE: Cerebral ischemia-reperfusion injury in the gerbil. J
Surg Res. 42:462–466. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tausompos C and Panoulis C: Tauοutouzas K
Zetaografos G and Papalois A: The effect of the antioxidant drug
“U-74389G” on oophoritis during ischemia reperfusion injury in
rats. Antiinflamm Antiallergy Agents Med Chem. 13:103–107. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lehrke M and Lebherz C: AAV-mediated gene
therapy for atherosclerosis. Curr Atheroscler Rep. 16:4342014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schlegel A, Kron P, Graf R, Dutkowski P
and Clavien PA: Warm vs. Histopathology. J Hepatol. 61:1267–1275.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Brines ML, Ghezzi P, Keenan S, Agnello D,
de Lanerolle NC, Cerami C, Itri LM and Cerami A: Erythropoietin
crosses the blood-brain barrier to protect against experimental
brain injury. Proc Natl Acad Sci USA. 97:10526–10531. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shin T, Ahn M, Moon C and Kim S:
Erythropoietin and autoimmune neuroinflammation, Lessons from
experimental autoimmune encephalomyelitis and experimental
autoimmune neuritis. Anat Cell Biol. 45:215–220. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
McCook O, Georgieff M, Scheuerle A, Möller
P, Thiemermann C and Radermacher P: Erythropoietin in the
critically ill, Do we ask the right questions? Crit Care.
16:3192012. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Grewer C, Gameiro A, Zhang Z, Tao Z,
Braams S and Rauen T: Glutamate forward and reverse transport, From
molecular mechanism to transporter-mediated release after ischemia.
IUBMB Life. 60:609–619. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Akbar MT, Torp R, Danbolt NC, Levy LM,
Meldrum BS and Ottersen OP: Expression of glial glutamate
transporters GLT-1 and GLAST is unchanged in the hippocampus in
fully kindled rats. Neuroscience. 78:351–359. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Persson M and Rönnbäck L: Microglial
self-defence mediated through GLT-1 and glutathione. Amino Acids.
42:207–219. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Romera C, Hurtado O, Mallolas J, Pereira
MP, Morales JR, Romera A, Serena J, Vivancos J, Nombela F, Lorenzo
P, et al: Ischemic preconditioning reveals that GLT1/EAAT2
glutamate transporter is a novel PPARgamma target gene involved in
neuroprotection. J Cereb Blood Flow Metab. 27:1327–1338. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Digicaylioglu M and Lipton SA:
Erythropoietin-mediated neuroprotection involves cross-talk between
Jak2 and NF-kappaB signalling cascades. Nature. 412:641–647. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Um M and Lodish HF: Antiapoptotic effects
of erythropoietin in differentiated neuroblastoma SH-SY5Y cells
require activation of both the STAT5 and AKT signaling pathways. J
Biol Chem. 281:5648–5656. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Byts N, Samoylenko A, Fasshauer T,
Ivanisevic M, Hennighausen L, Ehrenreich H and Sirén AL: Essential
role for Stat5 in the neurotrophic but not in the neuroprotective
effect of erythropoietin. Cell Death Differ. 15:783–792. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hosseinzadeh Z, Bhavsar SK, Sopjani M,
Alesutan I, Saxena A, Dërmaku-Sopjani M and Lang F: Regulation of
the glutamate transporters by JAK2. Cell Physiol Biochem.
28:693–702. 2011. View Article : Google Scholar : PubMed/NCBI
|