Introduction
Hepatocellular carcinoma (HCC) is one of the most
prevalent types of cancer worldwide, accounting for 85–90% of
primary liver cancers (1). In China,
there were estimated to be >390,000 new cases of HCC during 2012
and 380,000 HCC-related mortalities; in the same year, HCC was the
second most common cause of mortality from cancer worldwide,
estimated to be responsible for ~740,000 mortalities (International
Agency for Research on Cancer: http://www-dep.iarc.fr/) (2). Hepatectomy is the most effective
treatment for HCC at present; however, the tumor recurrence rate at
5 years after resection is ~70% (3,4). As the
molecular mechanisms underlying the development and progression of
HCC have not yet been clarified, the identification of
HCC-associated molecules may contribute to improvements in the
prevention and treatment of HCC.
The nitric oxide synthase (NOS) enzyme, which has
neuronal (NOS-1), inducible (NOS-2) and endothelial (NOS-3)
isoforms, catalyzes the oxidation of L-arginine to generate nitric
oxide (NO), a highly reactive radical that exerts a wide range of
biological activities, including smooth muscle relaxation,
inhibition of platelet aggregation and neurotransmission (5). The production of excessive quantities
of NO by NOS-2 has been implicated in the pathogenesis of various
types of human malignant tumors, including breast cancer (6), colon cancer (7), melanoma (8) and lung cancer (9). However, it remains unclear whether NO
functions as a pro-neoplastic or anti-neoplastic effector. Our
previous study indicated that decreased levels of NO/NOS-2 in liver
tissues may partly contribute to the development and metastasis of
HCC (10). In the present study, the
human hepatocellular carcinoma cell line HepG2 was selected to
investigate the effects of exogenous NO on cell proliferation, cell
cycle, apoptosis, migration and invasion.
Materials and methods
Cell culture
The HepG2 human hepatocellular carcinoma cell line
(American Type Culture Collection, Manassas, VA, USA) was cultured
in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10%
fetal calf serum (Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) and 1% penicillin/streptomycin (Wuhan Boster
Bio-Engineering Co., Ltd., Wuhan, China) at 37°C in a 5%
CO2 incubator.
Measurement of NO production
HepG2 cells were seeded in 6-well plates at a
density of 3×104 cells/well in growth medium until they
reached a confluence of 80% and were then exposed to 0, 0.5, 1.0,
1.5 or 2.0 mM sodium nitroprusside (SNP; Beyotime Institute of
Biotechnology, Haimen, China) for 24 h. NO levels were determined
by measuring stable NO derivatives (total nitrites), in the cell
culture supernatant with a commercially available kit according to
the manufacturer's protocol (Beyotime Institute of Biotechnology).
Briefly, 50 µl culture supernatant was mixed with 100 µl Griess
reagent (Beyotime Institute of Biotechnology, Haimen, China) in a
96-well plate. The absorbance was measured using a 680 model
microplate spectrophotometer (Bio-Rad Laboratories, Inc., Hercules,
CA, USA) at 540 nm, and nitrite was calculated from a standard
curve derived from NaNO2 (0–100 µM). Each experiment was performed
in triplicate.
Cell viability assay
Cell viability was assessed using a methyl thiazolyl
tetrazolium (MTT) conducted with a commercially available kit
according to the manufacturer's protocol (Beyotime Institute of
Biotechnology). Cells were seeded at a density of 5×103
cells/well in 96-well plates in 10% fetal calf serum-DMEM overnight
to allow attachment. The next day, the wells were washed 3 times
with DMEM and the medium was changed to serum-free DMEM.
Thereafter, the cells were incubated in 0, 0.5, 1.0 or 1.5 mM SNP
for 24 h. Subsequently, 10 µl MTT solution (5.0 mg/ml) was added to
each well and the plate was incubated for 4 h at 37°C, 5%
CO2. Then, the MTT solution was removed, and 100 µl
dimethylsulfoxide was added to dissolve the colored formazan
product. The intensity of the formazan product was measured at 570
nm using the microplate spectrophotometer (Bio-Rad Laboratories,
Inc.). Each experiment was performed in triplicate. Results were
reported in units of optical density (OD) and the relative growth
rate was calculated using the following formula: Relative growth
rate (%) = (ODtreatment/ODcontrol) × 100.
Cell cycle analysis
The cell cycle was analyzed with the aid of
propidium iodide (PI) staining. HepG2 cells were seeded into 6-well
plates at a density of 3×105 cells/well and exposed to
SNP at various concentrations (0, 0.5, 1.0 and 1.5 mM) for 24 h.
They were then collected and washed with 0.01 M ice-cold
phosphate-buffered saline (PBS, pH 7.4) before fixing with 70%
ethanol at 4°C overnight. After washing and resuspending the fixed
cells in 100 µl PBS, they were treated with 100 µl RNase A (DNase
free, 100 µg/ml; Solarbio Science & Technology Co., Ltd.,
Beijing, China) at 37°C for 30 min and stained with 400 µl PI (20
µg/ml; Beyotime Institute of Biotechnology) in the dark at 4°C for
30 min. The cell cycle was measured using a FACScan flow cytometer
(BD FACSCalibur; BD Biosciences, San Jose, CA, USA). Each
experiment was performed in triplicate.
Cell apoptosis assay
The apoptotic rate of the cells was detected by flow
cytometry after staining with an Annexin V-fluorescein
isothiocyanate (FITC) Apoptosis Detection kit (Beyotime Institute
of Biotechnology). HepG2 cells (5×104 cells/well) were
seeded into 6-well plates and incubated with SNP (0, 0.5, 1.0 or
1.5 mM) for 24 h. Subsequently, the cells from each sample were
harvested and washed with PBS. Staining was conducted with 195 µl
Annexin V-FITC binding buffer, and 5 µl Annexin V-FITC in the dark
at room temperature for 10 min. Each sample was then centrifuged at
445xg for 5 min, resuspended in 190 µl Annexin V-FITC binding
buffer and 10 µl PI was added. The samples were then analyzed using
a FACScan flow cytometer, with ≥10,000 events recorded for each
sample. Each experiment was performed in triplicate.
Cell migration assay
Cell migration was determined using a wound healing
assay. HepG2 cells were seeded at a density of 5×105
cells/well in 6-well plates and incubated at 37°C for 24 h. After
that, a linear wound was created using a standard 1-ml pipette tip.
Cells were washed with PBS to remove cell debris, cultured in
serum-free DMEM medium and exposed to SNP (0, 0.5, 1.0 or 1.5 mM)
for 24 h. The wound spaces were imaged under a CK31 model inverted
microscope (at ×100 magnification; Olympus Corporation, Tokyo,
Japan). Wound healing was analyzed using Image-Pro Plus software,
version 6.0 (Media Cybernetics, Inc., Rockville, MD, USA),
according to the following formula: Wound healing area (%) =
[cell-free area (0 h) - cell-free area (24 h)]/cell-free area (0 h)
× 100 (11). Each experiment was
performed in triplicate.
Cell invasion assay
Transwell plates (24-well insert, 8 µm pore size;
Corning Costar; Corning Incorporated, Lowell, MA, USA) were used to
examine the ability of cells to invade through a Matrigel-coated
filter following the protocol provided by the manufacturer. HepG2
cells were detached with trypsin (Wuhan Boster Bio-Engineering Co.,
Ltd.) and resuspended in serum-free DMEM medium. The lower chamber
was filled with medium supplemented with 10% fetal calf serum as a
chemoattractant, and 1×105 cells/well in 200 µl
serum-free DMEM medium were then seeded on the upper side of the
chamber of the Transwell and incubated with SNP (0, 0.5, 1.0 or 1.5
mM) for 24 h. Following incubation, non-migrating cells on the
upper chambers were removed with a cotton swab, and cells that had
migrated to the lower side of the membrane were fixed with methanol
and stained with 0.1% crystal violet. The membrane was visualized
with an inverted microscope (at ×200 magnification; Olympus
Corporation). Invading cells were scored by counting at ≥5 randomly
selected fields per membrane, and expressed as the average number
of cells/field of view. Each experiment was performed in
triplicate.
Statistical analysis
All quantitative data are represented as mean ±
standard deviation. Statistical significance was analyzed by
one-way analysis of variance followed by Newman-Keuls test. A
P-value <0.05 was considered statistically significant. All
statistical analyses were performed using SPSS software, version
17.0 (SPSS, Inc., Chicago, IL, USA) and GraphPad Prism, version 5.0
(GraphPad Software Inc., San Diego, CA, USA).
Results
Increased NO production in SNP-treated
HepG2 cell cultures
As shown in Fig. 1A,
HepG2 cells treated with SNP (0.5, 1.0, 1.5 and 2.0 mM) for 24 h
exhibited a significant increase in NO production compared with the
control (P<0.05). The levels of NO in the control and
SNP-treated (0.5, 1.0 and 1.5 mM) groups were as follows:
18.17±3.40, 47.67±2.52, 75.27±4.61 and 89.33±3.79 µmol/l,
respectively (Fig. 1A). However, no
significant difference in NO production was observed between the
1.5 and 2.0 mM groups (89.33±3.79 vs. 94.13±5.33 µmol/l, P>0.05;
Fig. 1A), indicating that the
increase in NO production levels was not completely dependent upon
SNP concentration in HepG2 cultures.
Effect of NO on the viability of HepG2
cells
To assess whether NO regulates cell viability, HepG2
cells were incubated with various concentrations of SNP (0, 0.5,
1.0 and 1.5 mM) for 24 h and an MTT assay was subsequently
performed. The relative growth rates consistently decreased as the
concentration of SNP increased, as shown in Fig. 1B; for 0, 0.5, 1.0 and 1.5 mM SNP, the
relative growth rates were 98.33±2.26, 79.50±9.04, 62.67±3.06 and
50.81±5.02%, respectively. For all concentrations of SNP, the
difference in growth rate between the SNP-treated cells and the
control was significant (P<0.05; Fig.
1B). These data showed that NO clearly suppressed HepG2 cell
proliferation in a concentration-dependent manner.
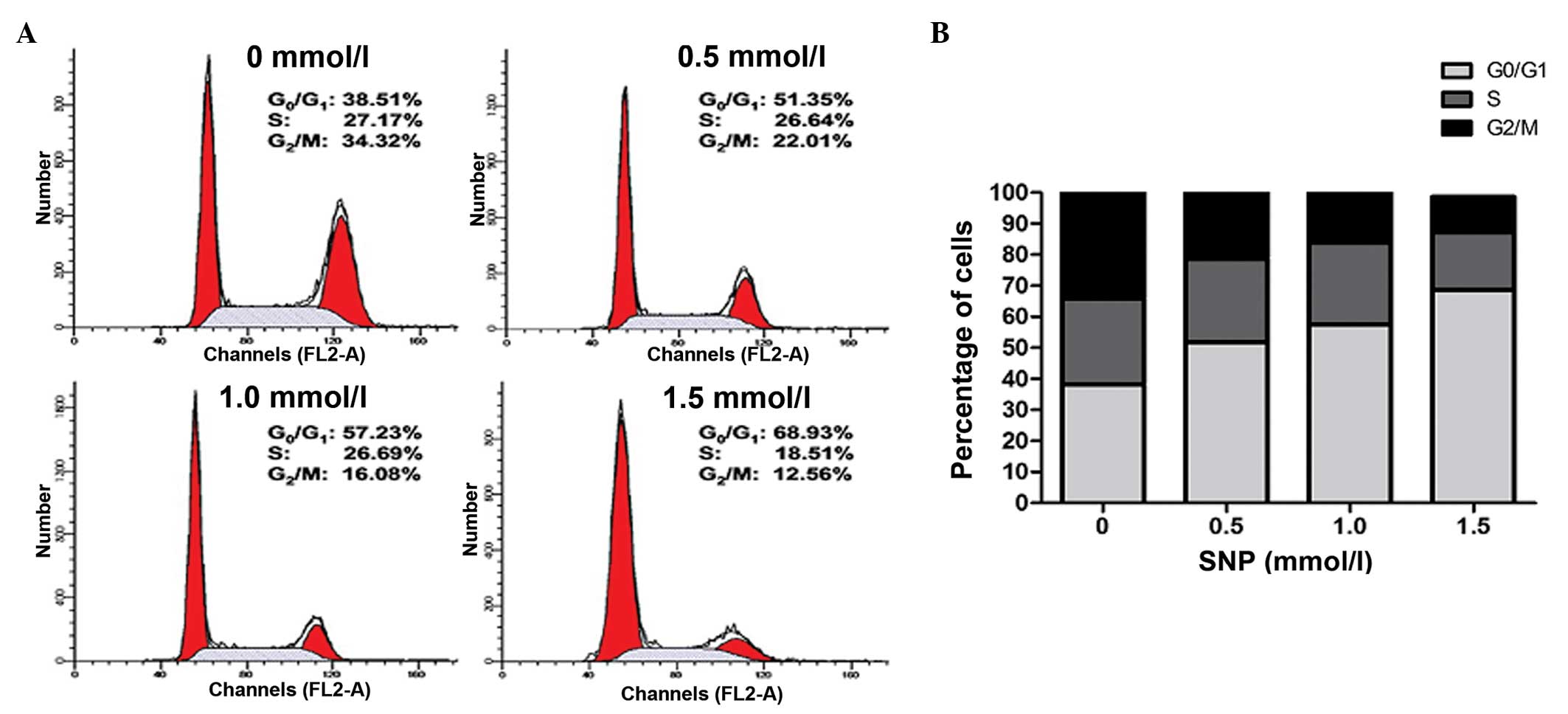
Effect of NO on the cell cycle of
HepG2 cells
A cell cycle assay was conducted to further examine
the effect of NO on HepG2 cell growth. Following exposure to
various concentrations of SNP (0, 0.5, 1.0 and 1.5 mM) for 24 h,
the cell cycle was analyzed by flow cytometry. NO significantly
increased the percentage of HepG2 cells in the G0/G1 phase in a
concentration-dependent manner; for 0, 0.5, 1.0 and 1.5 mM SNP, the
percentage of cells in the G0/G1 phase was 38.22±1.49, 51.82±1.83,
57.55±1.58 and 68.59±1.27%, respectively (P<0.05; Fig. 2). This was accompanied by a reduction
in the percentage of cells in the G2/M phase; for 0, 0.5, 1.0 and
1.5 mM SNP, the percentage in the G2/M phase was 34.16±2.54,
22.04±1.78, 16.83±2.15 and 11.47±1.20%, respectively (P<0.05;
Fig. 2). The percentage of cells in
the S phase remained unchanged following treatment with low
concentrations of SNP; for 0, 0.5 and 1.0 mM SNP, the percentage of
cells in the S phase was 27.77±1.56, 26.92±1.24 and 26.37±0.99%,
respectively (P>0.05; Fig. 2).
However, HepG2 cells treated with 1.5 mM SNP exhibited a marked
reduction in the percentage in the S phase compared with the
control (18.58±1.04 vs. 27.77±1.56%, P<0.05; Fig. 2). These results suggest that NO
inhibited the proliferation of HepG2 cells by inducing G0/G1 phase
arrest.
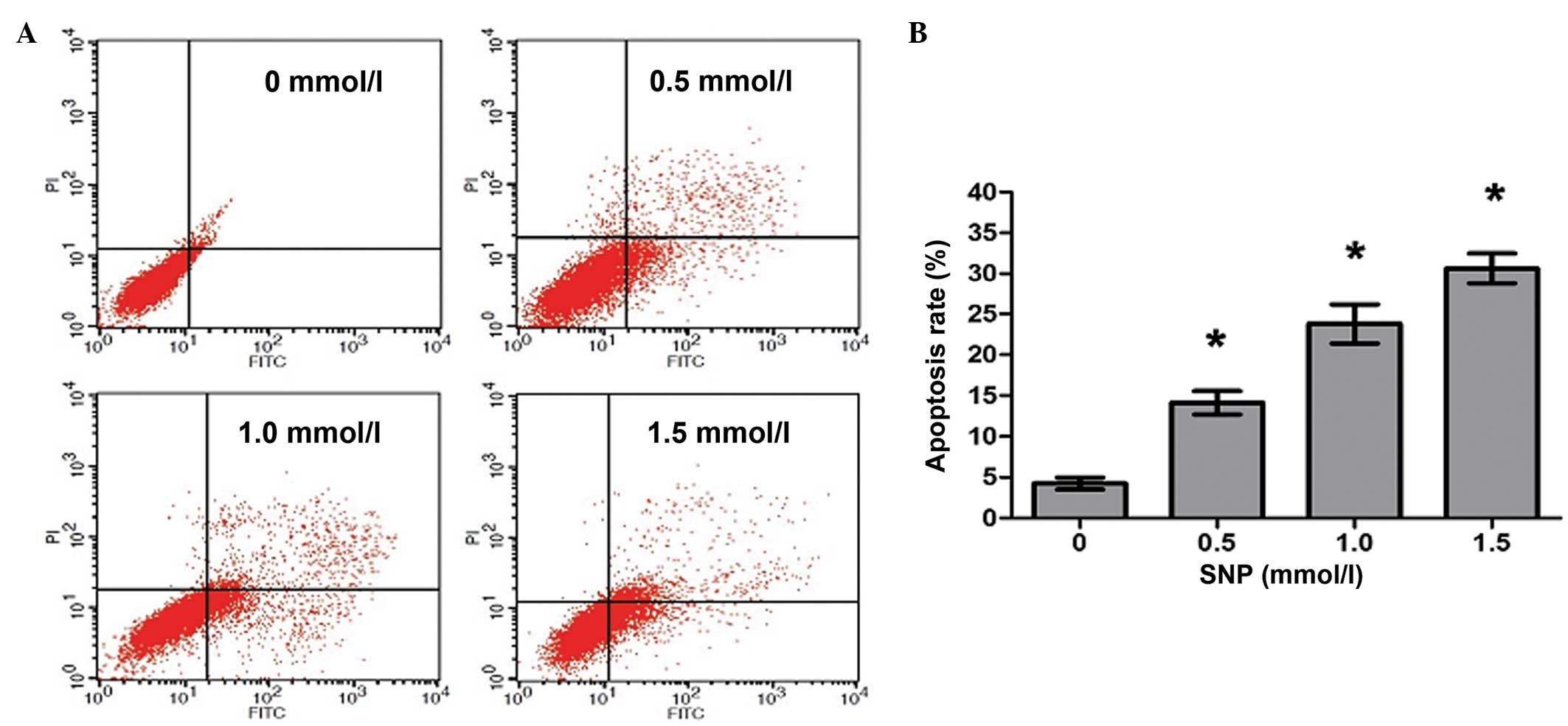
NO induces apoptosis of HepG2
cells
To investigate whether the decreased viability
observed following NO treatment was caused by increased apoptosis,
HepG2 cells were cultivated in the presence of SNP (0, 0.5, 1.0 and
1.5 mM) for 24 h, and then flow cytometric analysis was conducted
to quantify the apoptotic HepG2 cells. Following treatment with 0,
0.5, 1.0 and 1.5 mM SNP, the percentages of apoptotic cells were
4.25±1.26, 14.12±2.49, 23.78±4.13 and 30.62±3.22%, respectively,
with significant differences in apoptosis between the SNP-treated
and untreated cells (P<0.05, Fig.
3). The results revealed that NO induced apoptosis in HepG2
cells in a concentration-dependent manner.
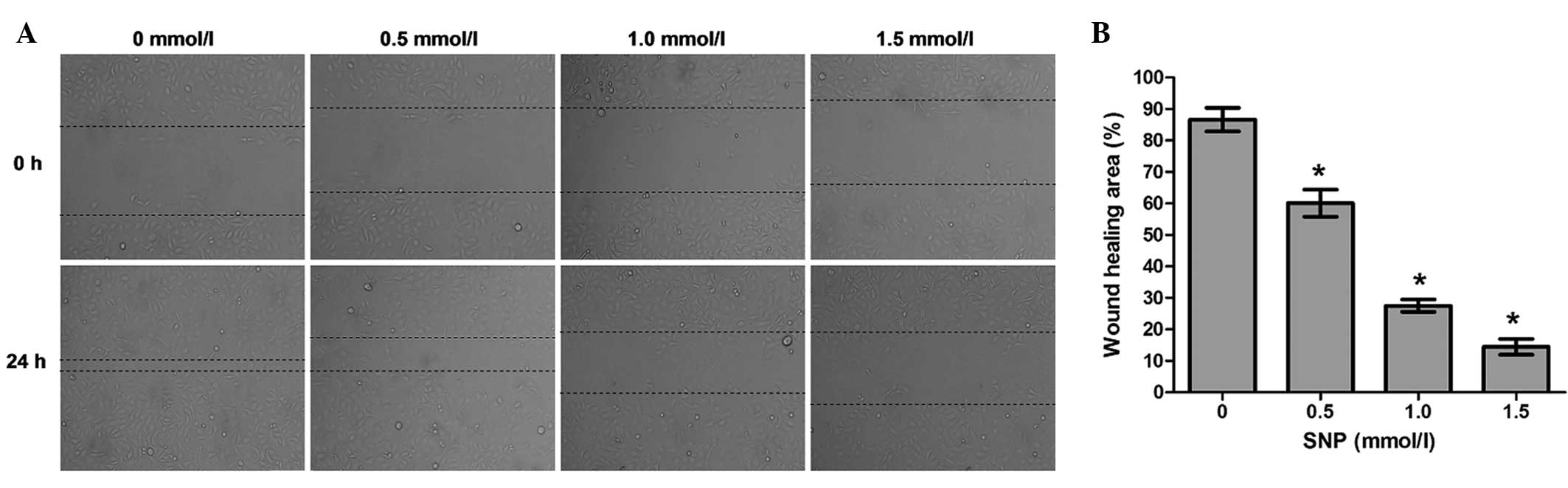
Effect of NO on the cell migration of
HepG2 cells
To explore whether NO affects the migration
potential of HepG2 cells, cell migration was examined using a wound
healing assay. The wound healing assay demonstrated that the
migration ability of HepG2 cells incubated with SNP (0.5, 1.0 and
1.5 mM) for 24 h was significantly impaired compared with that of
the negative control (0 mM; all P<0.05, Fig. 4). Following treatment for 24 h, the
percentage of wound healing area was 86.59±3.75% for the negative
control cells, 60.06±4.32% for the cells treated with 0.5 mM SNP,
27.51±1.97% for the cells treated with 1.0 mM SNP and 14.45±2.49%
for the cells treated with 1.5 mM SNP (Fig. 4). This indicates that the migration
capacity of HepG2 cells was inhibited by NO.
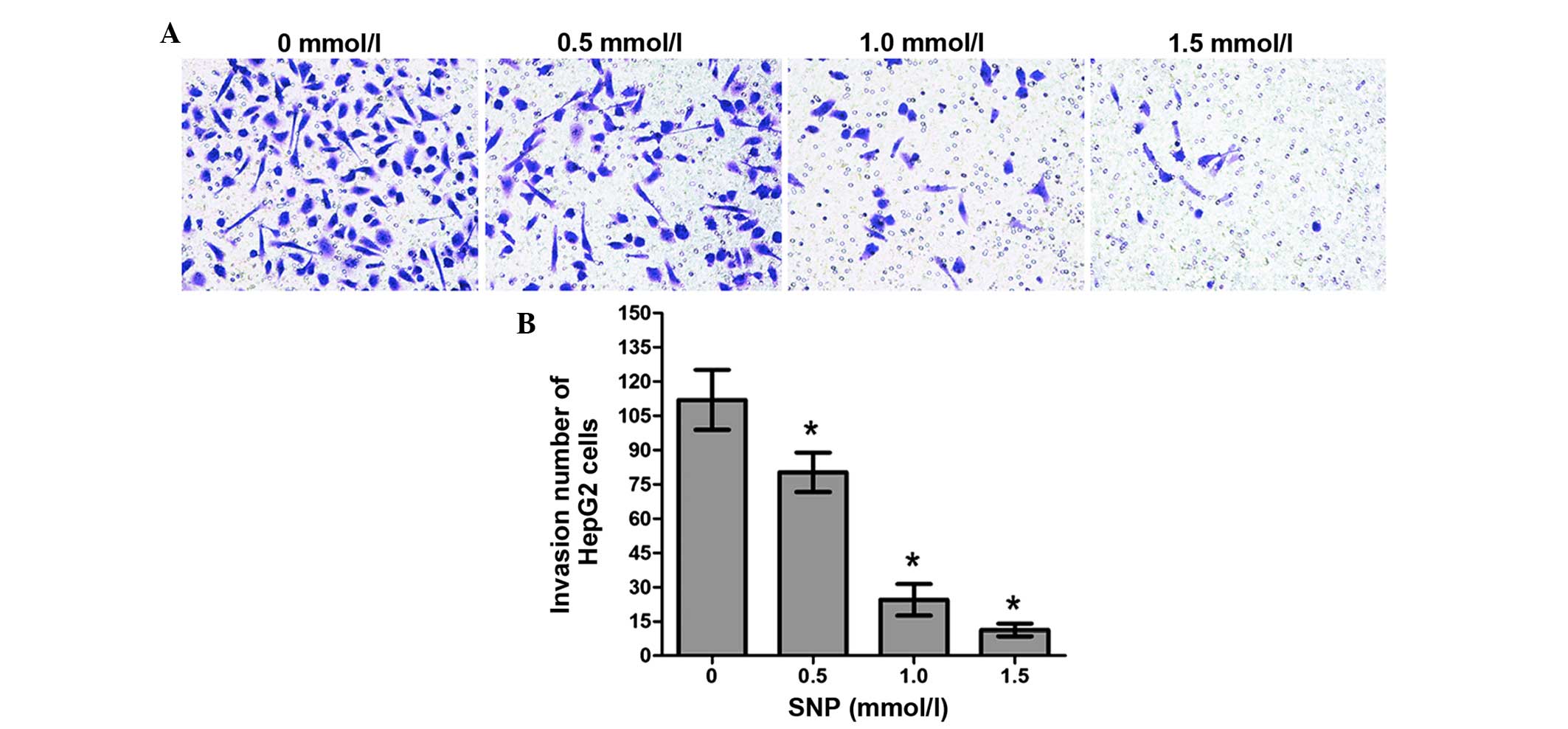
Effect of NO on the invasion of HepG2
cells
The invasion ability of HepG2 cells treated with SNP
(0, 0.5, 1.0 and 1.5 mM) for 24 h was also examined using a
Transwell assay. The numbers of invading cells in the control group
and the SNP-treated groups (0.5, 1.0 and 1.5 mM) were as follows:
112.0±13.11, 80.25±8.65, 24.47±6.91 and 11.30±2.79, respectively.
For all concentrations of SNP, the difference in the number of
invading cells between the SNP-treated cells and the control was
significant (P<0.05, Fig. 5). The
results of the Transwell chamber assay showed that NO inhibited
cell invasion in a concentration-dependent manner.
Discussion
NO is known to be involved in diverse processes in
numerous physiological and pathophysiological conditions; previous
studies have suggested that NO can exert a negative effect on the
regulation of tumor cell behavior and an anti-neoplastic effect
in vivo and in vitro (10,12–18).
However, studies concerning the effects of NO on HCC cells are
rare. In the present study, the effects of chemically derived NO on
the biological behavior of the human hepatocellular carcinoma cell
line HepG2 were investigated.
The typical characteristics of tumor cell
proliferation are out-of-control cell division and excessive
growth. Therefore, inhibiting tumor cell proliferation is an
important aspect of the control of tumorigenesis. In the present
study, the effect of NO on the proliferation of HepG2 cells was
evaluated. As shown in Fig. 1B, the
proliferation of HepG2 cells was significantly inhibited by NO,
with a greater degree of suppression when increasing concentrations
of SNP were used. This observation is consistent with a previous
study, which demonstrated that NO generated by treatment with SNP
inhibited the cell proliferation of gastric cancer cells in a
concentration-dependent manner (16).
In eukaryotic cells, cell cycle checkpoints are
regulatory pathways that control the order and timing of cell cycle
transitions and ensure that critical events, such as DNA
replication and chromosome segregation, are completed (19). During the cell cycle, the regulation
of cells transformation from the G1 phase into the S
phase is particularly important, as the G1-S checkpoint
allows for the replication of DNA (20). In the present study, cell cycle
analysis revealed that in SNP-treated HepG2 cells, a significant
increase in the proportion of G0/G1 phase
cells occurred, accompanied by a reduction in the proportion of
G2/M phase cells. These data indicate that NO arrested
HepG2 cells in the G1 phase. Sang et al observed
that SNP-derived NO inhibited the proliferation of gastric cancer
cells by blocking the conversion from the G1 to the S
phase, and the G1 arrest was mediated through the
regulation of cell cycle-related proteins, which was likely to be
associated with the inactivation of Akt signaling (16). These results suggest that NO can
regulate cell cycle transition, thereby suppressing cancer cell
proliferation.
Apoptosis is programmed cell death that plays a
major role during developmental processes and is circumvented
during the malignant transformation and progression of tumors
(21,22). The induction of apoptosis is a common
mechanism underlying the cytotoxic effects of anticancer agents, in
addition to cell cycle arrest (23,24). In
addition to cell proliferation, apoptosis in NO-treated HepG2 cells
was investigated in the present study. The results indicated that
NO induced cell apoptosis in a concentration-dependent manner.
Previously, SNP-derived NO has exhibited potent pro-apoptotic
effects on lung carcinoma cells and cutaneous T cell lymphoma cells
(13,14). The induction of apoptosis by NO
involves downregulation of the expression of survivin, constitutive
nuclear factor-κB and B-cell lymphoma-extra large (Bcl-xL)
(13,14).
Metastasis is a typical characteristic of HCC,
including intrahepatic metastasis and metastasis to extrahepatic
organs (25). Tumor metastasis
involves a series of interrelated events; cell migration and
invasion are two critical steps. In our previous study, it was
noted that the level of NO was higher in HCC tissues without
metastasis than those with metastasis (10). This implied that NO could partially
inhibit the metastatic cascade in HCC. Therefore, the effect of NO
on the migration and invasion of HepG2 cells was examined in the
present study using a wound healing assay and a Transwell invasion
assay. The results showed that NO significantly inhibited cell
migration and invasion in a concentration-dependent manner in
vitro. These results were concordant with those of other
studies, in which it was observed that NO suppressed migration and
invasion in different tumor cell lines, and the anti-metastasis
effects were shown to be associated with the upregulation of N-Myc
downstream-regulated gene 1 (NDRG1), inhibition of
hypoxia-inducible factor 1 (HIF-1) and impairment of mitochondria
(17,26). However, this is in contrast to
another study, which demonstrated that long-term (7 or 14 day)
treatment with NO significantly increased the migratory action of
human lung cancer cells through increased expression of caveolin-1
(Cav-1) and cell division cycle 42 (Cdc42) proteins (27). These conflicting results could be
explained by the observation that the final activity of NO in
oncology is dependent upon its working microenvironment, including
the type of cell exposed to the compound, the redox state of the
reaction, as well as the final intracellular concentration and the
duration of intracellular exposure to NO (5).
In conclusion, the present study indicated that NO
effectively suppressed proliferation, migration and invasion,
arrested the cell cycle and induced apoptosis in HepG2 cells;
however, the detailed mechanism underlying these effects is
unclear. Further investigation is required to identify cellular
targets of NO and the precise molecular mechanism of action
underlying the effects of NO in HepG2 cells, with the aim of
preventing and treating HCC with NO.
Acknowledgements
This study was supported by a starting fund from The
Central Hospital of Wuhan (grant no. YQ13B06).
References
|
1
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: Epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
International Agency for Research on
Cancer: Cancer incidence and mortality worldwide: sources, methods
and major patterns in GLOBOCAN 2012. http://www-dep.iarc.fr/Accessed. August
10–2014
|
|
3
|
Imamura H, Matsuyama Y, Tanaka E, Ohkubo
T, Hasegawa K, Miyagawa S, Sugawara Y, Minagawa M, Takayama T,
Kawasaki S, et al: Risk factors contributing to early and late
phase intrahepatic recurrence of hepatocellular carcinoma after
hepatectomy. J Hepatol. 38:200–207. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kamiyama T, Nakanishi K, Yokoo H, Kamachi
H, Tahara M, Suzuki T, Shimamura T, Furukawa H, Matsushita M, Todo
S, et al: Recurrence patterns after hepatectomy of hepatocellular
carcinoma: implication of Milan criteria utilization. Ann Surg
Oncol. 16:1560–1571. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huerta S, Chilka S and Bonavida B: Nitric
oxide donors: Novel cancer therapeutics (review). Int J Oncol.
33:909–927. 2008.PubMed/NCBI
|
|
6
|
Thomsen LL, Miles DW, Happerfield L,
Bobrow LG, Knowles RG and Moncada S: Nitric oxide synthase activity
in human breast cancer. Br J Cancer. 72:41–44. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ambs S, Merriam WG, Bennett WP,
Felley-Bosco E, Ogunfusika MO, Ose SM, Klein S, Shields PG, Billiar
TR and Harris CC: Frequent nitric oxide synthase-2 expression in
human colon adenomas: Implication for tumor angiogenesis and colon
cancer progression. Cancer Res. 58:334–341. 1998.PubMed/NCBI
|
|
8
|
Ekmekcioglu S, Ellerhorst J, Smid CM,
Prieto VG, Munsell M, Buzaid AC and Grimm EA: Inducible nitric
oxide synthase and nitrotyrosine in human metastatic melanoma
tumors correlate with poor survival. Clin Cancer Res. 6:4768–4775.
2000.PubMed/NCBI
|
|
9
|
Masri FA, Comhair SA, Koeck T, Xu W,
Janocha A, Ghosh S, Dweik RA, Golish J, Kinter M, Stuehr DJ, et al:
Abnormalities in nitric oxide and its derivatives in lung cancer.
Am J Respir Crit Care Med. 172:597–605. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou L, Wang Y, Tian DA, Yang J and Yang
YZ: Decreased levels of nitric oxide production and nitric oxide
synthase-2 expression are associated with the development and
metastasis of hepatocellular carcinoma. Mol Med Rep. 6:1261–1266.
2012.PubMed/NCBI
|
|
11
|
Hu J and Verkman AS: Increased migration
and metastatic potential of tumor cells expressing aquaporin water
channels. FASEB J. 20:1892–1894. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hussain SP, Trivers GE, Hofseth LJ, He P,
Shaikh I, Mechanic LE, Doja S, Jiang W, Subleski J, Shorts L, et
al: Nitric oxide, a mediator of inflammation, suppresses
tumorigenesis. Cancer Res. 64:6849–6853. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chao JI, Kuo PC and Hsu TS:
Down-regulation of survivin in nitric oxide-induced cell growth
inhibition and apoptosis of the human lung carcinoma cells. J Biol
Chem. 279:20267–20276. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Rishi L, Dhiman R, Raje M and Majumdar S:
Nitric oxide induces apoptosis in cutaneous T cell lymphoma
(HuT-78) by downregulating constitutive NF-kappaB. Biochim Biophys
Acta. 1770:1230–1239. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bonavida B, Baritaki S, Huerta-Yepez S,
Vega MI, Chatterjee D and Yeung K: Novel therapeutic applications
of nitric oxide donors in cancer: Roles in chemo-and
immunosensitization to apoptosis and inhibition of metastases.
Nitric Oxide. 19:152–157. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sang J, Chen Y and Tao Y: Nitric oxide
inhibits gastric cancer cell growth through the modulation of the
Akt pathway. Mol Med Rep. 4:1163–1167. 2011.PubMed/NCBI
|
|
17
|
Hickok JR, Sahni S, Mikhed Y, Bonini MG
and Thomas DD: Nitric oxide suppresses tumor cell migration through
N-Myc downstream-regulated gene-1 (NDRG1) expression: Role of
chelatable iron. J Biol Chem. 286:41413–41424. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bonavida B and Baritaki S: Dual role of NO
donors in the reversal of tumor cell resistance and EMT:
Downregulation of the NF-kB/Snail/YY1/RKIP circuitry. Nitric Oxide.
24:1–7. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Elledge SJ: Cell cycle checkpoints:
Preventing an identity crisis. Science. 274:1664–1672. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang Y, Ji P, Liu J, Broaddus RR, Xue F
and Zhang W: Centrosome-associated regulators of the G(2)/M
checkpoint as targets for cancer therapy. Mol Cancer. 13:82009.
View Article : Google Scholar
|
|
21
|
Jacobson MD, Weil M and Raff MC:
Programmed cell death in animal development. Cell. 88:347–354.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Onishi Y, Ueha T, Kawamoto T, Hara H, Toda
M, Harada R, Minoda M, Kurosaka M and Akisue T: Regulation of
mitochondrial proliferation by PGC-1α induces cellular apoptosis in
musculoskeletal malignancies. Sci Rep. 4:39162014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sun SY, Hail N Jr and Lotan R: Apoptosis
as a novel target for cancer chemoprevention. J Natl Cancer Inst.
96:662–672. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yin X, Zhang J, Li X, Liu D, Feng C, Liang
R, Zhuang K, Cai C, Xue X, Jing F, et al: DADS suppresses human
esophageal xenograft tumors through RAF/MEK/ERK and
mitochondria-dependent pathways. Int J Mol Sci. 15:12422–12441.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tang ZY: Hepatocellular carcinoma-cause,
treatment and metastasis. World J Gastroenterol. 7:445–454. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang F, Zhang R, Xia T, Hsu E, Cai Y, Gu Z
and Hankinson O: Inhibitory effects of nitric oxide on invasion of
human cancer cells. Cancer Lett. 257:274–282. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sanuphan A, Chunhacha P, Pongrakhananon V
and Chanvorachote P: Long-term nitric oxide exposure enhances lung
cancer cell migration. Biomed Res Int. 2013:1869722013. View Article : Google Scholar : PubMed/NCBI
|