Introduction
The liver is a vital organ that is highly
susceptible to fat accumulation, resulting in a condition known as
fatty liver disease or hepatic steatosis (1). Although chronic alcohol consumption is
a major cause of fatty liver disease (2), non-alcoholic fatty liver disease
(NAFLD) is also common and is strongly associated with central
obesity, insulin resistance, hyperlipidemia and the metabolic
syndrome (3). Insulin resistance
causes increased lipolysis and thus leading to high levels of
plasma free fatty acids (FFAs), as well as increased FFA uptake by
hepatocytes, which results in the formation of intracellular lipid
droplets (4). Hepatic lipid
accumulation can progress from simple steatosis to non-alcoholic
steatohepatitis (NASH), which includes hepatocellular injury,
inflammation and fibrosis (5). In
addition, further severe complications may occur, such as liver
cirrhosis and hepatocellular carcinoma (6).
In order to prevent and treat NAFLD, lifestyle
changes including weight reduction and increased physical activity
are considered as the first-line approach (7). Although pharmacologic therapy is mainly
directed toward increasing insulin sensitivity using
insulin-sensitizing medications, such as metformin and pioglitazone
(8), the use of other medications is
increasingly investigated. Medications used to treat dyslipidemia,
such as gemfibrozil (a triglyceride lowering agent) and statins
(HMG-CoA reductase inhibitors that reduce cholesterol synthesis in
the liver), are among the agents investigated (9). Gemfibrozil has been found to be
beneficial in the treatment of patients with NASH as it was able to
significantly reduce the elevated levels of hepatic
aminotransferases when compared with the control group (10). Similarly, atorvastatin was
efficacious in the treatment of patients with NAFLD and
dyslipidemia as it was effective in reducing hepatic
aminotransferases and improving lipid profile (11,12).
Although statins are administered with caution in patients with
elevated aminotransferases due to the risk of statin-induced
hepatotoxicity, this concern is not clinically important as
statin-associated hepatic adverse effects are of low incidence,
reversible and dose-dependent (13).
Thus, statins should not be contraindicated in patients with NAFLD
and elevated liver enzymes as they are promising medications for
these conditions (9).
Through a review of the relevant literature using
PubMed (http://www.ncbi.nlm.nih.gov/pubmed) and English as a
search language, the majority of studies were found to clinically
evaluate the effect of statin use on reducing the elevated hepatic
enzyme levels in patients with NAFLD (14–17).
A previous study evaluated the effect of statin
therapy on hepatic lipid accumulation by comparing the liver
density measured by computerized tomography prior to and following
statin use for a certain period of time (12). Changes in the density reflected
alterations in hepatic lipid accumulation in response to statin
therapy. However, to the best of our knowledge, the efficacy of
statins in reducing hepatic intracellular lipid accumulation has
not been previously assessed in vitro. Therefore, the aim of
the present study was to evaluate the effect of simvastatin on
hepatic intracellular lipid accumulation on an in vitro
model of NAFLD. A human hepatocellular carcinoma cell line
(HepG2) was exposed to oleic acid (OA), which is a
monounsaturated omega-9 fatty acid, and this served as a model of
NAFLD (18,19). Specifically, the study aimed to
visualize and quantify OA-induced lipid accumulation in
HepG2 cells treated with various concentrations of
simvastatin.
Materials and methods
Cell line
HepG2 cells were a gift of Professor
David Morris at the Department of Surgery at St. George Hospital
Clinical School (New South Wales, Australia). The cell line was
originally obtained from the European Collection of Authenticated
Cell Cultures (Salisbury, UK).
Cell culture method
HepG2 cells were maintained in Dulbecco's
modified Eagle's medium that contained 2 mM L-glutamine (both
purchased from Lonza Australia Pty., Ltd., Mount Waverley,
Australia) and 4.5 g/l glucose (Sigma-Aldrich, Castle Hill,
Australia), and was supplemented with 10% fetal bovine serum (FBS;
Sigma-Alrdich), 0.1% penicillin/streptomycin (Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA), 24 mM sodium hydrogen
carbonate and 25 mM HEPES [also known as
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid] (both purchased
from Lonza Australia Pty., Ltd.). Cells were incubated at 37°C in a
humidified atmosphere of 5% CO2 in air (v/v). Cell
growth and induction of cell death was monitored by phase contrast
microscopy (Olympus CKS; Olympus Corporation, Tokyo, Japan) and by
detecting the generation of HepG2 cell-derived Annexin
V+ microparticles. Digitized images were generated using
a SPOT CCD camera (Diagnostic Instruments, Inc., Sterling Heights,
MI, USA) using SPOT version 2.1.2 software.
Cell culture treatments
HepG2 cells were grown in 96-well plates
at a density of 1×104 cells/well until ~70% confluence
was reached. Next, cells were deprived from FBS for 24 h before
treatment with 0–10, 20 and 30 µM simvastatin (Sigma-Aldrich).
Oleic acid (OA; Sigma-Aldrich) was dissolved at a concentration of
12 mM in phosphate-buffered saline (PBS; 137 mM NaCl, 10 mM
phosphate, 2.7 mM KCl and pH 7.4) that contained 11% fatty
acid-free bovine serum albumin (BSA; MP Biomedicals, Santa Ana, CA,
USA) by sonification at a frequency of 10 KHz with two 5 min pulses
(Soniprep 150 fitted with an exponential probe; Thermo Fisher
Scientific, Inc.) prior to shaking at 37°C for 15 h (20) using an OM10 Orbital Shaking Incubator
(Ratek Instruments Pty, Ltd., Boronia, Australia). OA solution was
filtered using a 0.22 µm filter and stored at 4°C prior to use.
HepG2 cells were treated with increasing concentrations
of OA solution (0–1 mM) for 24 h to determine the optimal
concentration that induces cellular OA accumulation. To determine
the effect of simvastatin on OA-induced HepG2 cell
steatosis, cells were treated with increasing concentrations of
simvastatin (0–30 µM) for 24 h before treatment with 1 mM OA.
Simvastatin was activated prior to use with NaOH as previously
described (21), and according to
the manufacturer's instructions.
Oil Red O staining
A stock solution of 0.35% Oil Red O (BDH Chemicals,
Poole, England) in isopropanol was prepared, filtered twice using a
0.22 µm filter and diluted in double-distilled H2O
(ddH2O; 3:2) prior to use. To detect and quantify
cellular lipid accumulation, OA-treated HepG2 cells were
gently washed with PBS and fixed using 4% paraformaldehyde for 1 h
at room temperature (RT). Subsequently, the cells were washed twice
using ddH2O and stained with Oil Red O solution for 20
min at RT. In order to remove the background staining, the cells
were washed for 5 min with 60% isopropanol solution. Lipid droplet
accumulation was detected by watching under the microscope. To
quantify intracellular lipid accumulation, Oil Red O stain was
extracted using pure isopropanol and the optical density was
detected at 510 nm using a Spectramax 250 Plate reader; data
analysis was performed using SoftMax Pro version 5.0 software (both
purchased from Molecular Devices (UK), Ltd. (Wokingham, UK).
Microparticle quantification
HepG2 cell-derived microparticles were
stained and quantified as previously described (22,23). The
technique was performed according to the guidelines established by
the International Society of Thrombosis and Haemostasis on the
standardization of platelet-derived microparticle enumeration by
flow cytometry (24), along with
modifications suggested in the manufacturer instructions of the BD
FACSCanto flow cytometer (BD Biosciences, San Jose, CA, USA). The
flow cytometer was calibrated to set the lower microparticles'
detection limit as previously described (22). HepG2 cells in culture
media were centrifuged at 400 × g for 5 min at room temperature to
remove cellular debris. Subsequently, 10 µl samples were incubated
at RT for 30 min with Annexin V-APC (eBioscience, Inc., San Diego,
CA, USA) to detect phosphatidylserine microparticle expression as a
marker of vesicles derived from apoptotic cells. Appropriate
dilutions were made using calcium-rich binding buffer as supplied
by Annexin V-APC manufacturer. Absolute microparticle numbers were
also determined as previously described (22) using TruCount counting tubes (BD
Biosciences).
Statistical analysis
Data for continuous variables are expressed as the
mean ± standard deviation of three independent experiments for each
treatment. Data was analyzed using the Student's t-test (SPSS
version 18.0; IBM SPSS, Amronk, NY, USA). P<0.05 was considered
to indicate a statistically significant difference.
Results
OA induces dose-dependent
HepG2 cellular lipid accumulation
High levels of plasma FFAs are implicated in the
pathogenesis of NAFLD as they can accumulate in hepatocytes to form
lipid droplets (4). This condition
was recreated in vitro by treating HepG2 cells
with increasing concentrations of OA (0–1 mM). Intracellular lipid
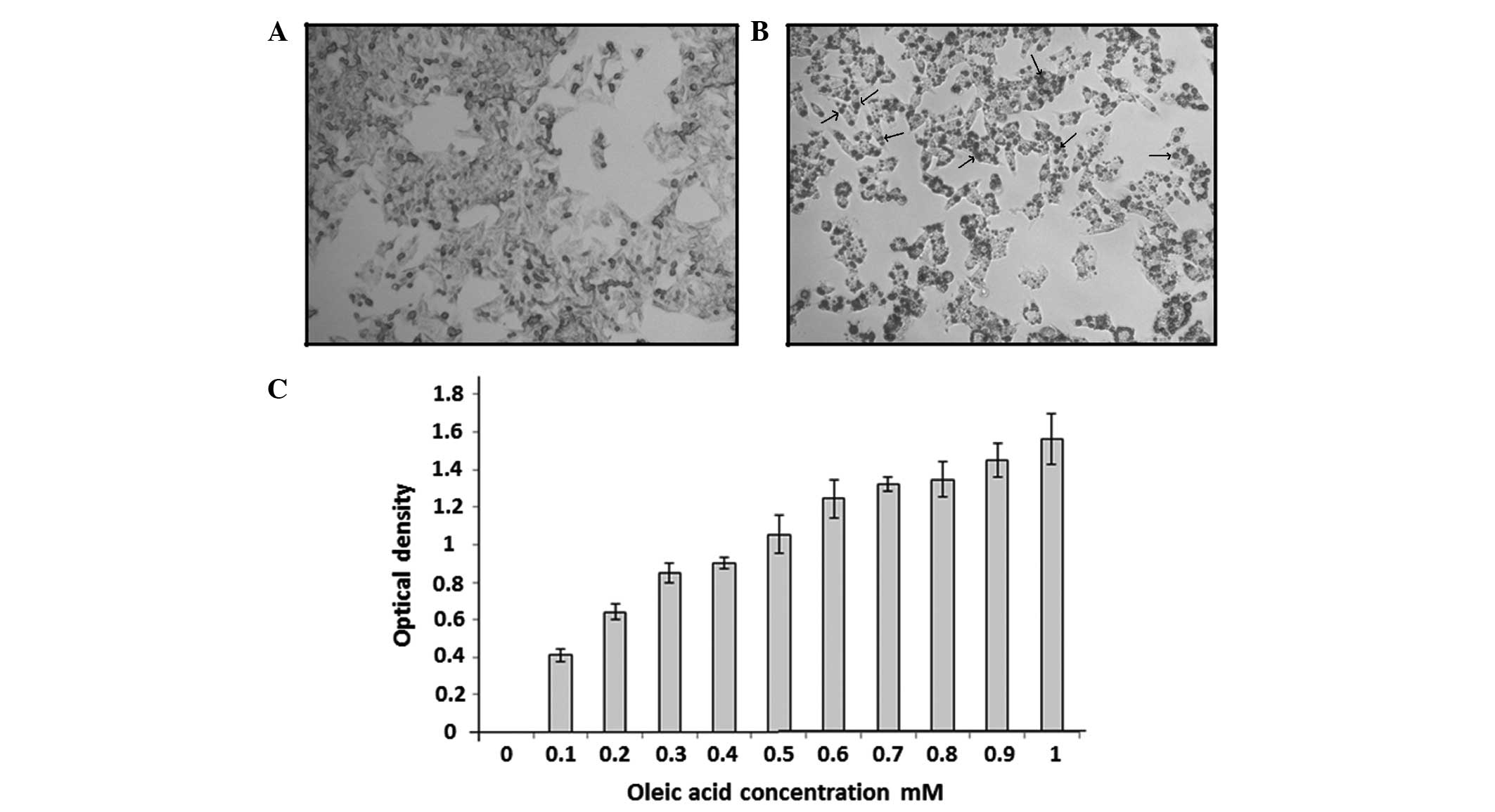
droplets were negatively stained for Oil Red O dye in the
OA-untreated HepG2 cells (Fig. 1A) and positively stained in the
OA-treated cells (Fig. 1B). As shown
in Fig. 1C, lipid accumulation in
OA-treated cells was dose-dependent and the concentration of 1 mM
OA was considered as an optimal concentration for the induction of
lipid accumulation in HepG2 cells as a model of
NAFLD.
Low simvastatin concentrations reduce
OA-induced steatosis in HepG2 cells
To evaluate the effect of simvastatin on
HepG2 cellular lipid accumulation, cultured
HepG2 cells were exposed to increasing concentrations of
simvastatin (0–10 µM) for 24 h before treatment with 1 mM OA.
Subsequently, Oil Red O staining and extraction for optical density
determination were performed in order to quantify the OA
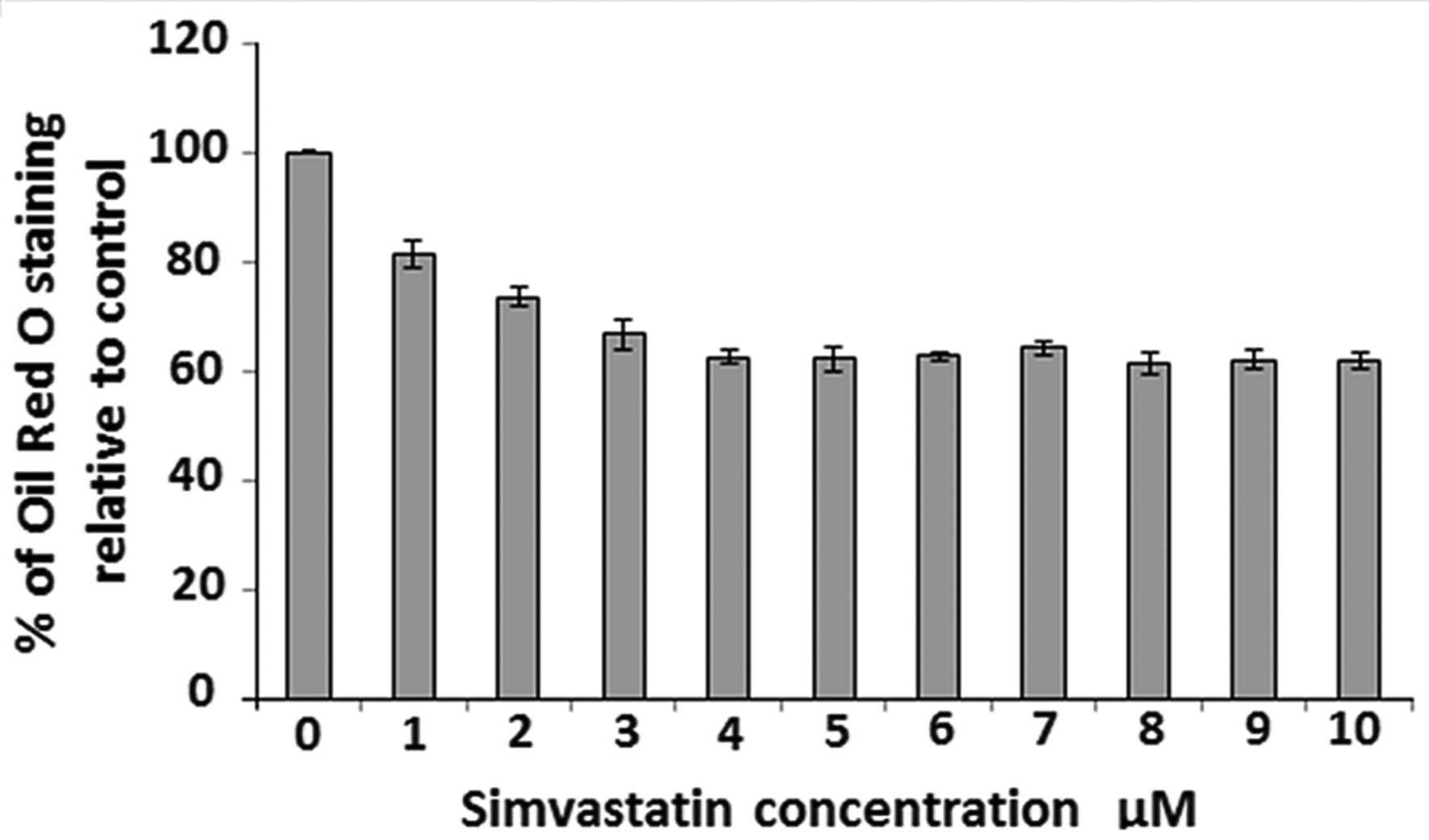
accumulation intracellularly. Fig. 2
shows that simvastatin was able to reduce OA accumulation in
HepG2 cells in a dose-dependent manner over a
concentration range of 1–4 µM. Further increase in simvastatin dose
(up to 10 µM) was not able to reduce OA accumulation more than
~40%. This effect was also confirmed by visualizing Oil Red O
staining using phase contrast microscopy (data not shown).
High simvastatin concentrations induce
the release of HepG2 cellular microparticles and
morphological characteristics of cell apoptosis
Microparticles are small (<1 µm in diameter)
vesicles that originate from the plasma membranes of cells
undergoing apoptosis (25). To
evaluate the effect of simvastatin on HepG2 cell growth
and induction of cellular apoptosis, cultured HepG2
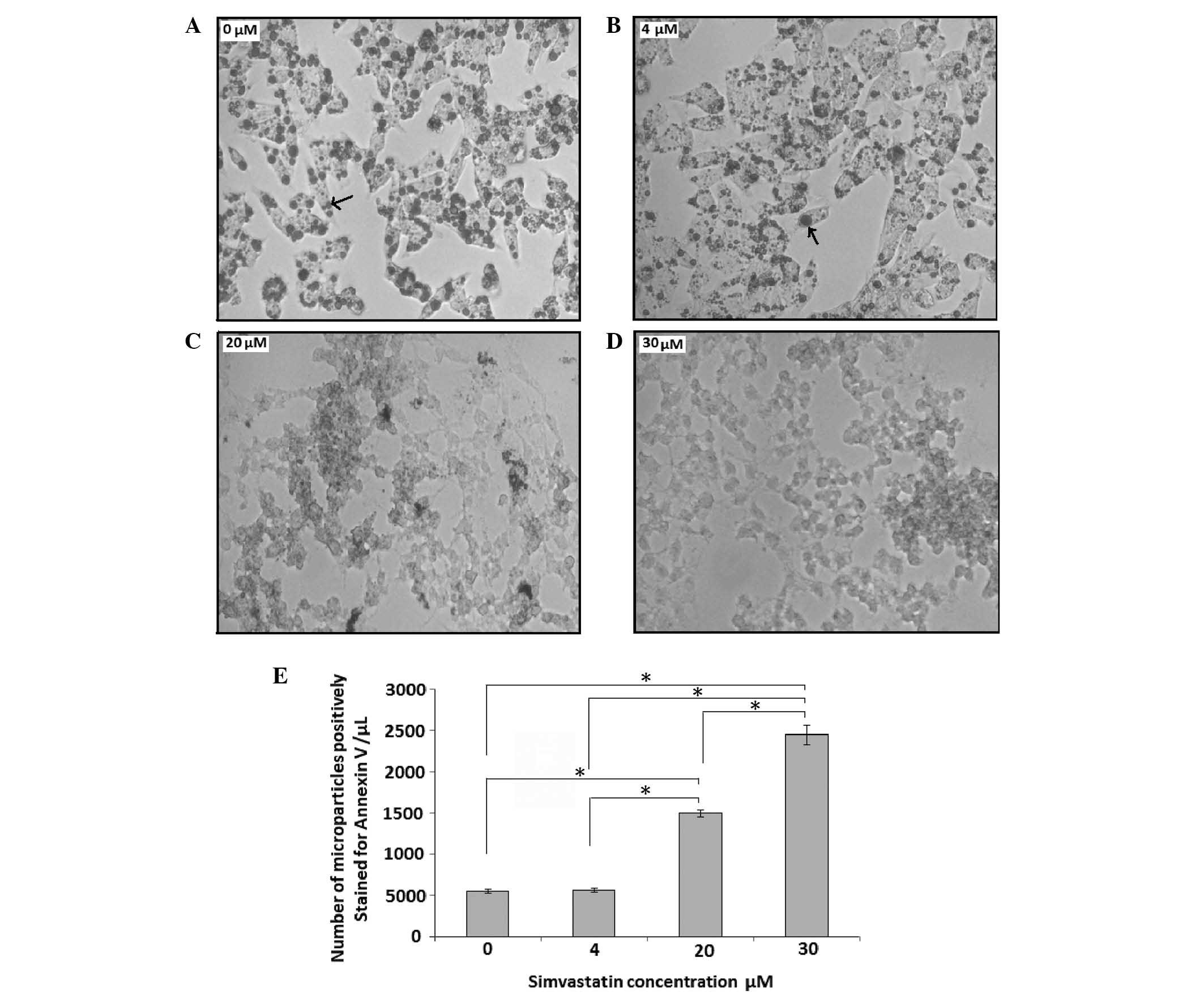
cells were treated with low-dose (4 µM) and high-dose (20 and 30
µM) simvastatin for 24 h and compared with the untreated control (0
µM simvastatin; Fig. 3A–E). Samples
from cell culture media were analyzed by flow cytometry to count
the number of Annexin V+ microparticles released by the
HepG2 cells. As shown in Fig.
3, low-dose simvastatin (4 µM) did not induce any change in
cell morphology (Fig. 3B) and did
not induce an increase in the number of Annexin V+
microparticles compared with that in the control group (Fig. 3E). By contrast, treatment high
simvastatin doses (20 and 30 µM; Fig. 3C
and D, respectively) induced changes in cellular morphology and
significantly increased the generation of cell-derived Annexin
V+ microparticles compared with the control (P<0.05;
Fig. 3E). As revealed by phase
contrast microscopy, HepG2 cells treated with 20 or 30
µM simvastatin prior to treatment with 1 µM OA showed reduced
staining for Oil Red O compared with cells treated with 4 µM and
the control cells (Fig. 3A–D).
Discussion
OA-induced lipid accumulation in HepG2
cells may act as an in vitro model for studying NAFLD.
Consistent with other studies (18,26), the
results of the present study demonstrated that lipid accumulation
in HepG2 cells in response to OA treatment was
dose-dependent and can be easily quantified using an Oil Red O
colorimetric technique. This technique is based on the staining of
intracellular lipid droplets by Oil Red O, followed by stain
extraction and measurement of optical density that is proportional
to the intracellular lipid content. In agreement with the findings
of Cui et al (18), the
present study also revealed that HepG2 intracellular
lipid quantification using the aforementioned staining technique
provided reliable measurements of intracellular lipid-droplet
levels. Assay results were able to reflect the dose-dependent
uptake of OA and appeared entirely consistent with lipid droplets
stained with Oil-Red O, as visualized using microscopy. In
addition, intracellular Oil Red O-stained lipid droplets can be
directly visualized using phase contrast microscopy. Therefore,
quantification of OA-induced HepG2 cell steatosis may
act as a valuable model to study the pathogenesis of NAFLD and
assess the effect of possible treatments for hepatic steatosis.
In the current study, pretreatment of
HepG2 cells with low simvastatin doses (4–10 µM) was
able to reduce the OA-induced intracellular lipid accumulation by
~40%. By contrast, pretreatment with high simvastatin doses (20 and
30 µM) induced HepG2 cellular apoptosis that was
detected by changes in cell morphology and production of
cell-derived Annexin V+ microparticles. To the best of
our knowledge, the present study quantified fatty acid-induced
lipid accumulation in human hepatocellular carcinoma cells in
response to simvastatin treatment for the first time. Previous
similar studies have clinically assessing the efficacy of statin
therapy in the treatment of patients with NAFLD by evaluating their
effect on reducing elevated hepatic enzymes including
aminotransferases and γ-glutamyl transferase (14–17).
Recently, de Keyser et al have also shown that statin
therapy for >2 years was associated with a lower prevalence of
hepatic steatosis among overweight subjects (27). This protective effect was considered
to be associated with the ability of statins to improve the lipid
profile by inhibiting the HMG-CoA reductase pathway and by acting
as anti-inflammatory, anti-oxidant and immune-modulatory agents
(27–29). However, no previous study has shown
any direct effect for statins on hepatic lipid accumulation at the
cellular level. At the organ level, statins, which can reduce
elevated serum triglyceride concentrations (30), may decrease hepatic lipid
accumulation in overweight and obese subjects by reducing the serum
levels of triglycerides and fatty acids (4).
Using an in vitro model of NAFLD to quantify
cellular lipid accumulation in response to simvastatin treatment is
considered the main strength of the current study in comparison
with other clinical studies. Although the mechanism through which
simvastatin can reduce intracellular HepG2 lipid
accumulation was not determined in the current study, this model
can be used in the future to study possible mechanisms or to
investigate other possible treatments for NAFLD. However, there are
certain limitations in the present study. Using HepG2
cells to investigate the effect of simvastatin or other HMG-CoA
reductase inhibitors on intracellular lipid accumulation may be
inappropriate or should be used carefully, since certain previous
studies have demonstrated that simvastatin can induce growth
inhibition and apoptosis in HepG2 cells (31,32).
However, this effect was dependent on the concentration of
simvastatin and the duration of cell exposure to simvastatin. For
instance, Huang et al (31)
and Kah et al (32) have
shown that treatment of HepG2 cells with 8 or 10 µM
simvastatin for 72 h induced ~50% decrease in cell viability. Huang
et al (31) have also shown
that lower simvastatin doses (2 and 4 µM) for shorter period of
time (24 h) induced ~20% decrease in HepG2 cell
viability. In the current study, HepG2 cell viability in
response to simvastatin treatment was not measured as an indicator
of cell apoptosis; by contrast, the number of Annexin V+
microparticles in the culture media was detected as an indicator of
cell apoptosis, beside examining the cell morphology under a
microscope as shown in Fig. 3.
Furthermore, treatment of HepG2 cells with low
simvastatin dose (4 µM) for 24 h in the current study was able to
reduce lipid accumulation by ~40% without increasing the number of
Annexin V+ microparticles or changing cell morphology,
which suggests that this dose reduces lipid accumulation without
inducing cell apoptosis. However, treatment of HepG2
cells with high doses of simvastatin (20 and 30 µM) for 24 h was
found to induce increased generation of Annexin V+
microparticles and changes in cell morphology that were suggestive
of cell apoptosis (Fig. 3). These
results are consistent with the findings of Relja et al
(33), which showed that
HepG2 cell apoptosis was induced by high doses of
simvastatin (32 and 64 µM).
Determining the underlying mechanism by which low
simvastatin concentrations can reduce HepG2
intracellular lipid accumulation was out of the scope of the
current study. However, more research is required to determine how
simvastatin can produce this effect and whether other statins have
similar effects on hepatic lipid accumulation. Possible roles for
statins that deserve further investigations include their effect on
fatty acid uptake by hepatic cells and their effect on hepatic
intracellular triglyceride formation, which is stored as lipid
droplets.
In conclusion, the present study demonstrated that
low simvastatin concentrations can reduce HepG2
intracellular OA-induced lipid accumulation without inducing cell
death, whereas high simvastatin concentrations induced
HepG2 cell apoptosis, as revealed by detecting
HepG2 cell-derived Annexin V+ microparticles
and changes in cell morphology. These findings support the results
of previous clinical studies that encourage administration of
statin therapy for the prevention of NAFLD (14–17).
Additionally, the current study has shown that OA-treated
HepG2 cells may act as a model for studying other
possible treatments for NAFLD.
Acknowledgements
The corresponding author would like to thank Dr.
Rick Thorne and Dr. Lisa Lincz as they did not hesitate to support
this work by providing sufficient lab space, equipment, cell line,
materials and advice to perform this study. The project was
supported by the HMRI Research Grant (grant no. 10–08), funded by
the Lions District 201 N3 Diabetes Foundation.
Glossary
Abbreviations
Abbreviations:
|
NAFLD
|
non-alcoholic fatty liver disease
|
|
OA
|
oleic acid
|
|
HepG2
|
human hepatocellular carcinoma cell
line
|
References
|
1
|
Green CJ and Hodson L: The influence of
dietary fat on liver fat accumulation. Nutrients. 6:5018–5033.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Baraona E and Lieber CS: Effects of
ethanol on lipid metabolism. J Lipid Res. 20:289–315.
1979.PubMed/NCBI
|
|
3
|
Souza MR, Diniz Mde F, Medeiros-Filho JE
and Araújo MS: Metabolic syndrome and risk factors for
non-alcoholic fatty liver disease. Arq Gastroenterol. 49:89–96.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Marchesini G, Brizi M, Morselli-Labate AM,
Bianchi G, Bugianesi E, McCullough AJ, Forlani G and Melchionda N:
Association of nonalcoholic fatty liver disease with insulin
resistance. Am J Med. 107:450–455. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pais R, Pascale A, Fedchuck L, Charlotte
F, Poynard T and Ratziu V: Progression from isolated steatosis to
steatohepatitis and fibrosis in nonalcoholic fatty liver disease.
Clin Res Hepatol Gastroenterol. 35:23–28. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Onnerhag K, Nilsson PM and Lindgren S:
Increased risk of cirrhosis and hepatocellular cancer during
long-term follow-up of patients with biopsy-proven NAFLD. Scand J
Gastroenterol. 49:1111–1118. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Centis E, Marzocchi R, Suppini A, Dalle
Grave R, Villanova N, Hickman IJ and Marchesini G: The role of
lifestyle change in the prevention and treatment of NAFLD. Curr
Pharm Des. 19:5270–5279. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Angelico F, Burattin M, Alessandri C, Del
Ben M and Lirussi F: Drugs improving insulin resistance for
non-alcoholic fatty liver disease and/or non-alcoholic
steatohepatitis. Cochrane Database Syst Rev.
CD0051662007.PubMed/NCBI
|
|
9
|
Tolman KG and Dalpiaz AS: Treatment of
non-alcoholic fatty liver disease. Ther Clin Risk Manag.
3:1153–1163. 2007.PubMed/NCBI
|
|
10
|
Basaranoglu M, Acbay O and Sonsuz A: A
controlled trial of gemfibrozil in the treatment of patients with
nonalcoholic steatohepatitis. J Hepatol. 31:3841999. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hyogo H, Tazuma S, Arihiro K, Iwamoto K,
Nabeshima Y, Inoue M, Ishitobi T, Nonaka M and Chayama K: Efficacy
of atorvastatin for the treatment of nonalcoholic steatohepatitis
with dyslipidemia. Metabolism. 57:1711–1728. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kiyici M, Gulten M, Gurel S, Nak SG, Dolar
E, Savci G, Adim SB, Yerci O and Memik F: Ursodeoxycholic acid and
atorvastatin in the treatment of nonalcoholic steatohepatitis. Can
J Gastroenterol. 17:713–718. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Calderon RM, Cubeddu LX, Goldberg RB and
Schiff ER: Statins in the treatment of dyslipidemia in the presence
of elevated liver aminotransferase levels: A therapeutic dilemma.
Mayo Clin Proc. 85:349–356. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Eslami L, Merat S, Malekzadeh R,
Nasseri-Moghaddam S and Aramin H: Statins for non-alcoholic fatty
liver disease and non-alcoholic steatohepatitis. Cochrane Database
Syst Rev. 12:CD0086232013.PubMed/NCBI
|
|
15
|
Egan M and Prasad S: PURLs: Statins for
patients with nonalcoholic fatty liver? J Fam Pract. 60:536–538.
2011.PubMed/NCBI
|
|
16
|
Nseir W and Mahamid M: Statins in
nonalcoholic fatty liver disease and steatohepatitis: Updated
review. Curr Atheroscler Rep. 15:3052013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dima A, Marinescu AG and Dima AC:
Non-alcoholic fatty liver disease and the statins treatment. Rom J
Intern Med. 50:19–25. 2012.PubMed/NCBI
|
|
18
|
Cui W, Chen SL and Hu KQ: Quantification
and mechanisms of oleic acid-induced steatosis in HepG2 cells. Am J
Transl Res. 2:95–104. 2010.PubMed/NCBI
|
|
19
|
Liu JF, Ma Y, Wang Y, Du ZY, Shen JK and
Peng HL: Reduction of lipid accumulation in HepG2 cells by luteolin
is associated with activation of AMPK and mitigation of oxidative
stress. Phytother Res. 25:588–596. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Susztak K, Ciccone E, McCue P, Sharma K
and Böttinger EP: Multiple metabolic hits converge on CD36 as novel
mediator of tubular epithelial apoptosis in diabetic nephropathy.
PLoS Med. 2:e452005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rossi J, Rouleau L, Tardif JC and Leask
RL: Effect of simvastatin on Kruppel-like factor2, endothelial
nitric oxide synthase and thrombomodulin expression in endothelial
cells under shear stress. Life Sci. 87:92–99. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Alkhatatbeh MJ, Enjeti AK, Acharya S,
Thorne RF and Lincz LF: The origin of circulating CD36 in type 2
diabetes. Nutr Diabetes. 3:e592013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Robert S, Poncelet P, Lacroix R, Arnaud L,
Giraudo L, Hauchard A, Sampol J and Dignat-George F:
Standardization of platelet-derived microparticle counting using
calibrated beads and a Cytomics FC500 routine flow cytometer: A
first step towards multicenter studies? J Thromb Haemost.
7:190–197. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lacroix R, Robert S, Poncelet P, Kasthuri
RS, Key NS and Dignat-George F: ISTH SSC Workshop: Standardization
of platelet-derived microparticle enumeration by flow cytometry
with calibrated beads: Results of the International Society on
Thrombosis and Haemostasis SSC collaborative workshop. J Thromb
Haemost. 8:2571–2574. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hargett LA and Bauer NN: On the origin of
microparticles: From platelet dust to mediators of intercellular
communication. Pulm Circ. 3:329–340. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gomez-Lechón MJ, Donato MT,
Martínez-Romero A, Jiménez N, Castell JV and O'Connor JE: A human
hepatocellular in vitro model to investigate steatosis. Chem Biol
Interact. 165:106–116. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
de Keyser CE, Koehler EM, Schouten JN,
Visser LE, Hofman A, Janssen HL and Stricker BH: Statin therapy is
associated with a reduced risk of non-alcoholic fatty liver in
overweight individuals. Dig Liver Dis. 46:720–725. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Endo A: The discovery and development of
HMG-CoA reductase inhibitors. J Lipid Res. 33:1569–1582.
1992.PubMed/NCBI
|
|
29
|
Liao JK and Laufs U: Pleiotropic effects
of statins. Annu Rev Pharmacol Toxicol. 45:89–118. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Stein EA, Lane M and Laskarzewski P:
Comparison of statins in hypertriglyceridemia. Am J Cardiol.
81:66B–69B. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huang X, Ma J, Xu J, Su Q and Zhao J:
Simvastatin induces growth inhibition and apoptosis in HepG2 and
Huh7 hepatocellular carcinoma cells via upregulation of Notch1
expression. Mol Med Rep. 11:2334–2340. 2015.PubMed/NCBI
|
|
32
|
Kah J, Wüstenberg A, Keller AD, Sirma H,
Montalbano R, Ocker M, Volz T, Dandri M, Tiegs G and Sass G:
Selective induction of apoptosis by HMG-CoA reductase inhibitors in
hepatoma cells and dependence on p53 expression. Oncol Rep.
28:1077–1083. 2012.PubMed/NCBI
|
|
33
|
Relja B, Meder F, Wilhelm K, Henrich D,
Marzi I and Lehnert M: Simvastatin inhibits cell growth and induces
apoptosis and G0/G1 cell cycle arrest in hepatic cancer cells. Int
J Mol Med. 26:735–741. 2010. View Article : Google Scholar : PubMed/NCBI
|