Introduction
Multiple renal cysts present clinical features in a
range of kidney diseases that may be inherited as dominant or
recessive traits (1). Autosomal
dominant diseases include autosomal dominant polycystic kidney
disease (ADPKD), which is by far the most common inherited cause of
kidney cysts worldwide, as well as HNF1B-associated kidney
disease, medullary cystic kidney disease (MCKD) and cystic
neoplasms (such as Von Hippel-Lindau disease and tuberous sclerosis
complex) (2). Although these
conditions may present in children, they often cause adult-onset
kidney disease, and a family history of kidney problems is
frequently reported (3). Recessive
conditions commonly present in childhood and often occur in the
absence of a relevant family history. They include autosomal
recessive polycystic kidney disease (ARPKD), nephronophthisis
(NPHP) and multi-system ciliopathies, such as Bardet-Biedl, Joubert
and Meckel-Gruber syndromes (4).
A range of other uncommon disorders
characteristically presents with congenital abnormalities of the
kidney and urinary tract (CAKUT), but may also manifest as cystic
kidney disease (5). These diseases
include: Townes-Brocks syndrome (TBS), which is caused by mutations
in SALL1; renal coloboma syndrome, which is associated with
PAX2 mutations; oral facial digital syndrome type 1, which
is due to mutations in gene OFD1; and other disorders that
frequently include extra-renal manifestations (6). Imaging features of these disorders are
variable and may overlap, while renal histology is frequently
unavailable or does not help the diagnosis (7). Thus, recognition of extra-renal
features is of critical importance in establishing the correct
diagnosis. This is particularly valuable in cases where no family
history is available, and therefore the differential diagnosis
includes both dominant and recessive conditions. The mode of
transmission determines whether there is a risk that other family
members may be affected, thus affecting reproductive
decision-making. Confirmation of the diagnosis by genetic testing
can therefore be clinically valuable in this context.
The current study presents the case of a 16-year-old
male patient with autosomal dominant TBS, who was initially
incorrectly diagnosed as having ARPKD. Appreciation of extra-renal
features prompted genetic testing that confirmed the correct
diagnosis of an autosomal dominant disease, which has significant
implications for the patient and family members.
Case report
The proband was a 16-year-old male who was referred
to XinHua Hospital (Shanghai, China) in May 2014 with end-stage
renal failure (ESRF) and a serum creatinine (Scr) level of 932
µmol/l (normal range, 35–97 µmol/l). The patient had been found to
have proteinuria of 30–300 mg/dl (normal range 0–20 mg/dl)) with no
hematuria following an upper respiratory infection at the age of 4
years, in April 2002. There was no family history of kidney disease
and his clinically healthy parents were not consanguineous.
Ultrasound examination at the age of 4 years demonstrated that the
patient's kidneys were a normal size for his age [left kidney,
69×33 mm; right kidney, 58×31 mm; reported normal kidney lengths
for a 4-year-old, 70.30±5.20 mm (8)]
with multiple bilateral cortical cysts (largest cyst, 10×17 mm).
The Scr level (45 µmol/l) and blood pressure were within the normal
range at that time. Liver function tests and hepatic imaging were
also normal. The lack of family history and childhood onset of
disease suggested a diagnosis of ARPKD, while ADPKD was also
considered possible by the treating clinicians. The renal function
gradually deteriorated and the Scr reached a level of 400 µmol/l at
the age of 15 years, with renal dialysis commencing at the age of
16 years at the Dialysis Center of XinHua Hospital.
Upon reviewing the medical history, it was found
that the patient had an imperforate anus at birth, which was
surgically repaired when he was 3 months old, as well as pre-axial
polydactyly of both hands, which was surgically corrected when the
patient was 3 years old. The patient was also reported to have
exhibited mild bilateral hearing loss since the age of 5 years.
However, no growth retardation or learning difficulties were
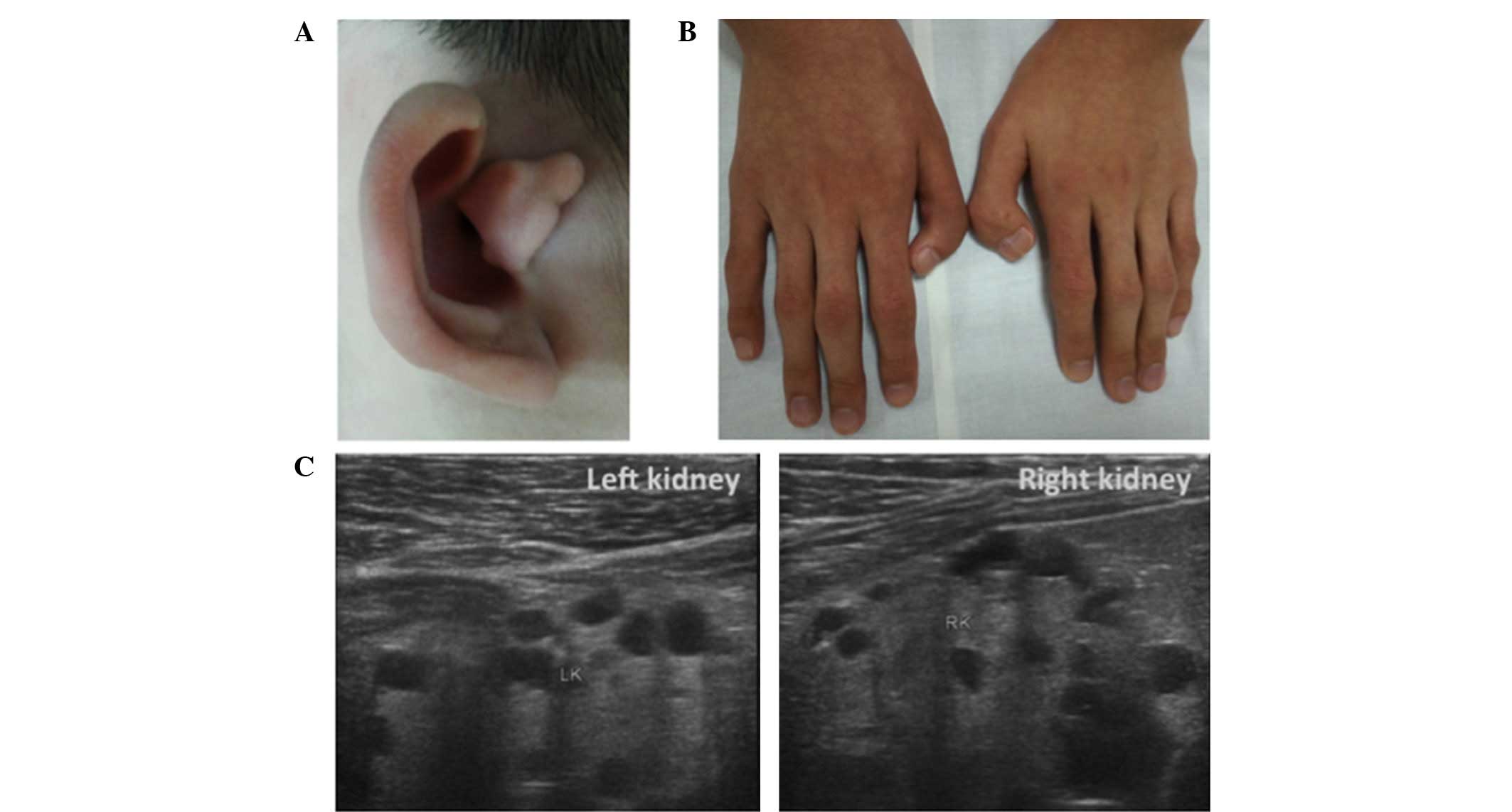
reported. Physical examination revealed a dysplastic right ear with
overfolded superior helices (Fig.
1A), as well as bilateral surgically corrected thumbs (Fig. 1B). Renal ultrasound examination
performed on an Ultrasound LOGIQ 500 (GE Healthcare Milwaukee, WI,
USA) showed bilateral small kidneys (left kidney, 74×36 mm; right
kidney, 59×31 mm) with multiple cortical and medullary cysts,
ranging between 10 and 17 mm (Fig.
1C). No other urinary tract structural abnormality was
identified, imaging assessments did not detect any abnormalities of
the liver, spleen or pancreas, and an echocardiogram was normal.
The parents were clinically unaffected and no family history of
genetic disease was reported.
Based on the aforementioned extra-renal features, a
diagnosis of TBS was suspected. Following approval by the Ethical
Committee of Shanghai XinHua Hospital according to the standards of
the Declaration of Helsinki and the signed informed consent
obtained from the proband, DNA was extracted from the proband's
peripheral blood. SALL1 gene coding regions and splice sites
were amplified and directly sequenced using an ABI PRISM 3730xl
Genetic Analyzer (Applied Biosystems; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) as reported previously (9), (SALL1 sequencing primer
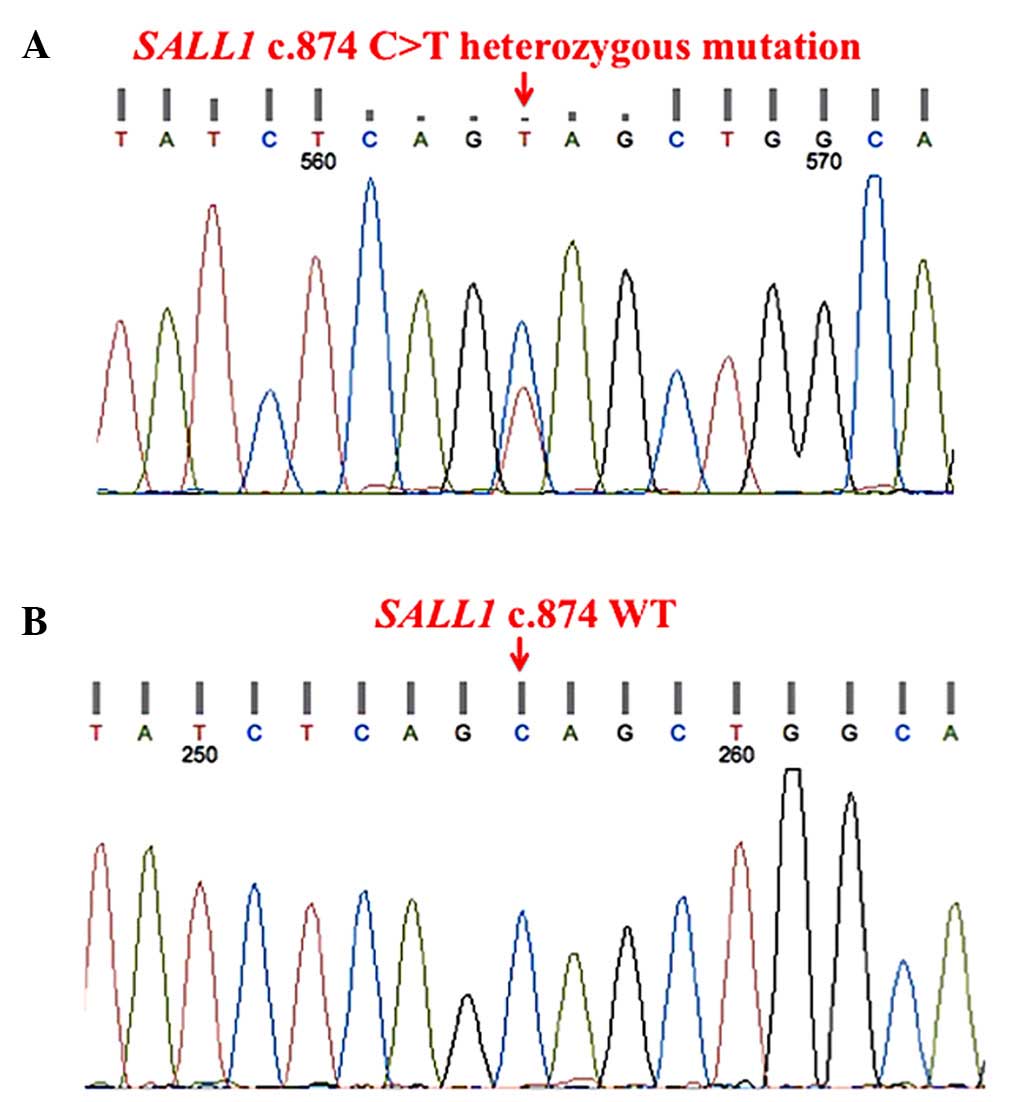
sequences are available upon request). Sequencing analysis
disclosed a novel SALL1 heterozygous nonsense mutation
located in the mutational ‘hotspot’ of exon 2 (c.874C>T,
p.Q292X; Fig. 2A), and this mutation
was not present in the 100 unrelated healthy adult volunteers (all
of whom provided informed consent, were free from renal disease and
matched geographically) used as controls (Fig. 2B). The parents of the proband
declined genetic testing, however it appears likely that this
represents a de novo mutation, which is estimated to occur
in ~50% of patients with TBS (10),
as the parents of the proband are clinically healthy. It is also
possible that one of the parents carries the mutation in a mosaic
state, as germline mosaicism in clinically unaffected parents has
been reported in TBS cases (11). As
the patient had reached ESRF at the time of genetic diagnosis, the
patient remained on maintenance hemodialysis three times per week
until the final follow-up in March 2015.
Discussion
TBS has a prevalence of 1 in 250,000 individuals in
the general population (12). It is
a rare autosomal dominant malformation syndrome characterized by
the core triad of anorectal malformation (imperforate anus,
anteriorly-placed anus and anal stenosis), hand malformations
(pre-axial polydactyly, triphalangeal thumbs and bifid thumb) and
external ear malformation (preauricular ear tags and overfolding of
ear helices) associated with deafness (13). TBS is confirmed by identification of
a pathogenic mutation in SALL1 with the detection rate of
64–83% (14). SALL1 is a
human gene homologous to the developmental regulator sal of
Drosophila melanogaster, which is considered to be an
essential organogenesis regulator for urological, renal, limb, ear,
brain and liver development (15).
Sall1 protein, encoded by SALL1, is
abundantly expressed in embryonic kidneys and plays a pivotal role
in mammalian nephrogenesis by controlling the expression of major
renal development genes, including PAX8, GDNF and
FOXD1 (16). Mice with
homozygous deletion of Sall1 (SALL1−/-) have been
observed to succumb to death in the perinatal period due to renal
agenesis or severe dysgenesis (17),
while another mouse model expressing a truncated N-terminal protein
(Sall1-N) recapitulated human TBS renal defects of renal hypoplasia
with or without cysts (18). The
incidence of renal anomalies in TBS patients ranges between 20 and
62.5%, with heterogeneous phenotypes including unilateral or
bilateral renal hypoplasia or dysplasia, renal agenesis,
multicystic kidneys, vesicoureteral reflux, posterior urethral
valve or meatal stenosis (19). In
addition, renal hypodysplasia has also been reported as an isolated
TBS phenotype (20). A study
involving 749 patients with CAKUT revealed that SALL1
mutations account for >20% of all the identified mutations (9
out of 37 mutations), indicating that this mutation is more common
than was expected (21). Therefore,
kidney imaging is recommended for TBS patients and in case
presenting renal anomalies. Furthermore, close monitoring of renal
function and appropriate medical interventions are indicated, as
TBS has been reported to be associated with early-onset renal
failure with nearly 15% of TBS patients with renal anomalies
progressing to ESRF between the ages of 1 and 23 years (19).
The patient reported in the present study harbored a
novel heterozygous nonsense mutation (c.874C>T, p.Q292X) in the
SALL1 mutational hotspot region. This mutational hotspot
region is located within exon 2 between nucleotide 765, 3′ of
nucleotides 687–750 encoding the glutamine-rich interaction domain,
and nucleotide 1565, 3′ of the region encoding the first double
zinc finger domain (22). Frameshift
and nonsense SALL1 mutations occur in this mutational
hotspot region, such as the most common SALL1 heterozygous
mutation, which is c.826C>T, p.R276X. These mutations are
usually associated with classical TBS and a more severe phenotype,
including renal failure (10,19,22),
which suggests a dominant-negative or gain-of-function mechanism
caused by the abnormal truncated Sall1 protein. In comparison,
haploinsufficiency for SALL1 caused by heterozygous large or
whole gene deletions have been described in milder TBS cases
(9). Only one previous study has
described renal insufficiency and ESRF in two Japanese adolescent
patients with 16q12 microdeletion including the entire SALL1
gene (23).
In the present TBS case, the correct diagnosis was
delayed until the patient reached ESRF, suggesting that recognition
of extra-renal features is of critical importance in making a
diagnosis in cystic kidney diseases, particularly when renal
imaging is atypical. The possibility that clinical features of TBS
may overlap with other rare developmental syndromes such as VACTERL
association (24), Goldenhar
syndrome (25) and branchiootorenal
(BOR) syndrome (26), should also be
considered. Additional features, such as vertebral abnormalities
and trachea-esophageal fistula in VACTERAL association,
craniofacial abnormalities in Goldenhar syndrome and branchial arch
anomalies in BOR syndrome, may aid the diagnosis of these
disorders.
In conclusion, the present case illustrated that TBS
may present with normal-sized cystic kidneys in childhood.
Recognition of extra-renal features of cystic kidney diseases, such
as TBS, and genetic testing may facilitate the correct diagnosis,
which is necessary to give the correct advice to patients and their
families regarding the mode of transmission and consequent risk to
relatives and offsprings of those affected. As TBS can cause ESRF
early in life, renal imaging and close monitoring of renal function
are recommended for TBS patients with renal anomalies.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81500507), the Young
Investigator Funding provided by the Shanghai Health Bureau (grant
no. 20114Y108) and the SMC-Young Investigator Funding (grant no.
2013SMCYIF01) provided by Shanghai Jiao Tong University.
References
|
1
|
Bergmann C: ARPKD and early manifestations
of ADPKD: The original polycystic kidney disease and phenocopies.
Pediatr Nephrol. 30:15–30. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ong AC, Devuyst O, Knebelmann B and Walz
G: ERA-EDTA Working Group for Inherited Kidney Diseases. Autosomal
dominant polycystic kidney disease: The changing face of clinical
management. Lancet. 385:1993–2002. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schrier RW, Brosnahan G, Cadnapaphornchai
MA, Chonchol M, Friend K, Gitomer B and Rossetti S: Predictors of
autosomal dominant polycystic kidney disease progression. J Am Soc
Nephrol. 25:2399–2418. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hoyer PF: Clinical manifestations of
autosomal recessive polycystic kidney disease. Curr Opin Pediatr.
27:186–192. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Devuyst O, Knoers NV, Remuzzi G and
Schaefer F: Board of the Working Group for Inherited Kidney
Diseases of the European Renal Association and European Dialysis
and Transplant Association: Rare inherited kidney diseases:
Challenges, opportunities, and perspectives. Lancet. 383:1844–1859.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hwang DY, Dworschak GC, Kohl S, Saisawat
P, Vivante A, Hilger AC, Reutter HM, Soliman NA, Bogdanovic R,
Kehinde EO, et al: Mutations in 12 known dominant disease-causing
genes clarify many congenital anomalies of the kidney and urinary
tract. Kidney Int. 85:1429–1433. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kiser RL, Wolf MT, Martin JL, Zalewski I,
Attanasio M, Hildebrandt F and Klemmer P: Medullary cystic kidney
disease type 1 in a large Native-American kindred. Am J Kidney Dis.
44:611–617. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Luk WH, Lo AX, Au-Yeung AW, Liu KK, Woo
YH, Chiang CC and Lo KK: Renal length nomogram in Hong Kong Asian
children: Sonographic measurement and multivariable approach. J
Paediatr Child Health. 46:310–315. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Miller EM, Hopkin R, Bao L and Ware SM:
Implications for genotype-phenotype predictions in Townes-Brocks
syndrome: Case report of a novel SALL1 deletion and review of the
literature. Am J Med Genet A. 158A:533–540. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Reardon W, Casserly LF, Birkenhäger R and
Kohlhase J: Kidney failure in Townes-Brocks syndrome: An under
recognized phenomenon? Am J Med Genet A. 143A:2588–2591. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kohlhase J, Taschner PE, Burfeind P,
Pasche B, Newman B, Blanck C, Breuning MH, ten Kate LP,
Maaswinkel-Mooy P, Mitulla B, et al: Molecular analysis of SALL1
mutations in Townes-Brocks syndrome. Am J Hum Genet. 64:435–445.
1999. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Eker HK and Balasar Ö: Variable
expressivity of renal involvement in a further family with
Townes-Brocks syndrome. Clin Dysmorphol. 24:24–25. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lawrence C, Hong-McAtee I, Hall B,
Hartsfield J, Rutherford A, Bonilla T and Bay C: Endocrine
abnormalities in Townes-Brocks syndrome. Am J Med Genet A.
161A:2266–2273. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Albrecht B, Liebers M and Kohlhase J:
Atypical phenotype and intrafamilial variability associated with a
novel SALL1 mutation. Am J Med Genet A. 125A:102–104. 2014.
View Article : Google Scholar
|
|
15
|
Kohlhase J, Wischermann A, Reichenbach H,
Froster U and Engel W: Mutations in the SALL1 putative
transcription factor gene cause Townes-Brocks syndrome. Nat Genet.
18:81–83. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Basta JM, Robbins L, Kiefer SM, Dorsett D
and Rauchman M: Sall1 balances self-renewal and differentiation of
renal progenitor cells. Development. 141:1047–1058. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nishinakamura R, Matsumoto Y, Nakao K,
Nakamura K, Sato A, Copeland NG, Gilbert DJ, Jenkins NA, Scully S,
Lacey DL, et al: Murine homolog of SALL1 is essential for ureteric
bud invasion in kidney development. Development. 128:3105–3115.
2001.PubMed/NCBI
|
|
18
|
Kiefer SM, Ohlemiller KK, Yang J, McDill
BW, Kohlhase J and Rauchman M: Expression of a truncated Sall1
transcriptional repressor is responsible for Townes-Brocks syndrome
birth defects. Hum Mol Genet. 12:2221–2227. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Faguer S, Pillet A, Chassaing N,
Merhenberger M, Bernadet-Monrozies P, Guitard J and Chauveau D:
Nephropathy in Townes-Brocks syndrome (SALL1 mutation): Imaging and
pathological findings in adulthood. Nephrol Dial Transplant.
24:1341–1345. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Weber S, Moriniere V, Knüppel T, Charbit
M, Dusek J, Ghiggeri GM, Jankauskiené A, Mir S, Montini G,
Peco-Antic A, et al: Prevalence of mutations in renal developmental
genes in children with renal hypodysplasia: Results of the ESCAPE
study. J Am Soc Nephrol. 17:2864–2870. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hwang DY, Dworschak GC , Kohl S, Saisawat
P, Vivante A, Hilger AC, Reutter HM, Soliman NA, Bogdanovic R,
Kehinde EO, et al: Mutations in 12 known dominant disease-causing
genes clarify many congenital anomalies of the kidney and urinary
tract. Kidney Int. 85:1429–1433. 2014.
|
|
22
|
Botzenhart EM, Bartalini G, Blair E, Brady
AF, Elmslie F, Chong KL, Christy K, Torres-Martinez W, Danesino C,
Deardorff MA, et al: Townes-Brocks syndrome: Twenty novel SALL1
mutations in sporadic and familial cases and refinement of the
SALL1 hot spot region. Hum Mutat. 28:204–205. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Morisada N, Sekine T, Ishimori S, Tsuda M,
Adachi M, Nozu K, Nakanishi K, Tanaka R and Iijima K: 16q12
microdeletion syndrome in two Japanese boys. Pediatr Int.
56:e75–e78. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Solomon BD: VACTERL/VATER association.
Orphanet J Rare Dis. 6:562011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ashokan CS, Sreenivasan A and Saraswathy
GK: Goldenhar syndrome-review with case series. J Clin Diagn Res.
8:ZD17–ZD19. 2014.PubMed/NCBI
|
|
26
|
Engels S, Kohlhase J and McGaughran J: A
SALL1 mutation causes a branchio-oto-renal syndrome-like phenotype.
J Med Genet. 37:458–460. 2000. View Article : Google Scholar : PubMed/NCBI
|