Introduction
Cardiac fibroblast differentiation, excessive
biosynthesis and destruction of the interstitial extracellular
matrix (ECM) in the ventricles of the heart are key features of
cardiac fibrosis, which is a consequence of cardiac remodeling
initiated by pathological events associated with various
cardiovascular disorders (1).
Induced by transforming growth factor (TGF)-β1 and other factors,
cardiac fibroblasts (CFs) differentiate into α-smooth muscle actin
(SMA) fiber-rich cardiac myofibroblasts that facilitate
contractility and increase ECM modulation ability (1). Although these changes are important for
wound repair and are beneficial for the maintenance of cardiac
function, continuous myocardial fibrosis may result in abnormal
myocardial stiffness and, ultimately, ventricular dysfunction
(1).
Curcumin,
1,7-bis(4-hydroxy-3-methoxyphenyl)-1,6-heptadiene-3,5-dione, is a
natural polyphenolic compound from the Curcuma longa herb,
possessing multiple biological and medicinal activities (2). The pharmacological safety of curcumin
has previously been demonstrated in various animal models, and
curcumin is nontoxic even at high doses (3). Previous studies have demonstrated that
curcumin has antioxida, anti-inflammatory, anti-proliferative,
pro-apoptotic and anti-carcinogenic properties (4–8).
Furthermore, previous investigations into the effects of curcumin
in heart disease have indicated that curcumin has a regulatory role
in the cardiac remodeling process; curcumin has been demonstrated
to ameliorate and reverse cardiac fibrosis, cardiac hypertrophy and
heart failure in animal models (9–13).
Therefore, curcumin may represent a novel therapeutic strategy for
the treatment of cardiac remodeling.
TGF-β1 is a key mediator of the differentiation of
fibroblasts to myofibroblasts (14),
and this TGF-β1-induced effect has been demonstrated to be
associated with the Smad2 and p38 mitogen-activated protein kinase
(MAPK) signaling pathways (15–17).
Although it has previously been demonstrated that curcumin has an
inhibitory effect on ECM secretion in cultured CFs (10), the effect of curcumin on the
differentiation of CFs and the underlying mechanisms are yet to be
fully elucidated. In the present study, using cultured CFs from
neonatal rats, it was demonstrated that curcumin has an inhibitory
effect on TGF-β1-induced cardiac fibroblast differentiation.
Furthermore, the role of Smad2 and p38 MAPK signaling in the
activation of CFs and the anti-fibrotic mechanism of curcumin in
the modulation of the TGF-β1 induced effects was addressed.
Materials and methods
Reagents
TGF-β1 was purchased from R&D Systems, Inc.
(Minneapolis, MN, USA). Curcumin and rabbit monoclonal anti-α-SMA
antibody (SP171) were purchased from Sigma-Aldrich (St. Louis, MO,
USA). Rabbit polyclonal anti-collagen type I (ColI; ab34710) and
-von Willebrand factor (ab6994) primary antibodies were purchased
from Abcam (Cambridge, MA, USA), whereas mouse monoclonal
anti-vimentin antibody (zm-0260), -glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) antibody (TA-08), and horseradish peroxidase
(HRP)-conjugated goat anti-rabbit and goat anti-mouse
immunoglobulin (Ig)G secondary antibodies (ZB-2301 and ZB-2305)
were purchased from Zhongsan Jinqiao Biotechnology Co., Ltd.
(Beijing, China). Phosphorylated and non-phosphorylated monoclonal
rabbit anti-rat Smad2 (#3108 and #5339) and p38 (#9215 and #2371)
primary antibodies were purchased from Cell Signaling Technology,
Inc. (Danvers, MA, USA). SB431542, a TGF-βR-Smad2/3 inhibitor was
purchased from Cayman Chemical Company (Ann Arbor, MI, USA) and
SB20358, a p38 inhibitor, was purchased from Merck Millipore
(Darmstadt, Germany). Cell culture materials and TRIzol® reagent
were purchased from Thermo Fisher Scientific, Inc. (Waltham, MA,
USA). The DNase and primers used for the reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) were
purchased from Sunbiotech (Beijing, China).
Cell culture and treatment
The present study was conducted with the approval of
the Ethics Committee of Experimental Animal Center of Shanxi
Cardiovascular Hospital (Taiyuan, China), according to the
regulations outlined by the National Institutes of Health
Guidelines on the Use of Laboratory Animals. CFs were isolated from
neonatal (1–3-days-old) Sprague-Dawley rats via trypsin digestion
methods, as previously described (18). CFs were cultured in Dulbecco's
Modified Eagle's Medium supplemented with 10% fetal bovine serum,
and were passaged using trypsin (1:3). Second passage CFs were used
in the present study and were serum-starved for 24 h at 80%
confluence in order to induce quiescence. Immunocytochemical
analysis demonstrated that the purity of the CFs used was >95%,
according to positive staining for vimentin and negative staining
for von Willebrand factor.
In order to elucidate the potential effects of
curcumin on the various signaling pathways in TGF-β1-induced CFs,
CFs were pretreated with 20 µmol/l curcumin, 10 µM TGF-βR-Smad2
inhibitor (SB431542) or 10 µM p38 MAPK inhibitor (SB203580) for 30
min, prior to treatment with recombinant 10 ng/ml TGF-β1 for 24 h.
In order to investigate the activation of Smad2 and p38, CFs were
treated with 10 ng/ml TGF-β1 for 1 h. Cells were subsequently
harvested and stored at −80°C prior to the determination of protein
and mRNA expression levels.
Immunofluorescent staining
CFs were cultured on coverslips in 6-well plates
(2.5×105 cells/well). Growth was arrested and CFs were
treated as described earlier. Following a 24-h treatment, CFs were
fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton
X-100 (Applygen Technologies, Inc., Beijing, China). Non-specific
binding was blocked via incubation with 10% normal goat serum
(Applygen Technologies, Inc.). Subsequently, α-SMA was detected
using a Cy5-conjugated goat anti-rabbit α-SMA polyclonal antibody
(Abcam; ab6564) and stained cells were visualized using the BX51
Fluorescence Microscope (Olympus Corporation, Tokyo, Japan).
Western blotting
CFs were harvested in lysis buffer (Applygen
Technologies, Inc.) containing 20 mM Tris, 150 mM NaCl, 1 mM EDTA,
1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM
β-glycerol phosphate, 1 mM Na3VO4 (pH 7.5), 1
mM phenylmethanesulfonyl fluoride, 1 mM benzamidine, 10 µg/ml
leupeptin and 10 µg/ml aprotinin. Protein concentrations were
determined using a bicinchoninic acid protein assay kit (Pierce
Biotechnology, Inc., Rockford, IL, USA), according to the
manufacturer's protocol. Following boiling for 1 min to denature,
20 µg protein was separated by 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis and transferred to
polyvinylidene difluoride membranes (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). Membranes were blocked with 5% fat-free milk in
Tris-buffered saline with Tween-20 (TBST; 10 mM Tris HCl, 150 mM
NaCl and 0.1% Tween-20) for 1 h at room temperature, and incubated
overnight with primary antibodies (1:2,500) at 4°C. Following each
incubation, membranes were washed three times for 10 min with TBST
and subsequently incubated with HRP-conjugated secondary antibody
(1:5,000) for 1 h at room temperature. Using chemiluminescence
(Applygen Technologies, Inc.), the membranes were scanned and
quantified using Quantity-One software, version 4.2 (Bio-Rad
Laboratories, Inc.), and the results were presented as the optical
density of phosphorylated-protein/total protein or of the target
protein/GAPDH.
RNA isolation and RT-qPCR
Total RNA was extracted from CFs using TRIzol®
reagent and treated with DNase, according to the manufacturer's
protocols. RT-qPCR analyses were performed using an ABI 7300 system
(Applied Biosystems; Thermo Fisher Scientific, Inc.). For each
sample, 1 µg RNA was utilized to synthesize cDNA using the Reverse
Transcriptase kit (Promega Corporation, Madison, WI, USA),
according to the manufacturer's protocol. The specific primer
sequences used were as follows: α-SMA forward,
5′-CATCAGGAACCTCGAGAAGC-3′, and reverse,
5′-TCGGATACTTCAGGGTCAGG-3′; ColI forward,
5′-CATAAAGGGTCATCGTGGCTTC-3′, and reverse,
5′-GTGATAGGTGATGTTCTGGGAG-3′; and GAPDH forward,
5′-AACCTGCCAAGTATGATGACATCA, and reverse,
5′-TTCCACTGATATCCCAGCTGCT-3′. Target genes were amplified using
SYBR Green PCR Master Mix (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The PCR cycling conditions were as follows: 95°C
for 5 min, followed by 40 cycles of 95°C for 35 sec and 60°C for 1
min, with a final extension step of 72°C for 5 min. Relative mRNA
levels were calculated using the 2−ΔΔCq method (19), where ΔCq was the difference between
GAPDH and target gene critical threshold cycle (Cq) values.
Statistical analyses
Statistical analyses in this study were conducted
using the SPSS software, version 16.0 (SPSS, Inc., Chicago, IL,
USA). The results are presented as the mean ± standard error of the
mean. Between-groups differences were assessed using one-way
factorial analysis of variance. P<0.05 was considered to
indicate a statistically significant difference. Each assay in the
present study was performed in triplicate.
Results
Treatment with curcumin impairs
TGF-β1-induced cardiac fibroblast differentiation
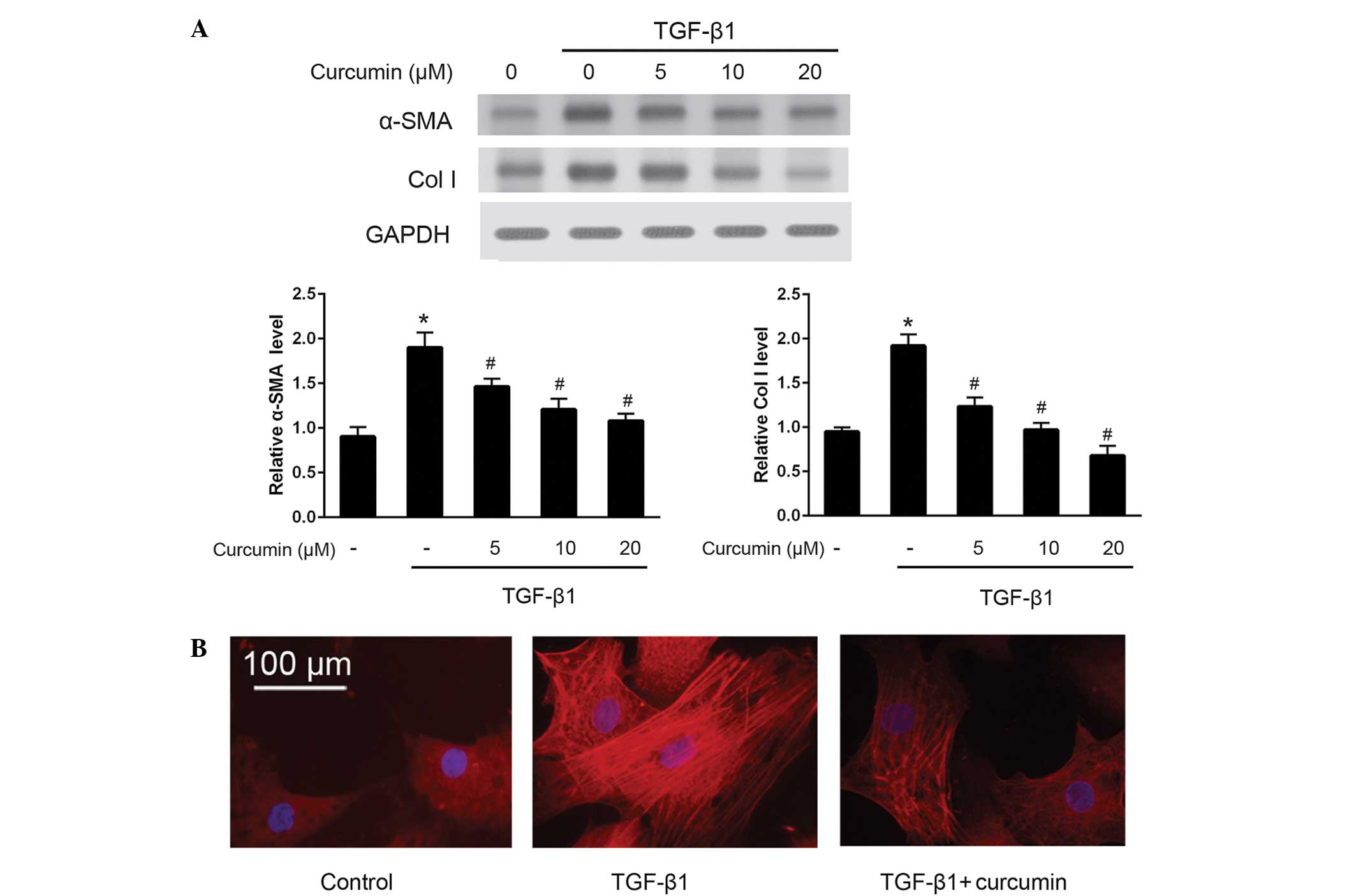
To investigate the effects of curcumin on the
differentiation of CFs into myofibroblasts, western blotting was
used to detect the protein expression levels of α-SMA and ColI
(Fig. 1A). The results demonstrated
that curcumin significantly suppressed the TGF-β1-induced protein
expression of α-SMA and ColI in CFs (P<0.05), in a
dose-dependent manner. These anti-differentiation effects were
further validated by immunofluorescence staining of α-SMA (Fig. 1B). As compared with untreated CFs,
cells induced by 10 ng/ml TGF-β1 exhibited bright fluorescence
staining signals for α-SMA and prominent stress fibers; by
contrast, treatment with 20 µM curcumin significantly attenuated
α-SMA fluorescence signals and morphological characteristics of CFs
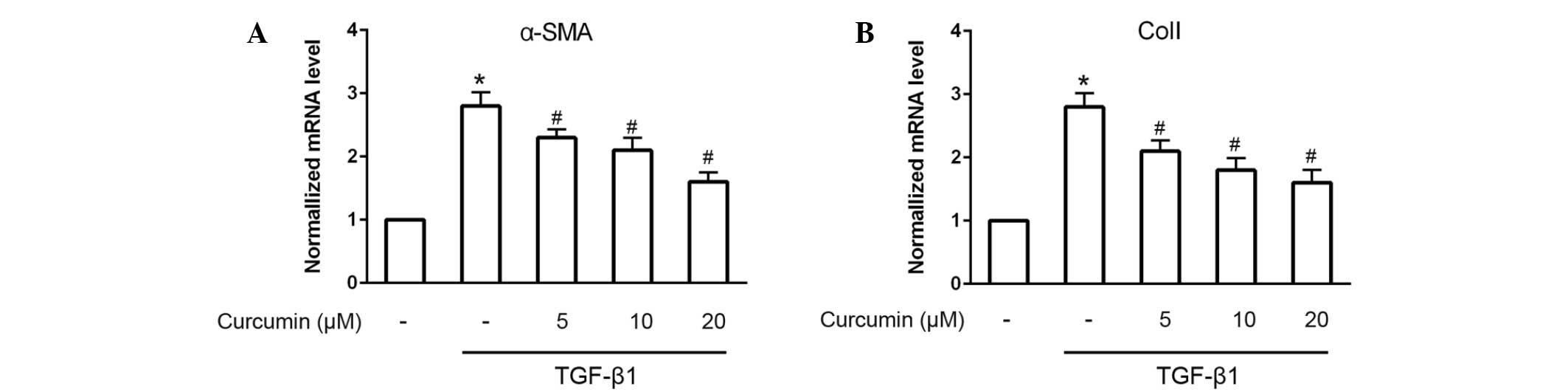
induced by 10 ng/ml TGF-β1 (P<0.05). Furthermore, RT-qPCR was
performed to analyze the mRNA expression levels of α-SMA and ColI.
The results demonstrated that α-SMA and ColI mRNA expression levels
in TGF-β1-induced CFs were also significantly and dose-dependently
decreased following treatment with curcumin (P<0.05; Fig. 2).
TGF-β1-activated Smad2 and p38
signaling pathways in CFs
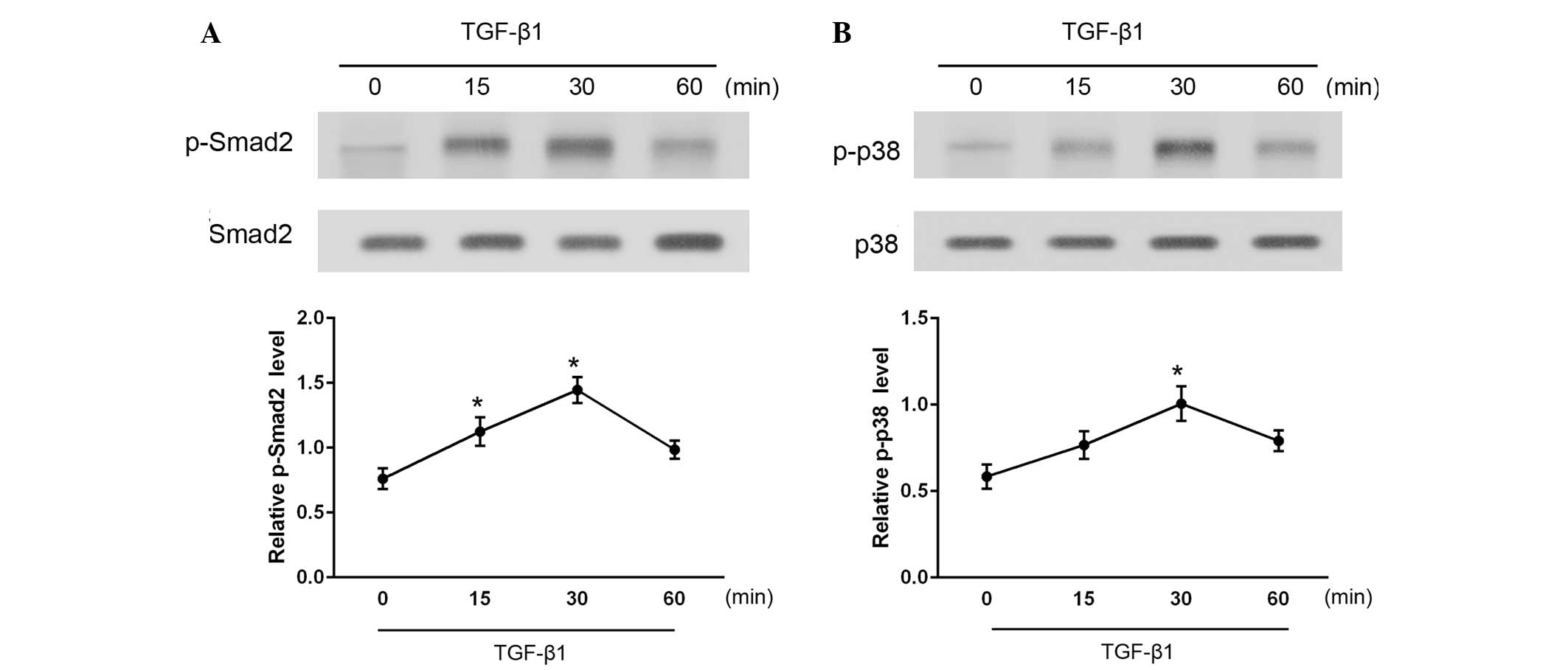
To investigate the downstream signaling pathways in
TGF-β1-stimulated CFs, the levels of activated (phosphorylated)
Smad2 and p38 were measured at various time points between 15 and
60 min after TGF-β1 administration (Fig.
3). As determined by western blotting, phosphorylated Smad2 and
p38 (p-Smad2 and p-p38) levels were significantly increased
following stimulation with TGF-β1 (P<0.05). Total Smad2 and p38
protein expression levels were also examined and found to be
unaffected by TGF-β1 administration. These results indicate that
treatment with TGF-β1 activates the Smad2 and p38 signaling
pathways in CFs.
Smad2 inhibitor, SB431542, and p38
inhibitor, SB203580, suppress TGF-β1-induced α-SMA and ColI
expression levels
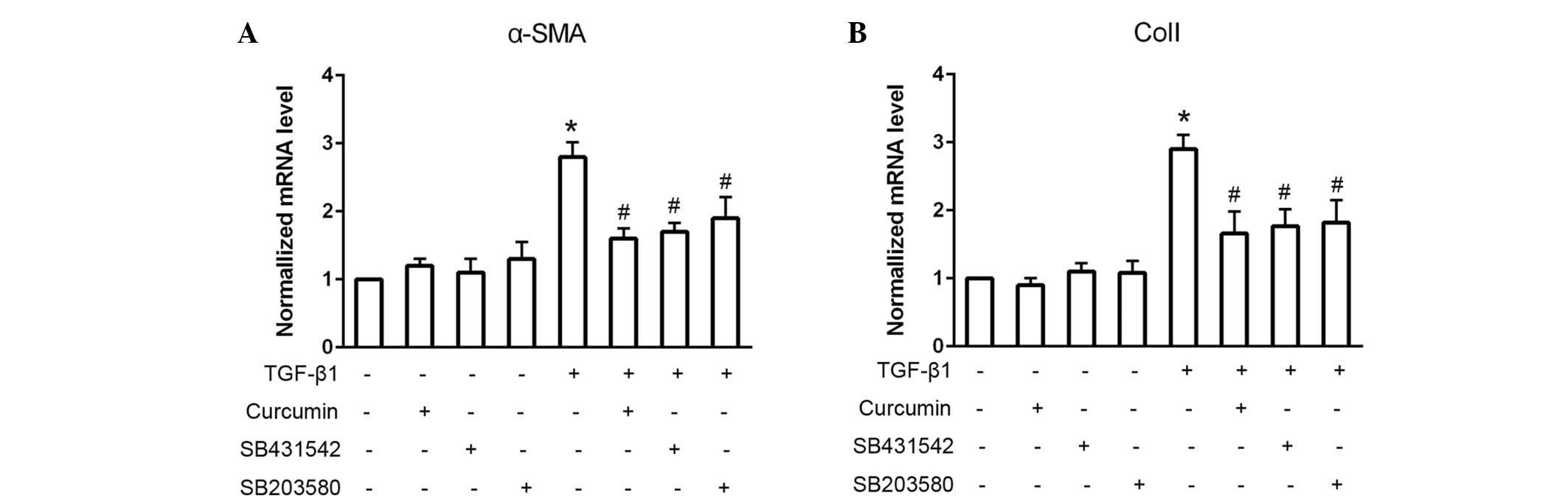
To assess whether Smad2 and p38 were associated with
the TGF-β1-induced differentiation of CFs, the Smad2 and p38
inhibitors, SB431542 and SB203580, respectively, were used to block
these signaling pathways (Fig. 4).
When administered alone, SB431542 and SB203580 exhibited no
significant effect on cardiac fibroblast differentiation, as
detected by consistent mRNA α-SMA and ColI expression levels
(P>0.05). However, pretreatment with SB431542 or SB203580 prior
to TGF-β1 administration significantly suppressed the
TGF-β1-induced mRNA expression levels of α-SMA and ColI
(P<0.05).
Curcumin inhibits TGF-β1-induced
activation of the Smad2 signaling pathway
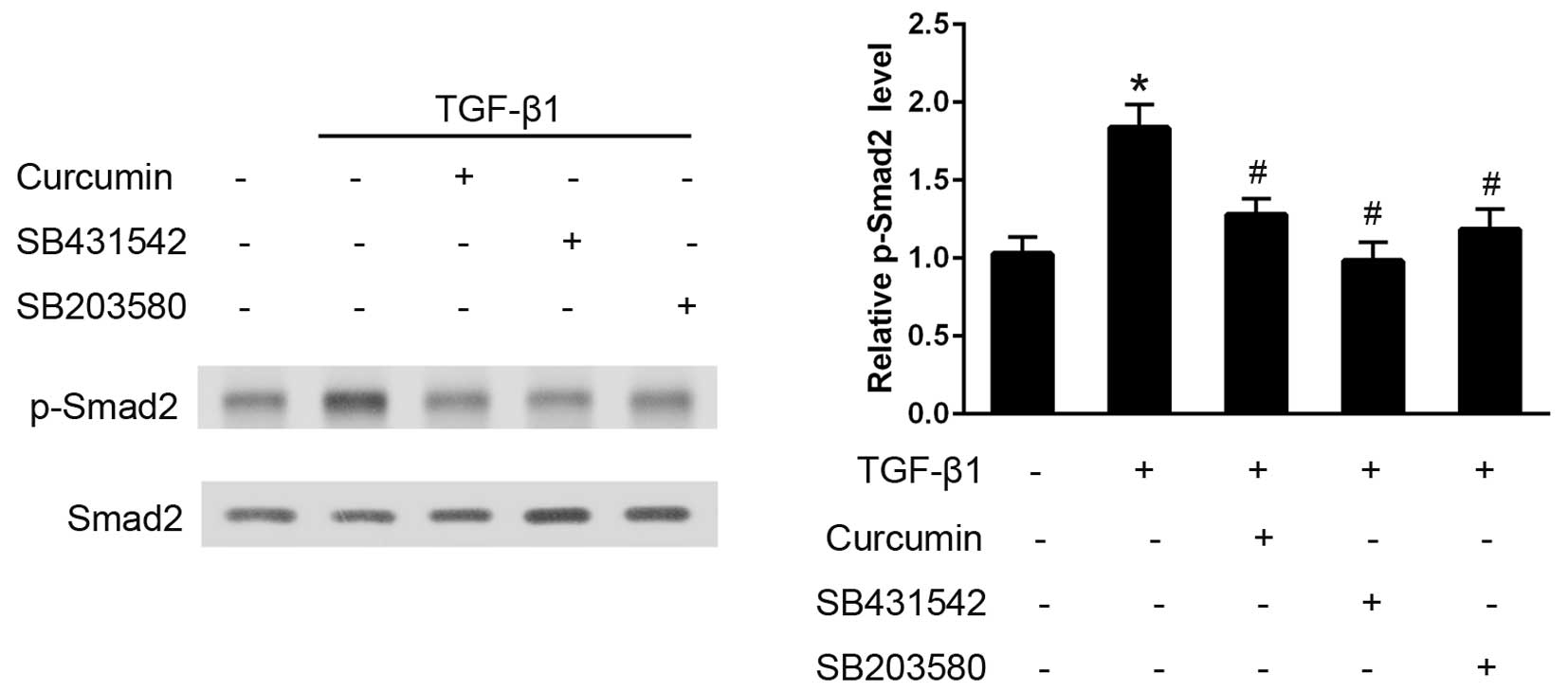
In CFs, TGF-β1 administration induced an early and
significant increase in p-Smad2 expression levels over the baseline
value in 30 min, as analyzed using western blotting (P<0.05;
Fig. 5). Pre-treatment with curcumin
induced a significant decrease in the degree of Smad2
phosphorylation and, as hypothesized, SB431542 (P<0.05), which
is a well established TGF-βR-Smad2 inhibitor, effectively
eliminated the activation of Smad2. Furthermore, pre-treatment with
the p38 inhibitor, SB203580, also significantly attenuated the
TGF-β1-induced Smad2 phosphorylation (P<0.05).
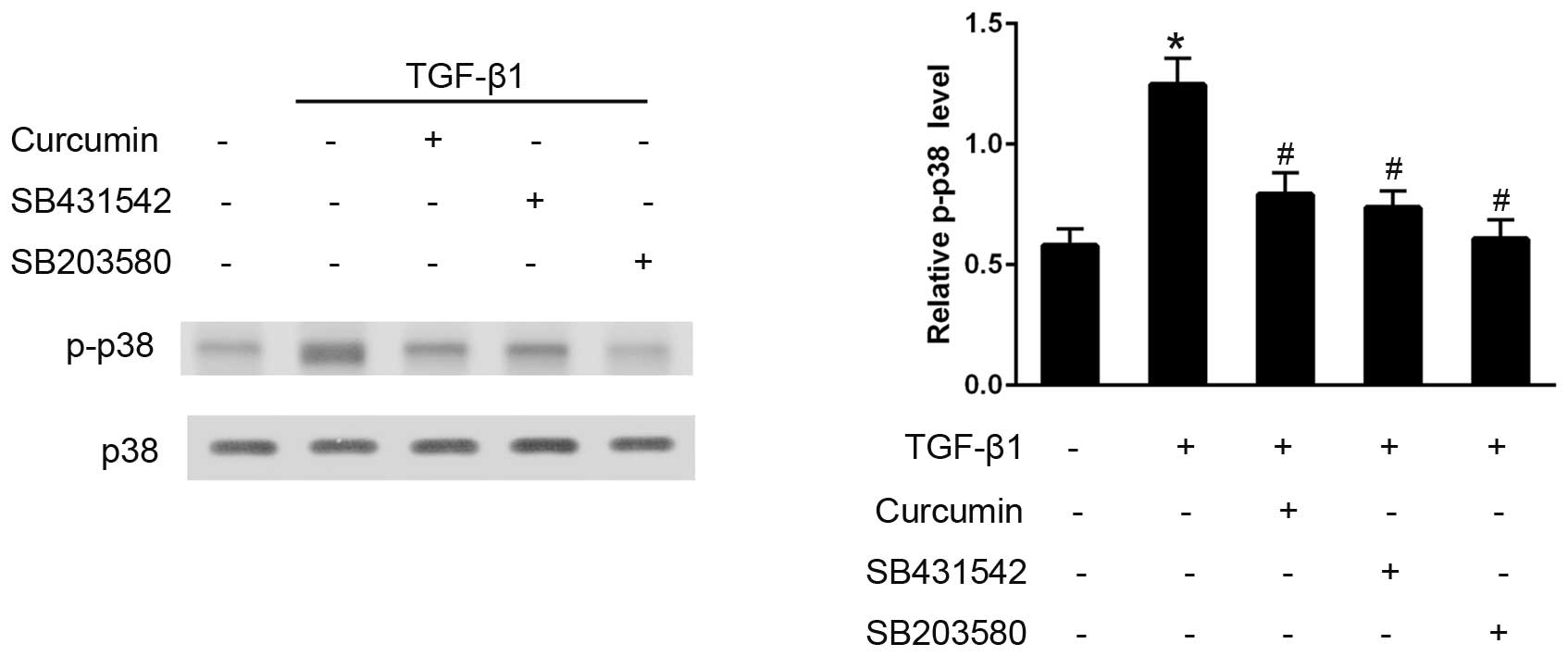
Curcumin inhibits TGF-β1-induced
activation of the p38 pathway
The effects of curcumin on the TGF-β1-induced
activation of the p38 signaling pathway were also examined. Western
blot analysis demonstrated that, in TGF-β1-stimulated CFs, curcumin
pre-treatment significantly decreased p-p38 expression levels, and
SB203580 effectively eliminated p38 activation (P<0.05).
Furthermore, SB431542 administration also significantly inhibited
the phosphorylation of p38, thus preventing its activation
(P<0.05; Fig. 6).
Discussion
In the present study, curcumin was demonstrated to
dose-dependently inhibit the TGF-β1-induced cardiac fibroblast
differentiation. Furthermore, the results indicated that these
effects may be mediated by the Smad2 and p38 signaling pathways.
Therefore, the present results suggested that curcumin may be a
potential therapeutic agent for the treatment of myocardial
fibrosis, which has been associated with the pathogenesis of heart
failure (1).
The transformation of CFs to cardiac myofibroblasts
is a key event in cardiac fibrosis. Cardiac myofibroblasts are
absent from normal myocardium, however, they are the predominant
source of excessive extracellular collagen in cardiac fibrosis
(20,21). In addition to aggravating cardiac
dysfunction, previous studies have demonstrated that the
persistence of cardiac myofibroblasts also contributes to malignant
arrhythmia (22–24). Therefore, factors associated with the
formation of cardiac myofibroblasts are of considerable clinical
interest.
High expression levels of α-SMA are a hallmark of
the formation of cardiac myofibroblasts (24,25).
Collagen, particularly fibrillar ColI, is the predominant component
of the ECM and is excessively synthesized by cardiac myofibroblasts
(26,27). The results of the present study
demonstrated that curcumin administration effectively suppressed
TGF-β1-induced cardiac fibroblast differentiation, as determined by
the decreased expression of α-SMA and ColI, at the protein and mRNA
levels. These findings are consistent with previous studies which
have demonstrated that curcumin is capable of inducing
anti-fibrotic effects in cultured CFs (10,28).
TGF-β1, which is the most well-characterized
cytokine, induces the differentiation of CFs to cardiac
myofibroblasts (29,30). Activation of the Smad cascade has an
essential role in the differentiation of myofibroblasts (15,16,31) and
is regarded as the classical mediator of TGF-β1. Following TGF-β1
stimulation, the TGF-β1RI serine-threonine kinase phosphorylates
the receptor-Smads (R-Smads), and Smad2 subsequently forms a
complex with Smad3 that, in turn, associates with a Co-Smad (Smad4)
and translocates into the nucleus, where it acts as a transcription
factor (32). In addition to
Smad-dependent pathways, previous studies have suggested that Smad
independent pathways, such as p38 MAPK, may also play a pivotal
role in TGF-β1-mediated differentiation (17,33). In
the present study, inhibition of Smad2 and p38 consistently
attenuated the TGF-β1-induced cardiac fibroblast differentiation,
whereas curcumin was able to reduce the activation of Smad2 and p38
molecular signaling. Therefore, curcumin may have inhibited
TGF-β1-induced activation via the Smad2 and p38 signaling pathways.
Furthermore, the Smad2 inhibitor SB431542 effectively prohibited
the phosphorylation of p38, and the p38 inhibitor SB203580 reduced
the phosphorylation of Smad2. These results demonstrated that there
is cross-talk or interaction between these two signaling pathways;
however, the detailed mechanism and biological consequences of
Smad2 and p38 activation remain unclear. This potential interaction
among Smad2, p38 and other associated pathways requires further
investigation.
In conclusion, the results of the present study
suggested that curcumin has fibrosis suppressor properties and an
inhibitory effect on TGF-β1-induced cardiac fibroblast
differentiation, which may be mediated via the suppression of the
Smad2 and p38 signaling pathways. Therefore, curcumin may be a
potential novel therapeutic agent for the treatment of cardiac
fibrosis.
Glossary
Abbreviations
Abbreviations:
|
CFs
|
cardiac fibroblasts
|
|
α-SMA
|
α-smooth muscle actin
|
|
ColI
|
collagen type I
|
References
|
1
|
Daskalopoulos EP, Janssen BJ and
Blankesteijn WM: Myofibroblasts in the infarct area: Concepts and
challenges. Microsc Microanal. 18:35–49. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gupta SC, Patchva S, Koh W and Aggarwal
BB: Discovery of curcumin, a component of golden spice and its
miraculous biological activities. Clin Exp Pharmacol Physiol.
39:283–299. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chiu J, Khan ZA, Farhangkhoee H and
Chakrabarti S: Curcumin prevents diabetes-associated abnormalities
in the kidneys by inhibiting p300 and nuclear factor-κB. Nutrition.
25:964–972. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kolodziejczyk J, Olas B, Saluk-Juszczak J
and Wachowicz B: Antioxidative properties of curcumin in the
protection of blood platelets against oxidative stress in vitro.
Platelets. 22:270–276. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Abe Y, Hashimoto S and Horie T: Curcumin
inhibition of inflammatory cytokine production by human peripheral
blood monocytes and alveolar macrophages. Pharmacol Res. 39:41–47.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Teiten MH, Gaascht F, Cronauer M, Henry E,
Dicato M and Diederich M: Anti-proliferative potential of curcumin
in androgen-dependent prostate cancer cells occurs through
modulation of the Wingless signaling pathway. Int J Oncol.
38:603–611. 2011.PubMed/NCBI
|
|
7
|
Hartojo W, Silvers AL, Thomas DG, Seder
CW, Lin L, Rao H, Wang Z, Greenson JK, Giordano TJ, Orringer MB, et
al: Curcumin promotes apoptosis, increases chemosensitivity, and
inhibits nuclear factor kappaB in esophageal adenocarcinoma. Transl
Oncol. 3:99–108. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chanvorachote P, Pongrakhananon V,
Wannachaiyasit S, Luanpitpong S, Rojanasakul Y and Nimmannit U:
Curcumin sensitizes lung cancer cells to cisplatin-induced
apoptosis through superoxide anion-mediated Bcl-2 degradation.
Cancer Invest. 27:624–635. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ghosh SS, Salloum FN, Abbate A, Krieg R,
Sica DA, Gehr TW and Kukreja RC: Curcumin prevents cardiac
remodeling secondary to chronic renal failure through deactivation
of hypertrophic signaling in rats. Am J Physiol Heart Circ Physiol.
299:H975–H984. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li HL, Liu C, de Couto G, Ouzounian M, Sun
M, Wang AB, Huang Y, He CW, Shi Y, Chen X, et al: Curcumin prevents
and reverses murine cardiac hypertrophy. J Clin Invest.
118:879–893. 2008.PubMed/NCBI
|
|
11
|
Wang NP, Wang ZF, Tootle S, Philip T and
Zhao ZQ: Curcumin promotes cardiac repair and ameliorates cardiac
dysfunction following myocardial infarction. Br J Pharmacol.
167:1550–1562. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Morimoto T, Sunagawa Y, Kawamura T, Takaya
T, Wada H, Nagasawa A, Komeda M, Fujita M, Shimatsu A, Kita T and
Hasegawa K: The dietary compound curcumin inhibits p300 histone
acetyltransferase activity and prevents heart failure in rats. J
Clin Invest. 118:868–878. 2008.PubMed/NCBI
|
|
13
|
Soetikno V, Sari FR, Sukumaran V,
Lakshmanan AP, Mito S, Harima M, Thandavarayan RA, Suzuki K, Nagata
M, Takagi R and Watanabe K: Curcumin prevents diabetic
cardiomyopathy in streptozotocin-induced diabetic rats: Possible
involvement of PKC-MAPK signaling pathway. Eur J Pharm Sci.
47:604–614. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Biernacka A, Dobaczewski M and
Frangogiannis NG: TGF-β signaling in fibrosis. Growth factors.
29:196–202. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cucoranu I, Clempus R, Dikalova A, Phelan
PJ, Ariyan S, Dikalov S and Sorescu D: NAD (P) H oxidase 4 mediates
transforming growth factor-β1-induced differentiation of cardiac
fibroblasts into myofibroblasts. Circ Res. 97:900–907. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dobaczewski M, Bujak M, Li N,
Gonzalez-Quesada C, Mendoza LH, Wang XF and Frangogiannis NG: Smad3
signaling critically regulates fibroblast phenotype and function in
healing myocardial infarction. Circ Res. 107:418–428. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Deaton RA, Su C, Valencia TG and Grant SR:
Transforming growth factor-β1-induced expression of smooth muscle
marker genes involves activation of PKN and p38 MAPK. J Biol Chem.
280:31172–31181. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yan-Hong F, Hui D, Qing P, Lei S,
Hai-Chang W, Wei Z and Yan-Jie C: Effects of arginine vasopressin
on differentiation of cardiac fibroblasts into myofibroblasts. J
Cardiovasc Pharmacol. 55:489–495. 2010.PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Porter KE and Turner NA: Cardiac
fibroblasts: at the heart of myocardial remodeling. Pharmacol Ther.
123:255–278. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
van den Borne SW, Diez J, Blankesteijn WM,
Verjans J, Hofstra L and Narula J: Myocardial remodeling after
infarction: the role of myofibroblasts. Nat Rev Cardiol. 7:30–37.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sabbah HN, Sharov VG, Lesch M and
Goldstein S: Progression of heart failure: A role for interstitial
fibrosis. Mol Cell Biochem. 147:29–34. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
McDowell KS, Arevalo HJ, Maleckar MM and
Trayanova NA: Susceptibility to arrhythmia in the infarcted heart
depends on myofibroblast density. Biophys J. 101:1307–1315. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rohr S: Myofibroblasts in diseased hearts:
new players in cardiac arrhythmias? Heart Rhythm. 6:848–856. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Leask A: Potential therapeutic targets for
cardiac fibrosis TGFβ, angiotensin, endothelin, CCN2 and PDGF,
partners in fibroblast activation. Circ Res. 106:1675–1680. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chapman D, Weber KT and Eghbali M:
Regulation of fibrillar collagen types I and III and basement
membrane type IV collagen gene expression in pressure overloaded
rat myocardium. Circ Res. 67:787–794. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cleutjens JP, Verluyten MJ, Smiths JF and
Daemen MJ: Collagen remodeling after myocardial infarction in the
rat heart. Am J Pathol. 147:325–338. 1995.PubMed/NCBI
|
|
28
|
Meng Z, Yu Xh, Chen J, Li L and Li S:
Curcumin attenuates cardiac fibrosis in spontaneously hypertensive
rats through PPAR-γ activation. Acta Pharmacol Sin. 35:1247–1256.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bujak M and Frangogiannis NG: The role of
TGF-β signaling in myocardial infarction and cardiac remodeling.
Cardiovasc Res. 74:184–195. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Leask A: TGFbeta, cardiac fibroblasts and
the fibrotic response. Cardiovasc Res. 74:207–212. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Carthy JM, Garmaroudi FS, Luo Z and
McManus BM: Wnt3a induces myofibroblast differentiation by
upregulating TGF-β signaling through SMAD2 in a β-catenin-dependent
manner. PloS one. 6:e198092011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Horowitz JC, Rogers DS, Sharma V, Vittal
R, White ES, Cui Z and Thannickal VJ: Combinatorial activation of
FAK and AKT by transforming growth factor-beta1 confers an
anoikis-resistant phenotype to myofibroblasts. Cell Signal.
19:761–771. 2007. View Article : Google Scholar : PubMed/NCBI
|