Introduction
Ammonia is produced in the gastrointestinal tract in
two ways: Colonic bacteria produce ammonia through the catabolism
of nitrogenous waste from dietary protein intake, and skeletal
muscle degrades and produces ammonia during seizures and intense
exercise (1). Ammonia is primarily
hepatically eliminated through the urea cycle, during which it is
converted to glutamine and renally excreted (2). Normal blood ammonia (BA) is 35 mmol/l,
and any increase in this concentration is typically due to its
increased production or decreased elimination.
Hepatic failure (HF), which is a serious disease
with a high mortality rate (70–80%), is predominantly caused by
hepatitis B and C viral infections (3,4).
Existing treatment strategies for hepatic failure include
artificial liver supporting systems and liver transplantation
(5). During HF, normal metabolism of
ammonia is blocked, which leads to an accumulation of ammonia in
the circulation. This results in an increase in hepatocyte necrosis
and apoptosis (6–8). However, it is uncertain if
hyperammonemia adversely affects residual hepatocyte function in HF
patients, which would cause a ‘vicious cycle’ by further
aggravating the hepatic injury (9).
In previous studies, a ‘two-hit theory’ was used to administer
antiviral therapy and liver function maintenance using
l-ornithine-l-aspartate to slow progression of hepatitis B
virus-related acute-on-chronic HF (10–13).
Therefore, the present study hypothesized that hepatic injury
induced by hyperammonemia may be activated via apoptotic signaling
pathways. In order to determine this, a rat model of hyperammonemia
was established using intragastric administration of ammonium
chloride solution, according to a previous study (14), and cell proliferation and apoptosis
of rat hepatic cells were observed.
Materials and methods
Animals, experimental design and
specimen collection
Male Sprague-Dawley (SD) rats (n=24; weight, 200±25
g; age, 6 weeks) were obtained from the Animal Experiment Center of
the Henan Province (Zhengzhou, China). The rats were maintained at
22±2°C under a 12-h light/dark cycle, with ad libitum access
to water. The rats were fed experimental particle feed provided by
the Animal Experimental Center of the Henan Province. The animal
protocol was approved by the Animal Care and Use Committee of the
Zhengzhou University (Zhengzhou, China; approval no. ZZ-83042-9).
Animals were cared for according to the institutional guidelines of
Zhengzhou University.
Experimental groups
The SD rats (n=24) were randomly divided into two
groups: i) The hyperammonemic group, in which hyperammonemia was
induced by treating the rats with 10% ammonium chloride solution
(10 ml/kg; Sangon Biotech Co., Ltd., Shanghai, China) via
intragastric administration twice daily for 30 days; and ii) the
control group, in which the rats were treated with saline (10
ml/kg) via the same route of administration and dosing regimen as
the hyperammonemic group. After 30 days, the rats were sacrificed
by cervical dislocation and blood and liver tissue samples were
collected, according to methods outlined in previous studies
(14,15).
Evaluating serum alanine transaminase
(ALT), aspartate transaminase (AST), direct bilirubin (DBil) and
blood ammonia (BA) concentration levels
Serum ALT, AST and DBil concentration levels were
assayed using the ALT Assay, AST Assay and Total Bilirubin
enzyme-linked immunosorbent assay kits obtained from Suzhou Calvin
Biotechnology (Suzhou, China). BA was measured using a standard
clinical automatic analyzer (AA-4120 Ammonia Checker II; Kyoto
Daiichi Kagaku Co., Ltd., Kyoto, Japan).
Histological analysis
Hepatic tissue blocks obtained from the rats in all
groups were dehydrated, embedded in paraffin, sectioned (5 µm) and
stained with hematoxylin and eosin (Nanjing Jiancheng
Bioengineering Institute, Nanjing, China). The histological
analysis of the tissue sections was conducted by two independent
pathologists who were blind to the study design and the specimen
identities. Sections were observed under a light microscope
(LSM-510; Carl Zeiss AG, Oberkochen, Germany).
Assessment of apoptosis
To quantify apoptosis, a terminal deoxynucleotidyl
transferase dUTP nick end-labeling (TUNEL) assay was performed on
paraffin-embedded tissue sections from each rat (16). Hepatic tissue sections were subjected
to a TUNEL assay, and analyzed with an In Situ Cell Death
Detection kit, POD (Roche Diagnostics, Shanghai, China). Briefly,
hepatic tissue sections were dewaxed, rehydrated and then incubated
with 50 µl TUNEL reaction mixture at room temperature in the dark
for 1 h. After washing three times with phosphate-buffered saline
(PBS), the tissue sections were counterstained with 0.0002%
4,6-diamidino-2-phenylindole (Sangon Biotech, Co., Ltd.). Positive
cells were scored under the Nikon Eclipse Ti-U fluorescence
microscope (Nikon Corporation, Tokyo, Japan). This was performed by
counting cells under low magnification (×100) in 10 separate
arbitrary fields in each tissue section. Cells with a brown nuclei
were interpreted as being positively apoptotic. Student's t-tests
were used to calculate any statistical significance. The apoptotic
index was expressed as a percentage of the TUNEL-positive
cells.
Cell cycle analysis
Cells were collected from hepatic tissue in a
humidified chamber at room temperature, and the cell concentration
was adjusted to 1×106 cells/ml. Cells were washed with
cold PBS and fixed in 70% cold ethanol (Tianjin Siyou Science and
Technology Development Co., Ltd., Tianjin, China) overnight at 4°C.
A fluorochrome solution (Sangon Biotech, Co., Ltd.), containing 3.4
mmol/l sodium citrate ion, 1% Triton X-100, 20 µg/ml RNase A and 50
µg/ml propidium iodide, was added and the mixture was incubated in
the dark at room temperature for 0.5 h (17). Cell cycle distribution was measured
using flow cytometry (FCM; Sysmex Partec GmbH, Münster, Germany).
FCM analysis was performed using CellQuest software, version 3.0
(BD Biosciences, Franklin Lakes, NJ, USA).
Immunohistochemistry (IHC)
To measure hepatic cyclin A and D1 expression, a
standard IHC protocol was followed to stain the rat liver tissue
samples (18). Briefly, 5-µm
paraffin-embedded tissue samples were de-paraffinized with xylene
(Sangon Biotech, Co., Ltd.), and endogenous peroxidase activity was
quenched with 3% H2O2 in methanol (Sangon
Biotech, Co., Ltd.) for 30 min in the dark. Tissue samples were
dehydrated through an alcohol gradient and subjected to antigen
retrieval using 10 mM sodium citrate (Sangon Biotech, Co., Ltd.).
Rabbit anti-rat cyclin A (1:500; sc-751), cyclin D1 (1:500; sc-753)
and β-actin (1:5,000; sc-130656) polyclonal antibodies (all Santa
Cruz Biotechnology, Inc., Dallas, TX, USA) were used as a primary
antibodies. Sections were then treated with a goat anti-rabbit
biotin-conjugated secondary antibody (1:1,000; sc-2004; Santa Cruz
Biotechnology, Inc.) followed by streptavidin (Sangon Biotech, Co.,
Ltd.). After washing three times with PBS, the tissue sections were
incubated with 3,3′-diaminobenzidine tetrahydrochloride
(Sigma-Aldrich, St-Louis, MO, USA) for 30 min, and immediately
washed under tap water following color development. Slides were
then counterstained with hematoxylin. Slides were mounted with
dibutyl phthalate xylene and were then observed under a light
microscope (LSM-510; Carl Zeiss AG). The status of nuclear
expression of cyclin A and D1 were determined using integrated
optical density, as measured with Biosens Digital Imaging System
software, version 1.6 (Shanghai Biotechnology Corporation,
Shanghai, China).
Semi-quantitative reverse
transcription-polymerase chain reaction (RT-PCR) assay
Three preliminary experiments were conducted prior
to semi-quantitative RT-PCR, in order to validate the linearity of
PCR product accumulation with increasing number of PCR cycles.
Briefly, aliquots containing fixed amounts of cDNA mixture were
subjected to amplification for varying numbers of PCR cycles
(between 20 and 40 cycles), using the S1000™ Thermal Cycler
(Bio-Rad Laboratories Co., Ltd, California, USA). Total RNA was
extracted from liver tissues using TRIzol® reagent (Thermo Fisher
Scientific, Inc., Waltham, MA, USA), according to the
manufacturer's protocol. Purified RNA treated with DNAase
(TURBODNA-free™ kit; Thermo Fisher Scientific, Inc.) was
reverse-transcribed into cDNA using random hexamers (Sangon
Biotech, Co., Ltd.) and the SuperScript First-Strand Synthesis
System for RT-PCR kit (Thermo Fisher Scientific, Inc.). PCR was
conducted using the S1000™ Thermal Cycler and the following cycling
conditions: 90°C denaturation for 1 min, followed by 30 cycles of
denaturation at 90°C for 60 sec, annealing at 60°C for 60 sec and
extension at 72°C for 60 sec, and a final extension step of 72°C
for 10 min. The PCR primers were as follows: Sense,
5′-CAAAGTGTGCCGTTGTCTCTT-3′ and antisense,
5′-ATCTGCGCTTGGAGTGATAGA-3′ for cyclin A; sense,
5′-CCACGATTTCATCGAACACTT-3′ and antisense for cyclin D1,
5′-CTCTGGAAAGAAAGTGCGTTG-3′; and sense, 5′-CAGTGCCAGCCTCGTCTCAT-3′
and antisense, 5′-AGGGGCCATCCACAGTCTTC-3′ for
glyceraldehyde-3-phosphate dehydrogenase (Sangon Biotech, Co.,
Ltd.). A negative control and an RT-minus control were used in
order to verify the results of the first strand cDNA synthesis
step. Experiments were performed in triplicate. Prior to
amplification of each gene, normalization was carried out with the
endogenous control gene, GADPH. All PCR products and the 100 bp DNA
ladder (cat no. MD109; Tiangen Biotech Co., Ltd., Beijing, China)
were separated by 2% agarose gel electrophoresis (Sangon Biotech,
Co., Ltd.). The amplified DNA bands on the agarose gels were
detected using ethidium bromide staining (Sangon Biotech, Co.,
Ltd.) and were quantified using the ImageQuant™ LAS 500 CCD imager
(GE Healthcare Life Sciences, Chalfont, UK).
Preparation of lysates and western
blotting
Hepatic tissues were frozen on dry ice upon
harvesting. Homogenates were sonicated in lysis buffer [150 mM
NaCl, 1.0% IGEPAL, 0.5% DOC, 0.1% SDS, 50 mM Tris (pH 8.0); Sangon
Biotech, Co., Ltd.] and centrifuged (Heraeus Biofuge Stratos
Centrifuge; Thermo Fisher Scientific, Inc.) at 21,890 × g and a
temperature of 4°C for 30 min. Supernatants were boiled in Laemmli
sample buffer (Santa Cruz Biotechnology, Inc.) for western blotting
at room temperature. The antibodies used were as follows:
Anti-cyclin D1 (cat no. H-295; dilution, 1:1,000; Santa Cruz
Biotechnology, Inc.) and anti-cyclin A (cat no. H-432; dilution,
1:1,000; Santa Cruz Biotechnology, Inc.). Western blots were
quantified using ImageJ software, version 1.48 (National Institutes
of Health, Bethesda, MD, USA).
Statistical analysis
Statistical analyses were conducted using SPSS
software, version 19.0 (IBM SPSS, Armonk, NY, USA). Data are
depicted as the mean ± standard deviation. Student's t-test was
used to examine the statistical significance between groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
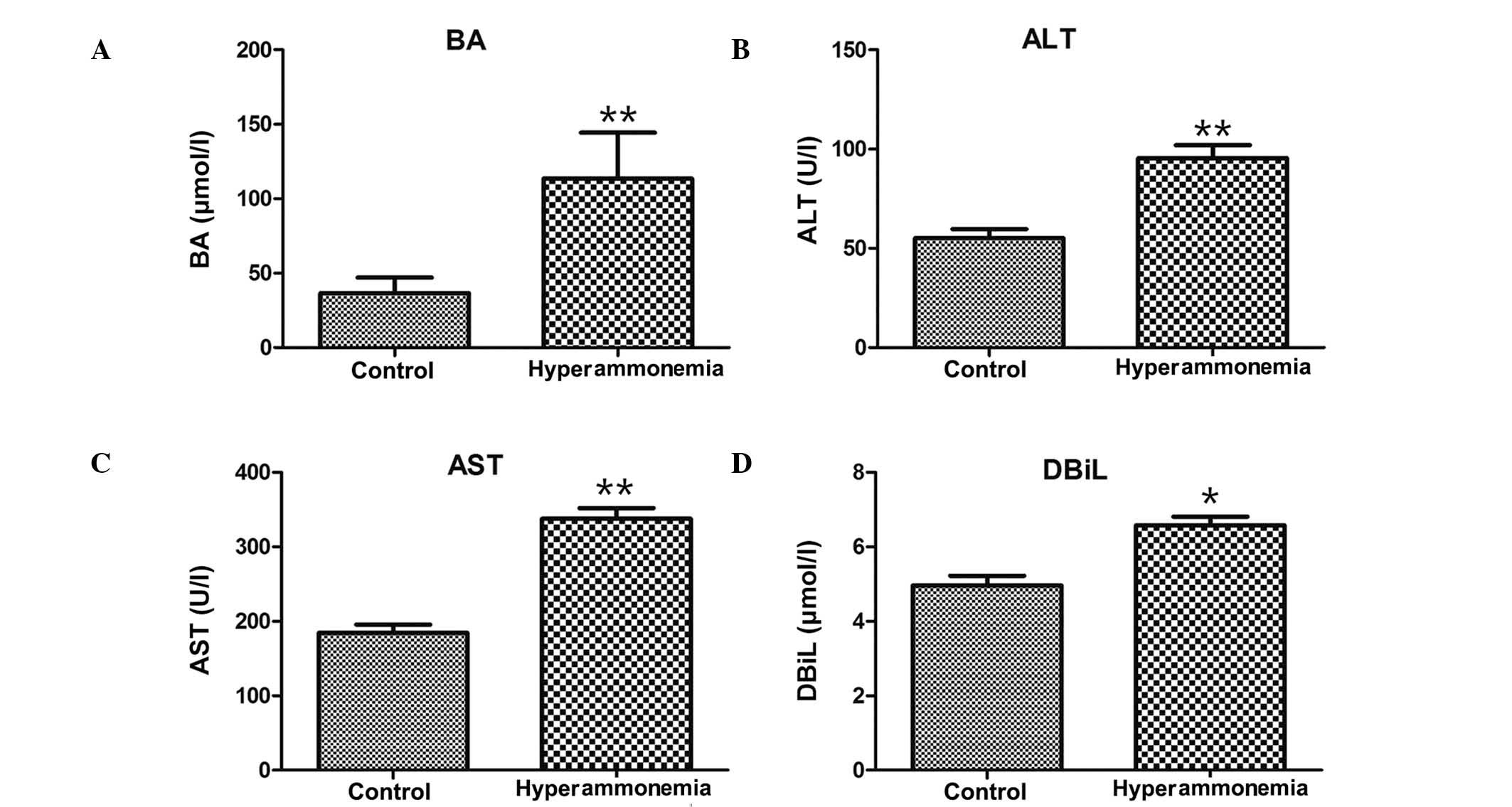
Hyperammonemia induces an increase in
the concentrations of BA, ALT, AST and DBil
The concentrations of BA, ALT, AST and DBit were
significantly increased in the hyperammonemic rats, as compared
with the controls (P<0.05; Fig.
1A–D), which indicated the occurrence of hepatic injury.
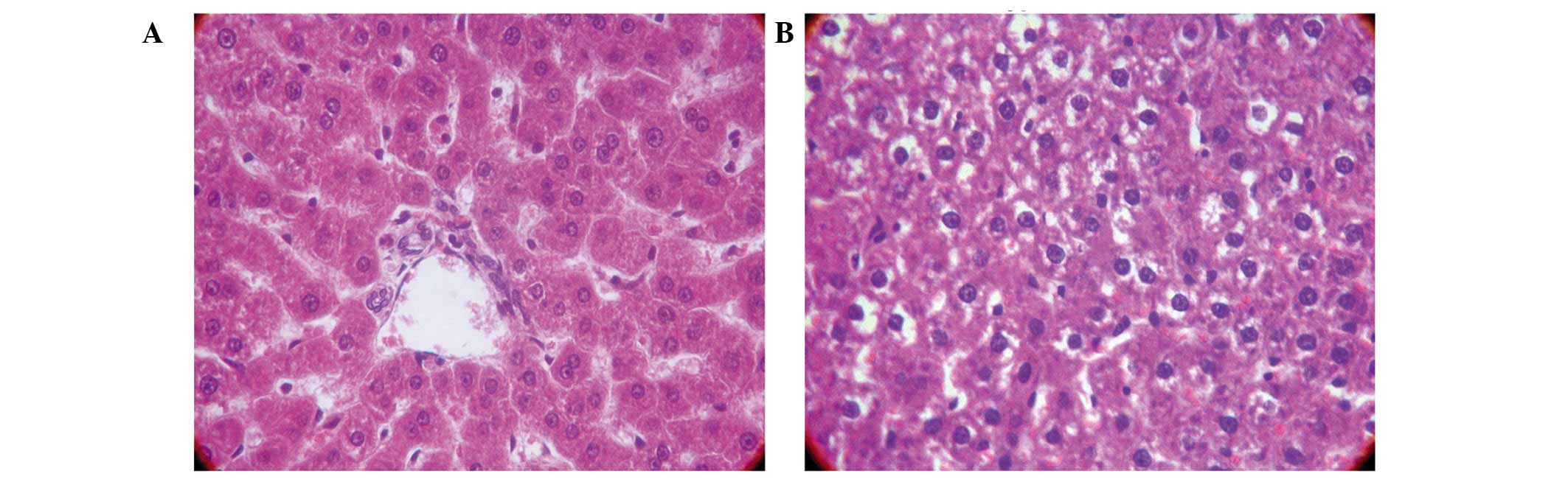
Hyperammonemia induces hepatic
injury
In the control group, the liver appeared to have a
normal histological structure; the majority of hepatic cells were
mononuclear and were arranged in rows that radiated out from the
center (Fig. 2A). Conversely, in the
hyperammonemic group, there were no signs of necrosis or
inflammatory cell infiltration. In addition, there were irregular
hepatic sinusoids and edema of a number of hepatocytes within the
hepatic lobule and portal areas, particularly around the central
vein (Fig. 2B).
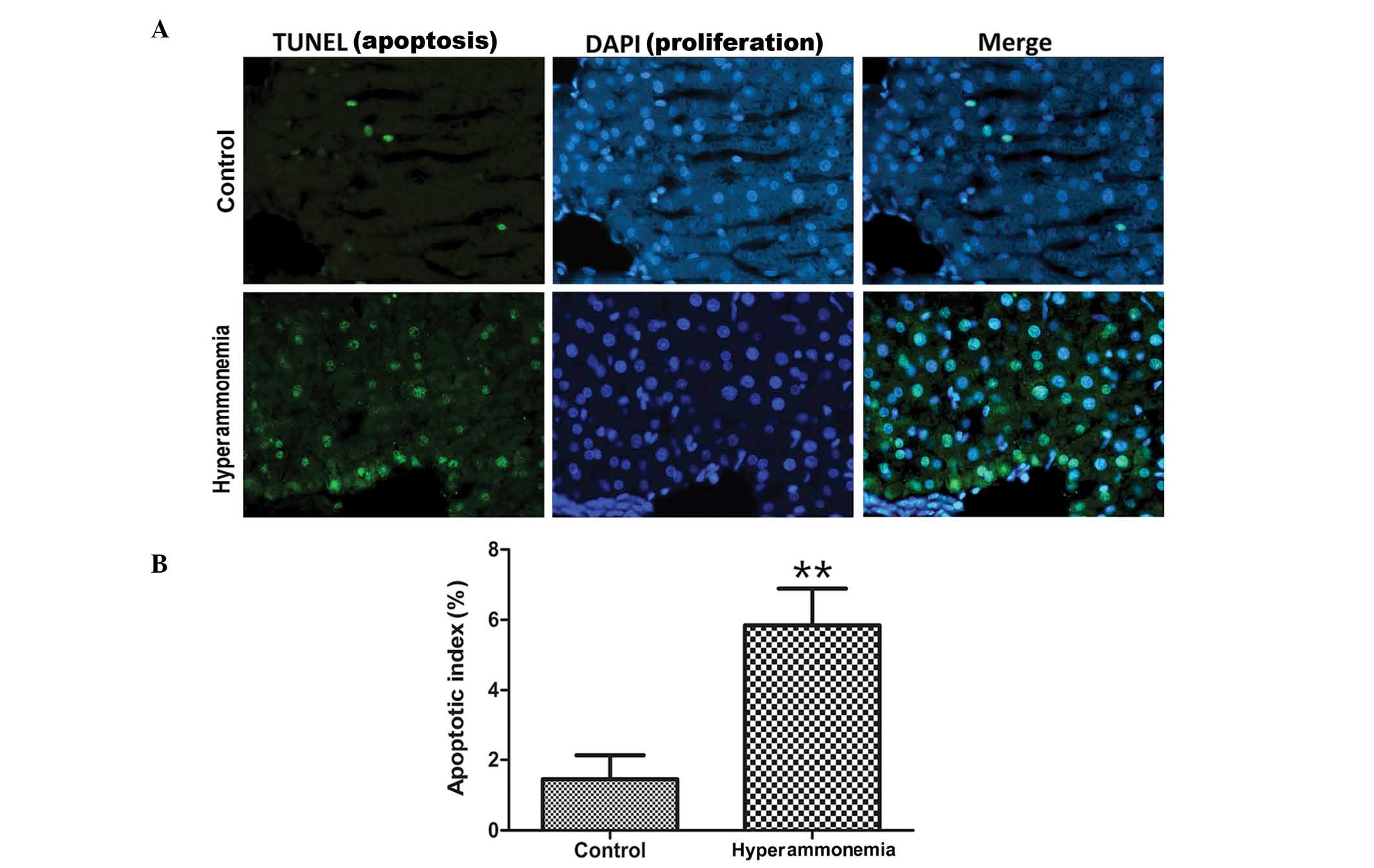
Hyperammonemia induces apoptosis of
hepatic cells
A TUNEL assay was used to evaluate cell apoptosis.
Apoptotic cells were observed in the hyperammonemic group, but not
in control group (Fig. 3A). In the
hyperammonemic group, apoptotic cells were scattered within the
hepatic portal and lobule areas. Quantitative analysis of
TUNEL-positive cells showed that the number of apoptotic cells in
the hyperammonemic group was higher than in the control group; with
a significantly higher apoptotic index calculated for the
hyperammonemic group (5.84±2.25%), as compared with the control
group (1.45±0.68%; P<0.01; Fig.
3B).
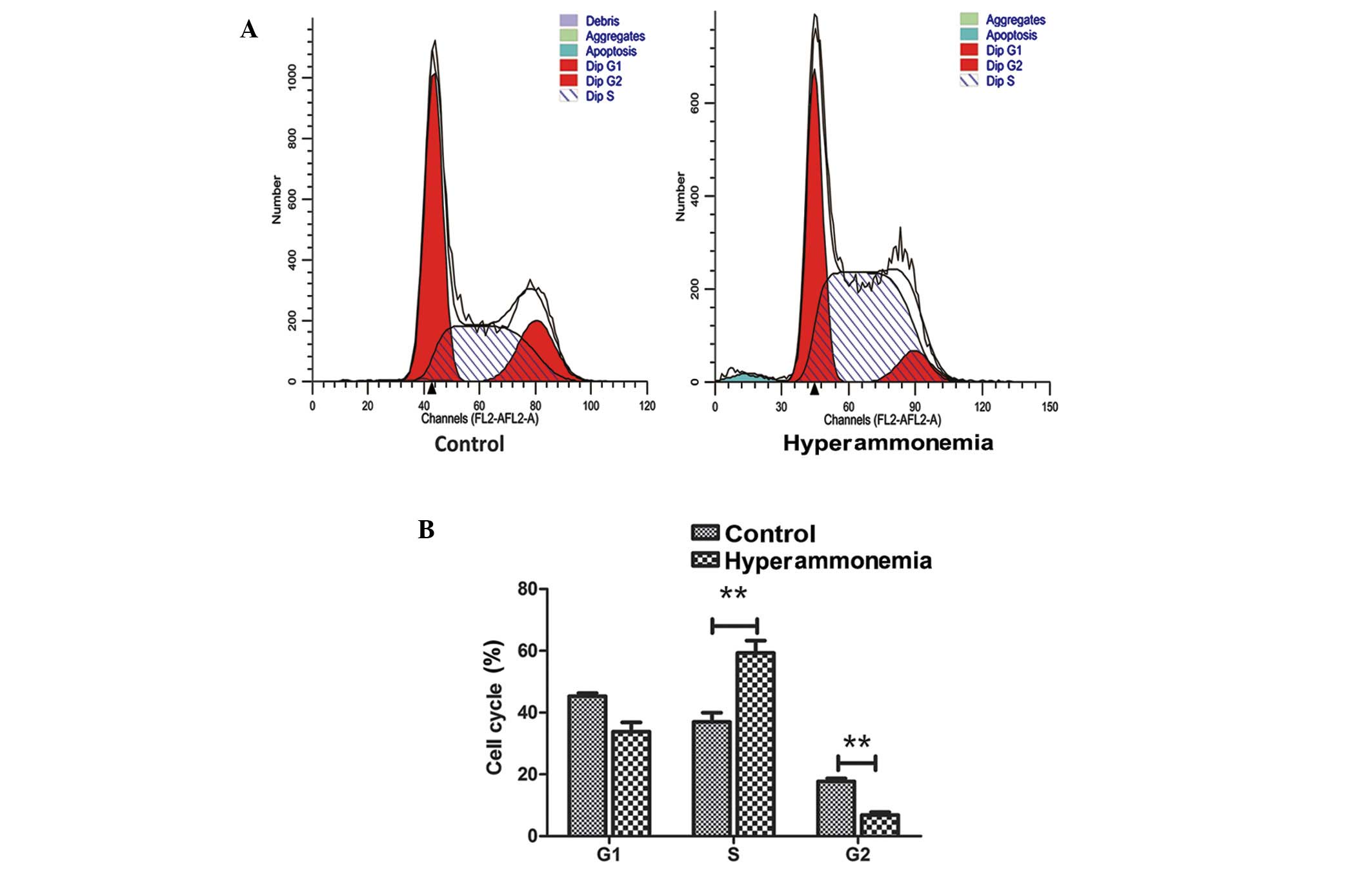
Hyperammonemia induces S phase cell
cycle arrest
Cell cycle distribution was analyzed using FCM. The
proportion of hyperammonemic hepatic cells in the S phase increased
from 36.7 to 59.3%, those in the sub-G1 phase (apoptotic
cells) increased from 0.68 to 1.89%, those in the
G0/G1 phase did not change, and the
proportion of cells in the G2 phase decreased from 17.7
to 6.9% (Fig. 4A). Thus,
hyperammonemia significantly increased the number of cells in the S
phase and significantly decreased the number of cells in the
G2/M phase (P<0.01; Fig.
4B).
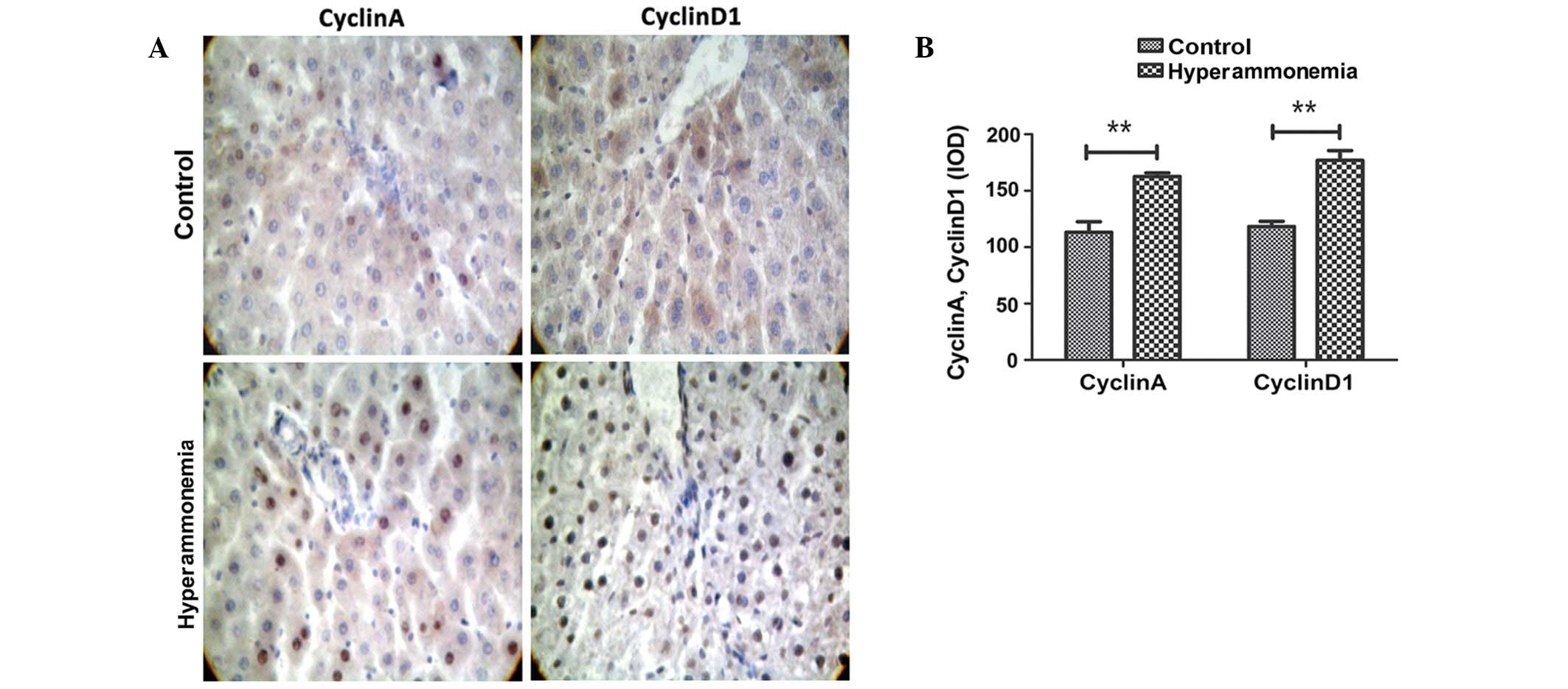
Hyperammonemia induces an increase in
the expression levels of cyclins A and D1
As compared with the controls, an increased number
of cells were stained positive for cyclin A and D1 in the
hyperammonemic group (Fig. 5A). The
integrated optical density values for cells staining positive for
cyclin A and D1 significantly increased from 113.2 and 118.3 to
162.6 and 176.9, respectively, as compared with the controls
(P<0.01; Fig. 5B).
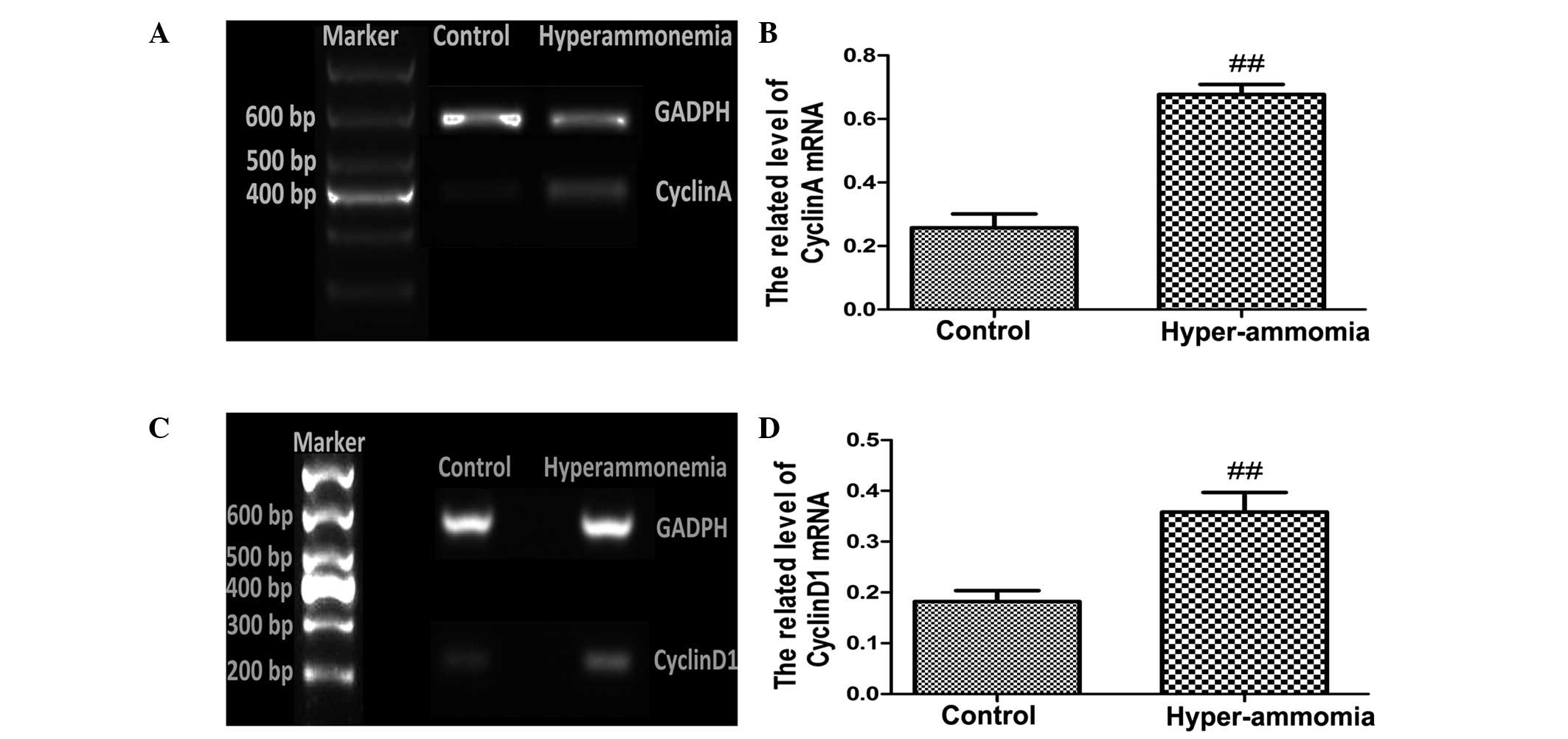
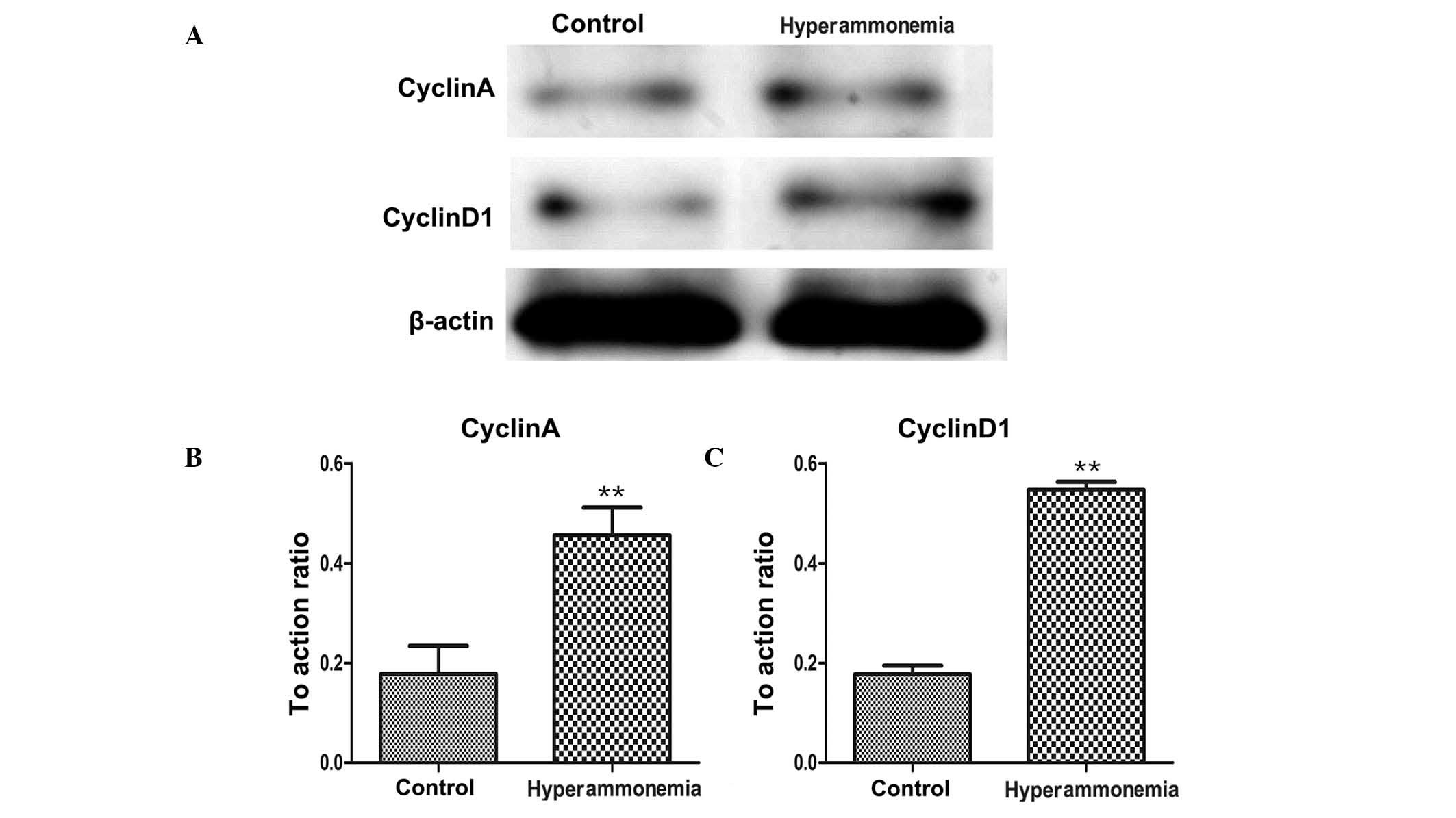
RT-qPCR (Fig. 6A–D)
and western blotting (Fig. 7A–D)
detected significantly increased mRNA and protein expression levels
of cyclin A and cyclin D1, respectively, in the livers of rats
subjected to hyperammonemia, as compared with the control group
(P<0.01).
Discussion
Hyperammonemia is primarily caused by disorders of
the urea cycle, and it is the most common symptom of HF (2,4,19–22).
Hyperammonemia has been identified as a possible cause of hepatic
encephalopathy following hepatic injury, where an excessive
quantity of ammonia is metabolized by astrocytes, causing cell and
brain swelling in vivo (11,22,23). In
support of this, exposing primary astrocytes to ammonia in
vitro has been documented to cause cell swelling and a loss of
cell viability at high ammonia concentrations (2,24–26). The
findings of the present study support previous reports
demonstrating the damaging effects of hyperammonemia on hepatic
function (4,14,15,27).
The present study measured changes in liver
histology in rats that had been treated with ammonia, as compared
with controls. Similar to a previous study (15), histological changes, including mild
hydropic degeneration, were detected in hyperammonemic livers;
however there was no presence of hepatic cell necrosis or
inflammatory cell infiltration. In addition to this, it was
demonstrated that hyperammonemia may cause hepatic injury by
inducing hepatic cell apoptosis, which suggested that ammonia may
target hepatic cells directly. Cell cycle arrest is closely
associated with apoptosis (28–30), and
the present study demonstrated that hyperammonemia significantly
increased the number of cells in the S phase and significantly
decreased the number of cells in the G2/M phase. These
results suggested that hyperammonemia arrests cells in the S phase,
which ultimately leads to a depletion of G2/M phase
cells.
Cyclin A and D1 have been reported to have an
important role in various cell processes (31–34). For
example, previous studies have demonstrated that cyclin
A-associated kinase mediates hypoxia-induced apoptosis in
cardiomyocytes (31,35,36).
Other findings indicated that cyclin D1 regulates cell progression
through the G1/S phase transition of the cell cycle, and
is a key factor in tumorigenesis (37–39). The
present study demonstrated that levels of cyclin A and D1 were
increased in hyperammonemic hepatic cells. In order to explore the
underlying mechanism of hyperammonemia-induced hepatic injury, the
expression levels of cyclin A and D1 were measured in association
with apoptotic signaling pathways, and it was demonstrated that
hyperammonemia enhanced the mRNA and protein expression levels of
cyclin A and D1. Furthermore, the present study demonstrated that
BA values were elevated in hyperammonemic rats. As hyperammonemia
is a consequence of severe HR, BA levels may be elevated due to the
decreased number of functional hepatocytes in HR and, therefore,
the ability of the liver to detoxify ammonia is compromised
(2,21–23).
However, the results of the present study suggested that
hyperammonemia-induced hepatic cell damage may occur as a result of
apoptosis signaling pathways being activated and altering cell
cycle progression. Until now, there have not been a great number of
studies published regarding the role of hyperammonemia in hepatic
cell apoptosis and cell cycle arrest (1,4,20). Therefore, the present study may offer
novel insights into hyperammonemia-induced hepatic injury.
In conclusion, the results of the present study
suggested that hyperammonemia-induced hepatic injury in rats may be
caused by changes in the expression levels of cyclin A and D1, cell
cycle arrest and cellular apoptosis. The present study also
concludes that hyperammonemia appeared to be a causative factor of
HF. These findings indicate possible mechanisms for hepatic injury
caused by hyperammonemia, and may provide potential targets for
treating or preventing further hepatic damage caused by
hyperammonemia, in particular, hepatic encephalopathy.
Acknowledgements
The authors would like to thank Dr Zujiang Yu for
support and excellent technical assistance during the experiments
and writing.
References
|
1
|
Adeva MM, Souto G, Blanco N and Donapetry
C: Ammonium metabolism in humans. Metabolism. 61:1495–1511. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wright G and Noiret L: OldeD amink SW and
Jalan R: Interorgan ammonia metabolism in liver failure: The basis
of current and future therapies. Liver Int. 31:163–175. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jones EA and Mullen KD: Theories of the
pathogenesis of hepatic encephalopathy. Clin Liver Dis. 16:7–26.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lee WM: Acute liver failure. N Engl J Med.
329:1862–1872. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Blackmore L and Bernal W: Acute liver
failure. Clin Med. 15:468–472. 2015. View Article : Google Scholar
|
|
6
|
Butterworth RF: Hepatic encephalopathy:. A
central neuroinflammatory disorder? Hepatology. 53:1372–1376.
2011.PubMed/NCBI
|
|
7
|
Trautwein C and Koch A: Mechanisms of
acute liver failure. Liver Immunology. Gershwin MR, Vierling J and
Manns MP: (2nd). (New York, NY). Springer International Publishing.
373–388. 2014. View Article : Google Scholar
|
|
8
|
Mouli Pratap V, Benjamin J, Singh Bhushan
M, Mani K, Garg SK, Saraya A and Joshi YK: Effect of probiotic
VSL#3 in the treatment of minimal hepatic encephalopathy, A
non-inferiority randomized controlled trial. Hepatol Res.
45:880–889. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jover-Cobos M, Khetan V and Jalan R:
Treatment of hyperammonemia in liver failure. Curr Opin Clin Nutr
Metab Care. 17:105–110. 2014.PubMed/NCBI
|
|
10
|
Lange CM, Bojunga J, Hofmann WP, Wunder K,
Mihm U, Zeuzem S and Sarrazin C: Severe lactic acidosis during
treatment of chronic hepatitis B with entecavir in patients with
impaired liver function. Hepatology. 50:2001–2006. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Fuhrmann V, Drolz A, Jaeger B, Wewalka M,
Saxa R, Horvatits T, Perkmann T, Zauner C and Ferenci P: Prognostic
relevance of arterial ammonia levels in different acute and
acute-on-chronic liver diseases. Crit Care. 16(Suppl 1): P3902012.
View Article : Google Scholar
|
|
12
|
Liu CP and Yu ZJ: Study on
L-Ornithine-L-Aspartate in the treatment of acute-on-chronic liver
failure. Zhonghua Gan Zang Bing Za Zhi. 19:63–64. 2011.(In
Chinese). PubMed/NCBI
|
|
13
|
Schmid M, Peck-Radosavljevic M, König F,
Mittermaier C, Gangl A and Ferenci P: A double-blind, randomized,
placebo-controlled trial of intravenous L-ornithine-L-aspartate on
postural control in patients with cirrhosis. Liver Int. 30:574–582.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yu ZJ, Sun R, Liu XR, Yan JY, Gao XJ and
Kan QC: Hyperammonemia-induced hepatic injury in rats,
Characterization of a new animal model. Zhonghua Gan Zang Bing Za
Zhi. 21:467–472. 2013.(In Chinese). PubMed/NCBI
|
|
15
|
Li J, Yu Z, Wang Q, Li D, Jia B, Zhou Y,
Ye Y, Shen S, Wang Y, Li S, et al: Hyperammonia induces specific
liver injury through an intrinsic Ca2+-independent
apoptosis pathway. BMC Gastroenterol. 14:1512014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhu Q, Gu L, Wang Y, Jia L, Zhao Z, Peng S
and Lei L: The role of alpha-1 and alpha-2 adrenoceptors in
restraint stress-induced liver injury in mice. PLoS One.
9:e921252014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bartek J and Lukas J: Mammalian G1- and
S-phase checkpoints in response to DNA damage. Curr Opin Cell Biol.
13:738–747. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Myklebust MP, Li Z, Tran TH, Rui H,
Knudsen ES, Elsaleh H, Fluge Ø, Vonen B, Myrvold HE, Leh S, et al:
Expression of cyclin D1a and D1b as predictive factors for
treatment response in colorectal cancer. Br J Cancer.
107:1684–1691. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Esposti Degli D, Hamelin J, Bosselut N,
Saffroy R, Sebagh M, Pommier A, Martel C and Lemoine A:
Mitochondrial roles and cytoprotection in chronic liver injury.
Biochem Res Int. 2012:3876262012.PubMed/NCBI
|
|
20
|
Jalan R, Gines P, Olson JC, Mookerjee RP,
Moreau R, Garcia-Tsao G, Arroyo V and Kamath PS: Acute-on chronic
liver failure. J Hepatol. 57:1336–1348. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sarin SK, Kumar A, Almeida JA, Chawla YK,
Fan ST, Garg H, de Silva HJ, Hamid SS, Jalan R, Komolmit P, et al:
Acute-on-chronic liver failure: Consensus recommendations of the
Asian Pacific Association for the study of the liver (APASL).
Hepatol Int. 3:269–282. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Aldridge DR, Tranah EJ and Shawcross DL:
Pathogenesis of hepatic encephalopathy, Role of ammonia and
systemic inflammation. J Clin Exp Hepatol. 5((Suppl 1)): S7–S20.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bobermin LD, Quincozes-Santos A, Guerra
MC, Leite MC, Souza DO, Gonçalves CA and Gottfried C: Resveratrol
prevents ammonia toxicity in astroglial cells. PLoS One.
7:e521642012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rose CF: Ammonia: M ore than a neurotoxin?
Liver Int. 34:649–651. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang Q, Wang Y, Yu Z, Li D, Jia B, Li J,
Guan K, Zhou Y, Chen Y and Kan Q: Ammonia-induced energy disorders
interfere with bilirubin metabolism in hepatocytes. Arch Biochem
Biophys 555-556. 16–22. 2014. View Article : Google Scholar
|
|
26
|
Zhang LJ, Qi R, Zhong J, Xu Q, Zheng G and
Lu GM: The effect of hepatic encephalopathy, hepatic failure, and
portosystemic shunt on brain volume of cirrhotic patients, A
voxel-based morphometry study. PLoS One. 7:e428242012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jia B, Yu ZJ, Duan ZF, Lü XQ, Li JJ, Liu
XR, Sun R, Gao XJ, Wang YF, Yan JY and Kan QC: Hyperammonaemia
induces hepatic injury with alteration of gene expression profiles.
Liver Int. 34:748–758. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Johnson DG and Walker CL: Cyclins and cell
cycle checkpoints. Annu Rev Pharmacol Toxicol. 39:295–312. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hartwell LH and Kastan MB: Cell cycle
control and cancer. Science. 266:1821–1828. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Stewart ZA, Mays D and Pietenpol JA:
Defective G1-S cell cycle checkpoint function sensitizes cells to
microtubule inhibitor-induced apoptosis. Cancer Res. 59:3831–3837.
1999.PubMed/NCBI
|
|
31
|
Chibazakura T, Kamachi K, Ohara M, Tane S,
Yoshikawa H and Roberts JM: Cyclin A promotes S-phase entry via
interaction with the replication licensing factor Mcm7. Mol Cell
Biol. 31:248–255. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kang DS, Hong K-M, Park J and Bae CD:
Cyclin A regulates a cell-cycle-dependent expression of CKAP2
through phosphorylation of Sp1. Biochem Biophys Res Commun.
420:822–827. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pardo FS, Su M and Borek C: Cyclin D1
induced apoptosis maintains the integrity of the G1/S checkpoint
following ionizing radiation irradiation. Somat Cell Mol Genet.
22:135–144. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ha SY, Kim HK, Im JS, Cho HY, Chung DH and
An J: Expression of Cyclin A, B1, D1, D3, and E in non-small lung
cancers. J Lung Cancer. 11:33–37. 2012. View Article : Google Scholar
|
|
35
|
Meng FJ, Jiao SM and Yu B: Picroside II
protects cardiomyocytes from hypoxia/reoxygenation-induced
apoptosis by activating the PI3K/Akt and CREB pathways. Int J Mol
Med. 30:263–270. 2012.PubMed/NCBI
|
|
36
|
Wu D, Jiang H, Chen S and Zhang H:
Inhibition of microRNA-101 attenuates hypoxia/reoxygenation-induced
apoptosis through induction of autophagy in H9c2 cardiomyocytes.
Mol Med Rep. 11:3988–3994. 2015.PubMed/NCBI
|
|
37
|
Hashimoto T, Yanaihara N, Okamoto A,
Nikaido T, Saito M, Takakura S, Yasuda M, Sasaki H, Ochiai K and
Tanaka T: Cyclin D1 predicts the prognosis of advanced serous
ovarian cancer. Exp Ther Med. 2:213–219. 2011.PubMed/NCBI
|
|
38
|
Pontano LL and Diehl JA: Speeding through
cell cycle roadblocks: Nuclear cyclin D1-dependent kinase and
neoplastic transformation. Cell Div. 3:122008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Peurala E, Koivunen P, Haapasaari KM,
Bloigu R and Jukkola-Vuorinen A: The prognostic significance and
value of cyclin D1, CDK4 and p16 in human breast cancer. Breast
Cancer Res. 15:R52013. View
Article : Google Scholar : PubMed/NCBI
|