Introduction
Ischemic stroke is induced by cerebral artery
occlusion, which can cause regional cerebral flow reduction or
interruption (1). The brain is
sensitive to ischemia, the effect of which may be exacerbated by
the reperfusion. Necrotic cells begin to die within a few minutes
of injury. Furthermore, the neuronal cells in the cerebral cortex,
striatum and hippocampus begin to die within several hours after
ischemic stroke and the process of cell death may last several days
(2). Although thrombolytic therapy
is considered to be the only beneficial treatment in clinical
practice, the majority of patients with ischemic stroke still fail
to receive adequate treatment in time (3,4).
Previous studies have revealed that enhancing angiogenesis and
reducing the apoptosis of nerve cells may improve clinical outcomes
during the recovery phase following an ischemic stroke.
Furthermore, increasing evidence has indicated that the cyclic
adenosine monophosphate (cAMP)-responsive element binding protein
(CREB) signaling pathway is intimately involved in a variety of
nerve protection mechanisms following ischemic stroke (5), and that the phosphorylation of CREB
plays a critical role in learning and memory function (6,7).
Phosphorylation of CREB can be achieved by a number of upstream
signaling cascades, including the cAMP-protein kinase A (PKA)
cascade (8,9), and the cAMP/CREB pathway exerts a
strong effect on the development, survival, maturation and
integration of new neurons (10,11).
This has prompted the theory that the cAMP/CREB pathway may provide
benefits for brain remodeling following ischemic injury and may be
a target of cerebral ischemia treatment. However, a limited number
of studies have investigated whether the cAMP/CREB pathway is
involved in the process of angiogenesis and apoptosis following
cerebral ischemia/reperfusion injury (12,13).
Rolipram typically acts as an antidepressant- and
anxiolytic-like agent (14);
however, a number of studies have revealed that it may reduce the
infarction area caused by cerebral ischemia (15) and also increase the phosphorylated-
(p-)CREB expression level in the hippocampus (14,16).
This study focused on the protective effect of rolipram on
transient cerebral ischemia/reperfusion injury in rats, and aimed
to investigate the hypothesis that rolipram acts through promoting
angiogenesis and reducing apoptosis following cerebral
ischemia.
Materials and methods
Experimental animals
Male Wistar rats, weighing 250–300 g, were obtained
from the Center of Experimental Animals, School of Medicine (Xi'an
Jiaotong University, Xi'an, China). The rats were maintained on a
12-h light/dark cycle and allowed free access to food and water.
All the experiments were approved and supervised by the Animal Care
Committee of Xi'an Jiaotong University Health Science Center.
Transient middle cerebral artery
occlusion (tMCAO)
Prior to the surgery, the rats were fasted overnight
but allowed free access to water. In brief, the rats were
anesthetized using chloral hydrate [350 mg/kg, intraperitoneal
(i.p.)]. The rectal temperature was monitored and maintained at
37.0±0.5°C, using a feedback-regulated heating system during the
surgery. tMCAO (17) was induced by
the method of intraluminal vascular occlusion. Briefly, a 4–0-nylon
monofilament suture with a slightly enlarged round tip was inserted
into the stump of the external carotid artery (ECA) and run across
the lumen of the internal carotid artery, until it reached and
occluded the MCA. The average distance between the bifurcation of
the common carotid artery and the tip of the suture inserted to
occlude the MCA was 18–20 mm. Two hours after MCAO, reperfusion was
achieved with the withdrawal of the suture until the tip cleared
the lumen of the ECA. Sham-operated animals were subjected to the
above-described procedures, with the exception of suture
insertion.
Rolipram treatment
Ischemic rats received injections of rolipram (3
mg/kg, vehicle i.p.; Sigma-Aldrich, St. Louis, MO, USA) from the
first day after ischemia. The treatment lasted three, seven and 14
consecutive days according to the group. The dosage and dosing
frequency of rolipram were selected on the basis of previous
studies (18). The rats were
randomly divided into five groups according to the tMCAO insult,
sham procedure and drug use: i) Sham group, rats underwent the
surgical procedure but without tMCAO; ii) vehicle group, rats
underwent tMCAO and received 0.9% saline treatments; iii) three
days group, rats underwent tMCAO and received a three-day rolipram
course; iv) seven days group, rats underwent tMCAO and received a
seven-day rolipram course; v) 14 days group, rats underwent tMCAO
and received a 14-day rolipram course. Food and water were freely
accessible throughout the experimental course.
Assessing cerebral infarction and
functional outcome
The functional outcome in the rats was evaluated by
the modified neurological severity score (mNSS), for which the rats
were assessed using several tests, including raising the rat by the
tail, placing the rat on the floor and beam balance walking. All
the test scores were incorporated into the mNSS (1). In addition, triphenyl tetrazolium
chloride (TTC) staining (1) was used
to evaluate the brain infarction size. The colorless TTC is reduced
to a red formazan produced by dehydrogenases, which are most
abundant in mitochondria. As such, TTC staining is a functional
test of dehydrogenase enzyme activity and is usually used for the
early histochemical diagnosis of infarction. Therefore, the rats
were sacrificed at three days post-surgery to assess the infarction
change using TTC. Subsequent to documenting the mNSS for 21 days,
the rats were all sacrificed and immunochemical staining was
performed. The brain tissue was rapidly removed, immersed in cold
saline for 10 min and sliced into 2.0 mm-sections. The brain slices
were incubated in 2% TTC dissolved in phosphate-buffered saline for
30 min at 37°C and then transferred to a 4% formaldehyde solution
for fixation.
Western blot analysis
p-CREB levels were estimated by western blot
analysis, and β-actin was utilized as a loading control. The
animals were sacrificed at three, seven and 14 days after surgery.
The right ischemic hemispheres were collected. Protein samples were
homogenized in radioimmunoprecipitation assay buffer. A total of 60
µg protein was loaded in each lane, and the proteins were separated
by 10% SDS-PAGE and electroblotted onto nitrocellulose membranes
(Millipore, Billerica, MA, USA). Subsequent to blockage with 5%
non-fat milk, the blots were incubated with rabbit anti-p-CREB
(1:400; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and
β-actin conjugated goat anti-rabbit (1:5,000; Santa Cruz
Biotechnology, Inc.) primary antibodies. β-actin was used as an
internal reference for relative quantification.
Immunohistochemistry
Animals were maintained for 20 days after MCAO and
then sacrificed with chloral hydrate (400 mg/kg, i.p.). The rat
brains were fixed by transcardial perfusion with saline, followed
by perfusion and immersion in 4% paraformaldehyde. Using a
microtome, serial coronal sections (4-µm-thick) were obtained for
immunostaining, terminal deoxynucleotidyl transferase-mediated dUTP
nick end labeling (TUNEL) assay and hematoxylin and eosin staining.
The study utilized goat anti-cluster of differentiation 34 (CD34)
primary antibodies (1:150; R&D Systems, Minneapolis, MN, USA),
a TUNEL detection kit (Roche, San Francisco, CA, USA) and secondary
antibodies coupled to biotin (1:200; Proteintech Group, Chicago,
IL, USA). Images were captured using an Olympus DP-72 confocal
microscope (Olympus Corporation, Tokyo, Japan).
Statistical analysis
Results are expressed as the mean ± standard error
of the mean for three or more independent experiments. To compare
data, the analysis of variance test was utilized. A value of
P<0.05 was considered to indicate a statistically significant
difference.
Results
Rolipram improves functional outcome
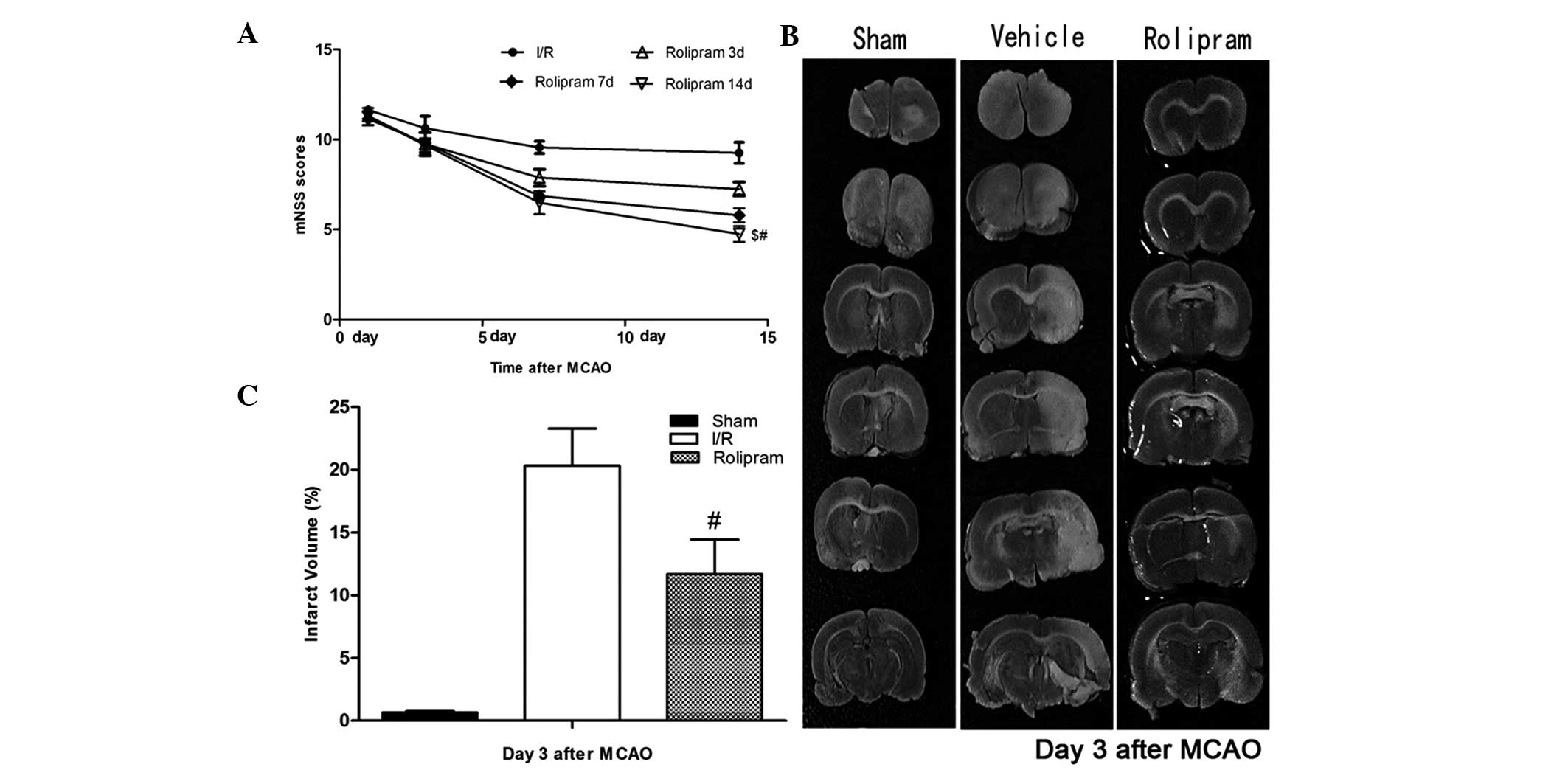
and decreases infarction size
To test whether rolipram affected functional
outcomes following ischemic stroke, the neurological functional
test was performed. Rats treated with rolipram in the 14 days group
showed significantly improved functional recovery (based on mNSS
testing) compared with the rats in the other groups (P<0.05)
between days 7 and 14. Rats in the three and seven days groups also
showed significantly lower neurological deficits (based on mNSS
testing) than the vehicle group on the third, seventh and 14th days
after reperfusion; however, the scores in these groups were
slightly higher than those in the 14 days group (Fig. 1A). Twenty-four hours after MCAO, the
rat brains were evaluated for infarction volume using TTC staining
and imaging software (Media Cybernetics, Silver Spring, MD, USA)
(19). Representative samples of
TTC-stained brain sections are shown in Fig. 1B. Increased areas of white were
observed in the brain tissue of the vehicle group compared with the
other groups; these areas were associated with increased ischemic
injury. The infarcted area shown by TTC staining was decreased in
the rolipram-treated groups from 23.4±1.72 to 10.34±2.25% (Fig. 1C). This indicated that rolipram may
attenuate cerebral ischemic injury in rats.
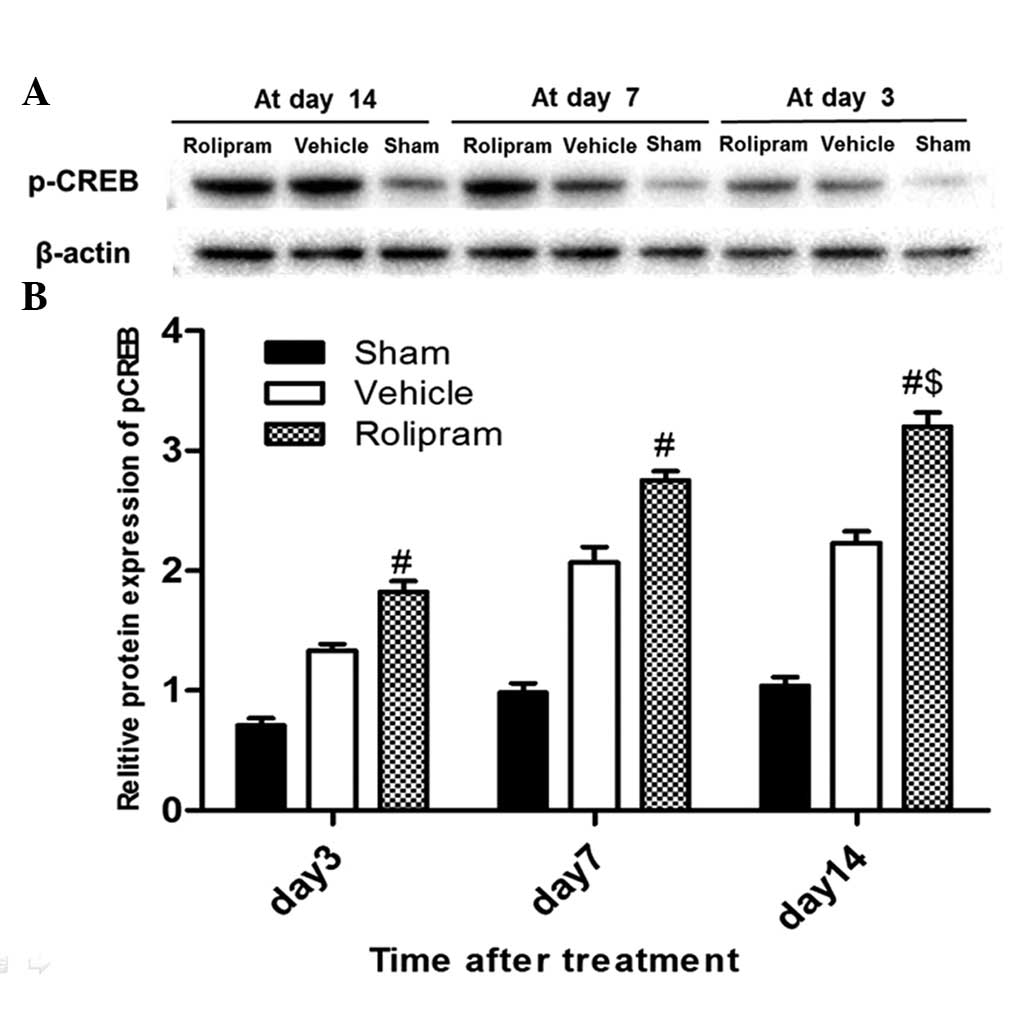
Rolipram increases the p-CREB
expression levels in the hippocampus
The ability of rolipram to increase p-CREB levels in
the ischemic hemisphere of the ischemic brain was examined by
western blot analysis. Ischemic tissues were isolated on the third,
seventh and 14th days after reperfusion. Western blotting was
performed with specific antibodies. Densitometric quantification of
immunoreactive p-CREB (43 kDa) band intensities was performed by
normalization to β-actin, an internal control. As shown in Fig. 2A, β-actin expression was not
different among the groups, and each p-CREB band intensity was
therefore corrected to that of β-actin. Fig. 2B shows that rolipram significantly
increased the expression of p-CREB on days 3, 7 and 14 in the
ischemic hemisphere compared with the sham and vehicle groups
(P<0.01, n=5 per group). A longer duration of rolipram treatment
further enhanced the increase in the p-CREB level, and the p-CREB
level in the 14 days group was higher than that in the other groups
(P<0.05). These results indicated that rolipram could activate
p-CREB in the ischemic brain as well as in cultured neurons.
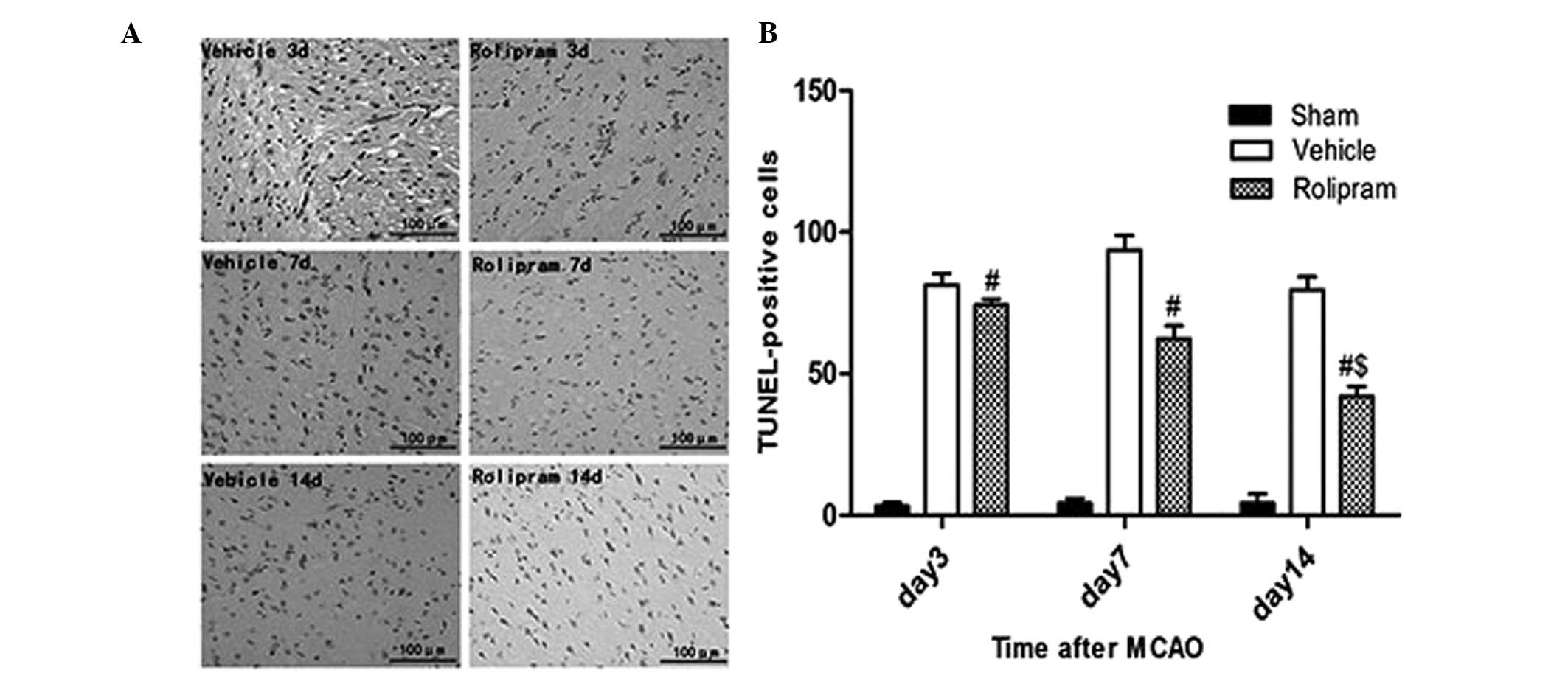
Rolipram attenuates neuronal apoptosis
and stimulates angiogenesis
To examine whether the improved functional outcome
induced by rolipram was mediated via a reduction in the number of
apoptotic cells, the number of TUNEL-positive cells in the ischemic
boundary region was measured. No apoptotic cells were detected in
the brains from the sham-operated rats (data not shown); however,
the rolipram group exhibited a significantly reduced number of
TUNEL-positive cells compared with the vehicle group (P<0.01,
Fig. 3). Additionally, the 14 days
group exhibited a reduced number of TUNEL-positive cells compared
with the seven and three days and vehicle groups (P<0.05,
Fig. 3). Collectively, these data
indicated that rolipram had the capability to attenuate neuronal
apoptosis in the ischemic brain.
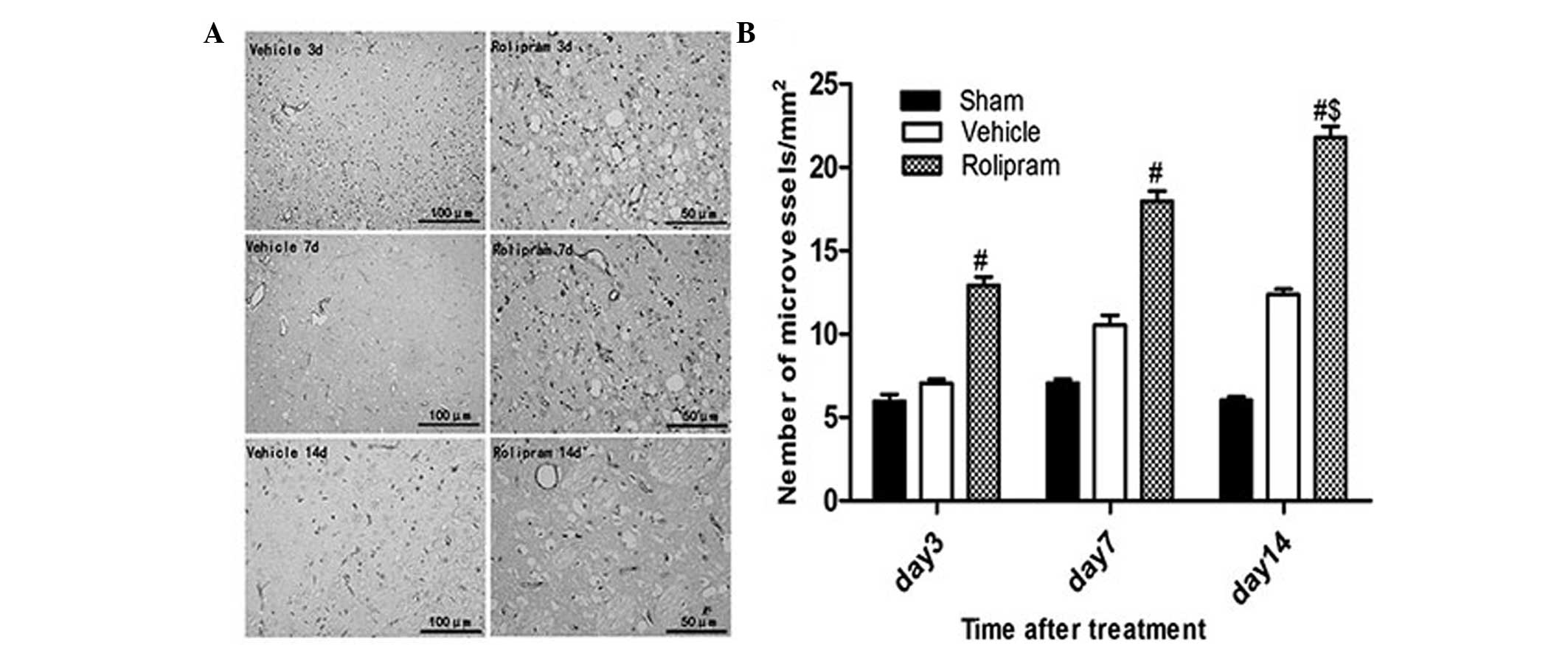
To determine whether rolipram increased angiogenesis
via recruiting CD34+ cells, which differentiate into
vascular endothelial cells, in the ischemic boundary region, the
number of CD34+ microvessels/mm2 in the
ischemic hemisphere was analyzed on day 14 after ischemic stroke.
The number of CD34+ microvessels/mm2 was
significantly greater in the rolipram-treated group as compared
with that in the vehicle-treated (21.78±0.47 vs. 16.27±0.32;
P<0.05) and the sham (21.78±0.47 vs. 7.23±0.47; P<0.01)
groups. Furthermore, in the rolipram group with the longest
treatment duration, the number of microvessels in the ischemic
boundary region appreciably increased when compared with that in
the groups treated for a shorter period of time (20.78±0.47 vs.
17.96±0.62 and 12.93±0.50; P<0.05) (Fig. 4). These results indicated that
rolipram treatment increases the microvessel density (MVD) in the
ischemic brain.
Discussion
Ischemic stroke is currently a significant worldwide
health issue and can lead to serious long-term disability. Ischemia
can stimulate an increase in CREB phosphorylation in neurons
(20). CREB belongs to the family of
leucine zipper transcription factors that are expressed in a
variety of tissues. CREB functions as an effector molecule that
initiates changes in the cellular response to extracellular
stimuli. Among various gene regulatory factors, CREB has been
suggested to be involved in the development and plasticity of
neurons, as well as numerous other neuronal processes. p-CREB is
the active form of CREB, and CREB phosphorylation at the serine 133
site is believed to be crucial in CREB-dependent transcription.
p-CREB regulates cell proliferation, differentiation and survival
in the developing brain, and mediates a number of responses,
including neuronal plasticity, learning and memory, in the adult
brain (21). Using a monkey model of
ischemia-enhanced hippocampus neurogenesis, Boneva and Yamashima
(20) recently revealed that the
expression of p-CREB was significantly upregulated between days 5
and 15 after transient global brain ischemia. In a number of
cellular contexts, CREB is transiently activated by its
phosphorylation, lasting only 30–60 min (22); however, CREB phosphorylation is
persistent in neurons in newborn animals and lasts for as long as
2–3 weeks in rodents (23,24) and ≥10 days in monkeys.
Phosphodiesterase-4 (PDE4) inhibitors may also promote p-CREB
expression and enhance the survival time of ischemic neurons
(5). In the present study, rolipram
was administered to rats that had undergone tMCAO, with the purpose
of exploring whether rolipram could promote angiogenesis and reduce
apoptosis following cerebral ischemia, and whether the protective
effect was exerted through the CREB signaling pathway.
Phosphorylation of CREB can be caused by a number of
upstream signaling cascades, including the cAMP-PKA cascade, the
mitogen-activated protein kinase signaling pathway, and the
calmodulin-dependent kinases II and IV and phospholipase C-PKC
signaling cascades (25,26). Among the above signaling pathways,
the CREB phosphorylation that is triggered by the cAMP-PKA cascade
has been well studied (27). As
described above, the level of p-CREB in the hippocampus increases
following hypoxic-ischemic injury; therefore, increasing p-CREB
levels may a potential strategy for the treatment of cerebral
ischemia. To date, the role of p-CREB in ischemic injury following
experimental tMCAO has been explored in diverse pharmacological
interventions (28,29). In the present study, rolipram
treatment lasting for three, seven and 14 days was utilized.
Fig. 2B showed that the
administration of rolipram in ischemic rats could induce CREB
phosphorylation in the right hemisphere (the ischemic region).
Furthermore, the expression level was higher in the 14 days than
the three days group, which indicated that rolipram had the ability
to enhance the level of p-CREB. The results showed that rolipram
administration for a longer period of time induced enhanced
protection in the ischemic rats, which was consistent with previous
results found in studies using donepezil (28) and resveratrol (29). These previous studies showed that
increasing the level of p-CREB could not only ameliorate focal
ischemia-induced neuronal death but also the level of the
downstream protein B-cell lymphoma 2 (Bcl-2) in the ischemic cortex
of rats with tMCAO. It has also been indicated that propofol and
ketamine can provide neuroprotection through the inhibition of
neuron-specific p-CREB dephosphorylation in the peri-infarct region
of mice with permanent MCAO (5). The
present study demonstrated that rolipram exerted neuroprotective
effects in brain ischemia through the induction of CREB production,
which was likely mediated by activation of the cAMP-PKA cascade; an
investigation using a CREB inhibitor is now required. Small
interfering (si)RNA or a repeated silencer would be necessary for
this investigation. Repeated silencer is an intervention measure
that is used on animals during the experimental process and can
eventually cause CREB in nerve cells to lose its function by
blocking its phosphorylation or lowering the expression of CREB
protein. A previous study showed that delayed hyperbaric oxygen
therapy (HBOT) could decrease the infarct size and cause a
neurobehavioral improvement. Furthermore, gene silencing with CREB
siRNA or protein phosphatase 1-γ siRNA attenuated the acute
beneficial effects of the HBOT (30). Therefore, we hypothesized that the
PDE4 inhibitor rolipram is an antagonistic treatment that could
induce angiogenesis and this was one of the experiments in the
present study.
PDE4, one of the 11 PDE families (PDE1–11), can
hydrolyze cAMP in neuronal tissue, which also plays an important
role in the neurochemical and pathological alterations of brain
ischemia (31,32). Therefore, we believe that targeting
PDE4 may be an innovative approach to treat cognitive disorders
associated with cerebral ischemia. Rolipram, as a prototypical PDE4
inhibitor, is widely used in ischemic stroke studies (15,27). A
previous study found that rolipram could increase cAMP
accumulation; cAMP accumulation activates cAMP-dependent PKA and
subsequently phosphorylates and activates CREB (27). Authors in a different study held the
view that rolipram could reduce the distracted platform searches
induced by cerebral ischemia (15).
In the present study, Fig. 1 showed
that rolipram could effectively reduce the infarct size and improve
neurobehavioral scores, as demonstrated by a lower mNSS and
decreased area of TTC staining in the tMCAO model following the use
of rolipram for two weeks.
A number of previous studies have demonstrated that
CD34 progenitor cells are involved in tissue repair, which can
restore the blood perfusion of the ischemic site in ischemic
diseases and traumatic injuries by vasculogenesis and angiogenesis
(33,34). Compared with the neuron which has
ischemic necrosis, the neuron in ischemic penumbra does not have
serious metabolic disturbance due to the collateral circulation
(35). By creating vascular pathways
following ischemia in order to recover the supply of oxygen and
sugar as soon as possible is likely to determine whether the neuron
can survive or not. This is why the present study focuses on the
combination of rolipram's promotion of revascularization and
inhibition of apoptosis. Figs. 3 and
4 showed that fewer TUNEL-positive
cells and an increased number of CD34+ microvessels were
present in the tissues obtained from rats that were treated with
rolipram. This meant that rolipram stimulated angiogenesis and
attenuated neuronal apoptosis in areas damaged by ischemia. The MVD
counted on day 14 demonstrated the association between neurological
and functional recovery and the early improvement in the number of
microvessels. In addition, the western blotting data (Fig. 2) were consistent with the
immunohistochemistry data (Figs. 3
and 4) in indicating a functional
link between angiogenesis/apoptosis resistance and p-CREB.
It is well known that enhancing angiogenesis and
reducing the apoptosis of nerve cells may improve brain function in
cerebral ischemic mammals. However, the occurrence of endogenous
neurogenesis following ischemic stroke is early, short-lived and
delays neuronal cell death for several days (2). The present results showed that rolipram
can attenuate neuronal apoptosis and increase cell proliferation
and survival rate in the peri-infarct region through the activation
of the CREB pathway, and may therefore be a novel therapeutic
strategy to promote brain function recovery following stroke.
Subsequent studies should investigate how rolipram enhances
angiogenesis, attenuates neuronal apoptosis and affects other
relevant transcription factors, including vascular endothelial
growth factor, hypoxia inducible factor 1 and
Bcl-2/Bcl-2-associated × protein, which are involved in the
ischemia-induced angiogenesis (5,36–38).
Acknowledgements
The authors would like to thank Professor Ying Mao
for the guidance provided during the study.
References
|
1
|
Lee YS, Chio CC, Chang CP, et al: Long
course hyperbaric oxygen stimulates neurogenesis and attenuates
inflammation after ischemic stroke. Mediators Inflamm.
2013:5129782013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Okuyama S, Shimada N, Kaji M, et al:
Heptamethoxyflavone, a citrus flavonoid, enhances brain-derived
neurotrophic factor production and neurogenesis in the hippocampus
following cerebral global ischemia in mice. Neurosci Lett.
528:190–195. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang J, Shi QD, Song TB, et al: Vasoactive
intestinal peptide increases VEGF expression to promote
proliferation of brain vascular endothelial cells via the cAMP/PKA
pathway after ischemic insult in vitro. Peptides. 42:105–111. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Genovese TI, Mazzon E, Paterniti I,
Esposito E and Cuzzocrea S: Neuroprotective effects of olprinone
after cerebral ischemia/reperfusion injury in rats. Neurosci Lett.
503:93–99. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Shu L, Li T, Han S, et al: Inhibition of
neuron-specific CREB dephosphorylation is involved in propofol and
ketamine-induced neuroprotection against cerebral ischemic injuries
of mice. Neurochem Res. 37:49–58. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bourtchuladze R, Frenguelli B, Blendy J,
et al: Deficient long-term memory in mice with a targeted mutation
of the cAMP-responsive element-binding protein. Cell. 79:59–68.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Josselyn SA, Shi C, Carlezon WA Jr, Neve
RL, Nestler EJ and Davis M: Long-term memory is facilitated by cAMP
response element-binding protein overexpression in the amygdala. J
Neurosci. 21:2404–2412. 2001.PubMed/NCBI
|
|
8
|
Yamashima T: ‘PUFA-GPR40-CREB signaling’
hypothesis for the adult primate neurogenesis. Prog Lipid Res.
51:221–231. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Carlezon WA Jr, Duman RS and Nestler EJ:
The many faces of CREB. Trends Neurosci. 28:436–445. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Merz K, Herold S and Lie DC: CREB in adult
neurogenesis - master and partner in the development of adult-born
neurons? Eur J Neurosci. 33:1078–1086. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wei Z, Belal C, Tu W, et al: Chronic
nicotine administration impairs activation of cyclic AMP-response
element binding protein and survival of newborn cells in the
dentate gyrus. Stem Cells Dev. 21:411–422. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chava KR, Tauseef M, Sharma T and Mehta D:
Cyclic AMP response element-binding protein prevents endothelial
permeability increase through transcriptional controlling
p190RhoGAP expression. Blood. 119:308–319. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ma W, Zheng WH, Powell K, Jhamandas K and
Quirion R: Chronic morphine exposure increases the phosphorylation
of MAP kinases and the transcription factor CREB in dorsal root
ganglion neurons: an in vitro and in vivo study. Eur J Neurosci.
14:1091–1104. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li LX, Cheng YF, Lin HB, Wang C, Xu JP and
Zhang HT: Prevention of cerebral ischemia-induced memory deficits
by inhibition of phosphodiesterase-4 in rats. Metab Brain Dis.
26:37–47. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li YF, Huang Y, Amsdell SL, Xiao L,
O'Donnell JM and Zhang HT: Antidepressant- and anxiolytic-like
effects of the phosphodiesterase-4 inhibitor rolipram on behavior
depend on cyclic AMP response element binding protein-mediated
neurogenesis in the hippocampus. Neuropsychopharmacology.
34:2404–2419. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li YF, Cheng YF, Huang Y, et al:
Phosphodiesterase-4D knock-out and RNA interference-mediated
knock-down enhance memory and increase hippocampal neurogenesis via
increased cAMP signaling. J Neurosci. 31:172–183. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen C, Hu Q, Yan J, et al: Multiple
effects of 2ME2 and D609 on the cortical expression of HIF-1alpha
and apoptotic genes in a middle cerebral artery occlusion-induced
focal ischemia rat model. J Neurochem. 102:1831–1841. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sasaki T, Kitagawa K, Omura-Matsuoka E, et
al: The phosphodiesterase inhibitor rolipram promotes survival of
newborn hippocampal neurons after ischemia. Stroke. 38:1597–1605.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shu L, Li T, Han S, Ji F, Pan C, Zhang B
and Li J: Inhibition of neuron-specific CREB dephosphorylation is
involved in propofol and ketamine-induced neuroprotection against
cerebral ischemic injuries of mice. Neurochem Res. 37:49–58. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Boneva NB and Yamashima T: New insights
into ‘GPR40-CREB interaction in adult neurogenesis’ specific for
primates. Hippocampus. 22:896–905. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lonze BE and Ginty DD: Function and
regulation of CREB family transcription factors in the nervous
system. Neuron. 35:605–623. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ji Y, Lu Y, Yang F, et al: Acute and
gradual increases in BDNF concentration elicit distinct signaling
and functions in neurons. Nat Neurosci. 13:302–309. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jagasia R, Steib K, Englberger E, et al:
GABA-cAMP response element-binding protein signaling regulates
maturation and survival of newly generated neurons in the adult
hippocampus. J Neurosci. 29:7966–7977. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Herold S, Jagasia R, Merz K, Wassmer K and
Lie DC: CREB signalling regulates early survival, neuronal gene
expression and morphological development in adult subventricular
zone neurogenesis. Mol Cell Neurosci. 46:79–88. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Johannessen M, Delghandi MP and Moens U:
What turns CREB on? Cell Signal. 16:1211–1227. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nair A and Vaidya VA: Cyclic AMP response
element binding protein and brain-derived neurotrophic factor:
molecules that modulate our mood? J Biosci. 31:423–434. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Barros DM, Izquierdo LA, Sant'Anna MK, et
al: Stimulators of the cAMP cascade reverse amnesia induced by
intra-amygdala but not intrahippocampal KN-62 administration.
Neurobiol Learn Mem. 71:94–103. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Min D, Mao X, Wu K, et al: Donepezil
attenuates hippocampal neuronal damage and cognitive deficits after
global cerebral ischemia in gerbils. Neurosci Lett. 510:29–33.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shin JA, Lee KE, Kim HS and Park EM: Acute
resveratrol treatment modulates multiple signaling pathways in the
ischemic brain. Neurochem Res. 37:2686–2696. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mu J, Ostrowski RP, Soejima Y, et al:
Delayed hyperbaric oxygen therapy induces cell proliferation
through stabilization of cAMP responsive element binding protein in
the rat model of MCAo-induced ischemic brain injury. Neurobiol Dis.
51:133–143. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang HT: Cyclic AMP-specific
phosphodiesterase-4 as a target for the development of
antidepressant drugs. Curr Pharm Des. 15:1688–1698. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhang HT: Phosphodiesterase targets for
cognitive dysfunction and schizophrenia - a New York Academy of
Sciences meeting. IDrugs. 13:166–168. 2010.PubMed/NCBI
|
|
33
|
Kao CH, Chen SH, Chio CC and Lin MT: Human
umbilical cord blood-derived CD34+ cells may attenuate
spinal cord injury by stimulating vascular endothelial and
neurotrophic factors. Shock. 29:49–55. 2008.PubMed/NCBI
|
|
34
|
Li S, Wei M, Zhou Z, Wang B, Zhao X and
Zhang J: SDF-1α induces angiogenesis after traumatic brain injury.
Brain Res. 1444:76–86. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Krupinski JI, Kaluza J, Kumar P, Kumar S
and Wang JM: Role of angiogenesis in patients with cerebral
ischemic stroke. Stroke. 25:1794–1798. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yang J, Shi QD, Song TB, Feng GF, Zang WJ,
Zong CH and Chang L: Vasoactive intestinal peptide increases VEGF
expression to promote proliferation of brain vascular endothelial
cells via the cAMP/PKA pathway after ischemic insult in vitro.
Peptides. 42:105–111. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Baugh JA, Gantier M, Li L, Byrne A,
Buckley A and Donnelly SC: Dual regulation of macrophage migration
inhibitory factor (MIF) expression in hypoxia by CREB and HIF-1.
Biochem Biophys Res Commun. 347:895–903. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Leung KW, Ng HM, Tang MK, Wong CC, Wong RN
and Wong AS: Ginsenoside-Rg1 mediates a hypoxia-independent
upregulation of hypoxia-inducible factor-1α to promote
angiogenesis. Angiogenesis. 14:515–522. 2011. View Article : Google Scholar : PubMed/NCBI
|