Introduction
Oral submucous fibrosis (OSF) has been described as
an insidious chronic disease, affecting any part of the oral cavity
and in certain cases the pharynx (1). The incidence rate of oral squamous cell
carcinoma in patients with OSF, after a follow-up period of 17
years, has been estimated to be 7.6% (2). Areca-associated oral squamous cell
carcinoma is the third most common malignancy in the developing
world (3). Despite the fact that OSF
is, at times, preceded by and/or linked to vesicle formation, it is
known to be associated with a juxta-epithelial inflammatory
reaction, which is followed by a fibroelastic change of the lamina
propria, with epithelial atrophy resulting in stiffness of the oral
mucosa and causing inability to eat and trismus (4).
Autophagy is a lysosomal degradation pathway that
turns superfluous or damaged cell components into basic
biomolecules, which are then recycled back into the cytosol
(5). Autophagy has been categorized
into the following three catabolic processes: Microautophagy,
macroautophagy and chaperone-mediated autophagy, which are
responsible for the degradation of cell components in the lysosome
(6,7). This morphological process was
originally described 50 years ago (8). A study focusing on determining the
pathogenesis of organic fibrosis emphasized the possibility that
the condition may be linked with autophagy, which triggers tissue
fibrogenesis (9). Markers of
oxidative stress have also been identified in patients with OSF
(1,10), and another study has suggested that
elevated reactive oxygen species or oxidative stress may activate
autophagic reaction (11). Autophagy
has also been shown to be sensitive to oxygen tension, and
hypoxia-inducible factor (HIF)-1α has been implicated as a major
regulator of autophagy under hypoxic conditions (12). Patients with OSF have been found to
exhibit elevated levels of HIF-1α (1), which may contribute to the progression
of the disease, suggesting that autophagy is likely to be induced
in OSF.
In the present study, human tissues and an in
vitro transforming growth factor (TGF)-β model were used to
investigate the association of autophagy with OSF. The results
demonstrated that autophagy may mediate the TGF-β-induced OSF.
Materials and methods
Patients
A total of 10 male volunteers were included in the
study (age, 20–45 years). Five OSF buccal mucosa specimens from
areca quid chewers and 5 healthy specimens from non-areca quid
chewers were provided by the Department of Pathology of Xiangya
Hospital, Central South University (Changsha, China). The diagnosis
was based on the histological examination of the sections following
staining with hematoxylin and eosin. Written informed consent from
all patients and approval from the Institutional Research Ethics
Committee were obtained for the use of these clinical materials in
the present study.
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR)
Total RNA from cell cultures was isolated using
TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. mRNA (1 µg) was
reverse-transcribed using a PrimarScript RT Regent kit (Takara
Biotechnology Co., Ltd., Dalian, China). RNA was isolated and
reverse transcribed into cDNA. RT-qPCR was performed using Applied
Biosystems 7500 Real-Time PCR System (Applied Biosystems, Thermo
Fisher Scientific, Inc., Foster City, CA, USA) by mixing equal
quantities of cDNA, iQ SYBR Green Supermix (Bio-Rad Laboratories
Inc., Hercules, CA, USA) and specific primers. PCR thermal cycling
conditions were as follows: 95°C For 30 sec, followed by 40 cycles
of 95°C for 5 sec, 60°C for 30 sec, 95°C for 15 sec, 60°C for 15
sec and 95°C for 15 sec. All quantitative data were normalized
against β-actin. The primers used in RT-qPCR screening were as
follows: Collagen type 1 alpha 2 (Col1A2) forward,
5′-AAGGTCATGCTGGTCTTGCT-3′ and reverse, 5′-GACCCTGTTCACCTTTTCCA-3′;
and microtubule-associated protein 1 light chain 3 (LC3) forward,
5′-GAGTGGAAGATGTCCGGCTC-3′ and reverse, 5′-CCAGGAGGAAGAAGGCTTGG-3′.
The were analyzed using the 2−ΔΔCq method (13), and presented as fold increases
relative to GAPDH.
Immunohistochemistry
Paraffin-embedded samples were obtained from 5
healthy volunteers with non-areca quid chewers for
immunohistochemical analysis. Paraffin-embedded sections (4 µm)
were deparaffinized and rehydrated. Endogenous peroxidase activity
was blocked using 3% H2O2 for 15 min at 95°C.
Following antigen retrieval, the sections were incubated with 5%
serum (Gibco; Thermo Fisher Scientific, Inc.) to avoid non-specific
binding. The sections were incubated overnight at 4°C with an
anti-LC3 rabbit polyclonal primary antibody at a dilution of 1:100
(cat. no. 3868; Cell Signaling Technology, Inc., Danvers, MA, USA).
Following washing of the primary antibody, the sections were
incubated with a peroxidase-conjugated AffiniPure goat anti-rabbit
antibody (1:500; cat. no. sc-45101; Santa Cruz Biotechnology, Inc.,
Santa Cruz, CA, USA) for 90 min at room temperature.
Immunoreactivity was visualized using 3′,3-diaminobenzidine
reaction, then hematoxylin was used to counter-stain the sections
(both purchased from ZSGB-BIO, Beijing., China). For the blank
control, the primary antibody was omitted. For the negative
control, the primary antibody was replaced with non-immune serum.
The stained slides were scored independently by two pathologists
blinded to the clinical data. Staining was graded
semi-quantitatively as follows: 0, Absent expression or nuclear
expression only; 1+, cytoplasmic faint, barely perceptible staining
not exceeding background in any percentage of cells; 2+,
cytoplasmic staining exceeding background in 0 to 50% of tumor
cells; and 3+, cytoplasmic staining exceeding background in >50%
tumor cells.
Cell culture
Human fibroblasts were prepared as outgrowth
cultures from 5 healthy oral mucosa biopsies, and were subsequently
cultured as previously described (14). In selected experiments, fibroblasts
were stimulated with recombinant TGF-β (10 ng/ml; R&D Systems,
Ambington, UK) and 5, 10 and 15 µM chloroquine (CQ; C6628;
Sigma-Aldrich, St. Louis, MO, USA), an autophagy inhibitor.
Stimulation experiments were performed in Dulbecco's modified
Eagle's medium/10% phosphate-buffered saline (PBS) (both purchased
from Gibco; Thermo Fisher Scientific, Inc.). Fibroblasts from
passages 4–8 were used for the experiments.
Western blot analysis
Protein was extracted from the fibroblast in
radio-immunoprecipitation assay buffer (50 mM Tris-hydrochloride,
pH 8, 150 mM NaCl, 0.1% SDS, 1% NP-40, 0.5% sodium deoxycholate,
0.57 mM phenylmethanesulfonyl fluoride and 1 µg/ml aprotinin),
consisting of the following: 50 mM Tris-hydrochloride, pH 8, 150 mM
NaCl, 0.1% SDS, 1% NP-40, 0.5% sodium deoxycholate, 0.57 mM
phenylmethanesulfonyl fluoride and 1 µg/ml aprotinin. Protein
samples (30 µg) were separated using 12% sodium dodecyl sulfate
polyacrylamide gel electrophoresis gels (Bio-Rad Laboratories,
Inc.) and transferred to polyvinylidene difluoride membranes (EMD
Millipore, Billerica, MA). The membranes were blocked with 5%
non-fat milk in Tris-buffered saline (Sigma-Aldrich) with 1%
Tween-20 for 1 h at room temperature, and immunoblotted with an
anti-LC rabbit polyclonal antibody (1:1,000; Cell Signaling
Technology, Inc.) and an anti-tubulin mouse monoclonal antibody
(cat. no. sc-69969; Santa Cruz Biotechnology, Inc.) at 4°C for 1 h.
The binding of the primary antibodies was detected following
incubation with goat anti-rabbit or anti-mouse horseradish
peroxidase-conjugated secondary antibodies (cat. no. sc-395758;
Santa Cruz Biotechnology, Inc.) at room temperature, and was
visualized using ECL Western Blotting Detection Reagents (Amersham
Pharmacia Biotech (UK) Ltd., Little Chalfont, UK) following
exposure to chemiluminescent film (Bio-Rad Laboratories, Inc.).
Densiometry analysis was performed using an ImageJ Gel Analysis
tool (National Institutes of Health, Bethesda, MA, USA).
MTS assay
Measurements were made according to the
manufacturer's protocol. Briefly, 20 µl MTS reagent (Promega
Corporation, Madison, WI, USA) was added directly to the wells of
the 96-well plates, and cells were incubated at 37°C for a minimum
of 2 h. Absorbance was measured using a SpectraMax Plus 384 reader
(Molecular Devices; Sunnyvale, CA, USA) at 490 nm. Initially,
background absorbance was subtracted using a set of wells that only
contained medium. It was then normalized against and expressed as a
relative percentage of the plate-averaged dimethyl sulfoxide
(Amresco, Inc., Farmingham, MA, USA) control.
Cell apoptosis assay
Cell apoptosis was analyzed using flow cytometry
(FCM) (Guava easyCyte; EMD Millipore). The apoptotic cells were
differentiated from the viable or necrotic ones by combined
application of annexin V-fluorescein isothiocyanate (FITC) and
propidium iodide (PI). The samples were washed twice and adjusted
to a concentration of 1×106 cells/ml with cold PBS.
Subsequently, 10 µl annexin V-FITC and 10 µl PI (both purchased
from BD Biosciences, Franklin Lakes, NJ, USA) were added into 100
µl cell suspension, and incubated for 15 min at room temperature in
the dark. Finally, 400 µl binding buffer was added to each sample
without washing, and analyzed using FCM. Each experiment was
performed at least in triplicate.
Statistical analysis
Data are expressed as the mean ± standard error. The
Wilcoxon signed-rank test was used for related samples, and the
Mann-Whitney U test was used for non-related samples. In subsets of
experiments, the mean value of the control groups was set to 1. All
other values were expressed as x-fold changes, compared with the
respective controls used as ‘comparison mean values’. P<0.05 was
considered to indicate a statistically significant difference.
Results
Overexpression of autophagy in
OSF
In order to determine the role of the autophagy in
OSF, the expression profiling of LC3 in OSF, which was commonly
used to monitor autophagy in cultured cells and organization
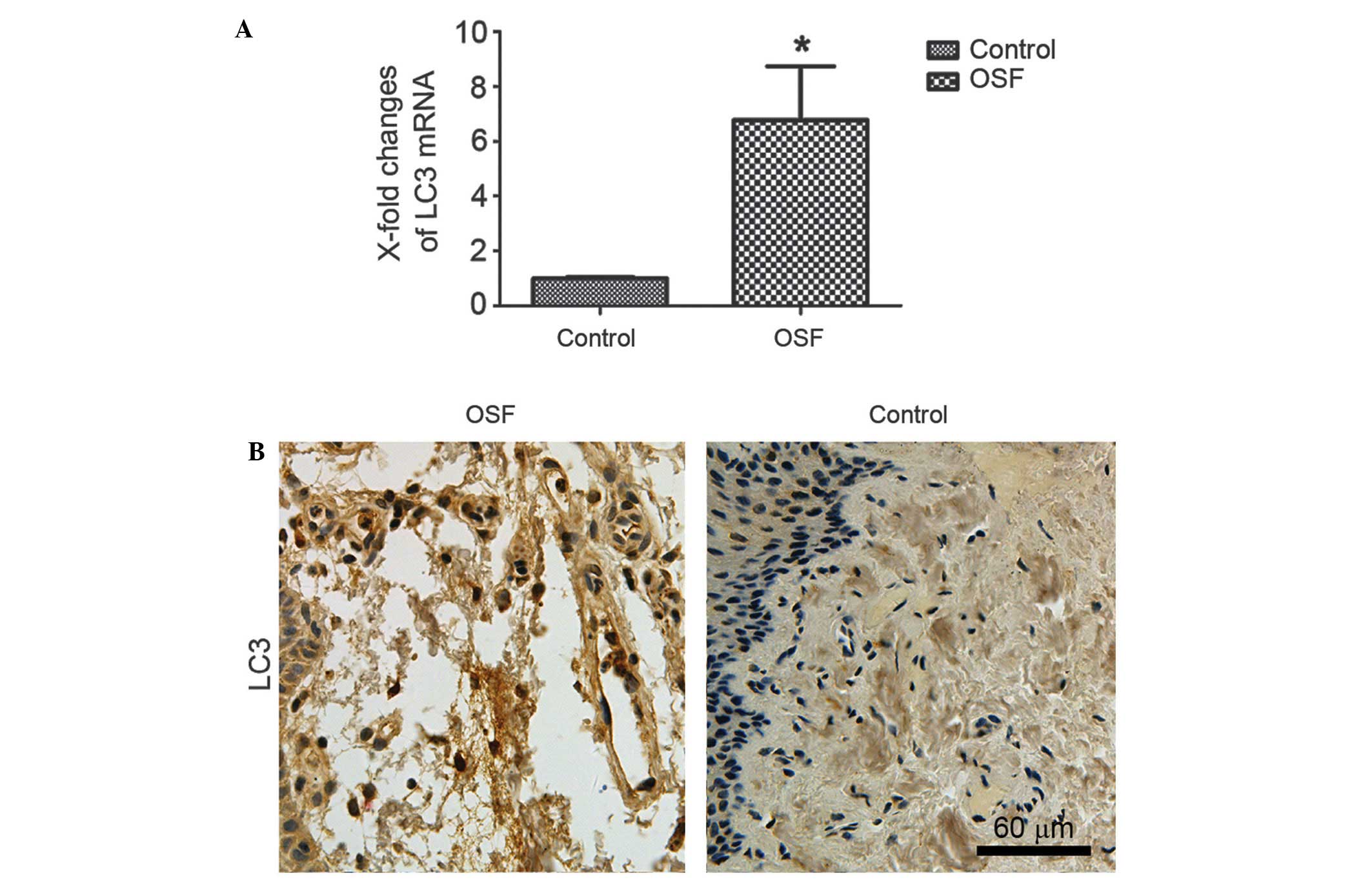
tissue, was investigated. As shown in Fig. 1A, LC3 was significantly upregulated
in OSF samples compared with normal oral mucosa tissues. Overall,
the paraffin-embedded OSF samples showed positive expression of
LC3. The representative immunostaining of LC3 in OSF samples was
shown in Fig. 1B. All the results
show that autophagy is activated in OSF.
TGF-β activates autophagy in
fibroblasts
Due to the crucial role of TGF-β in fibrotic
disease, we speculated that TGF-β signaling might contribute to the
activation of autophagy. To investigate the effect of TGF-β on
autophagic activity, the turnover of LC3 in lysosomes was directly
evaluated (15). Stimulation with
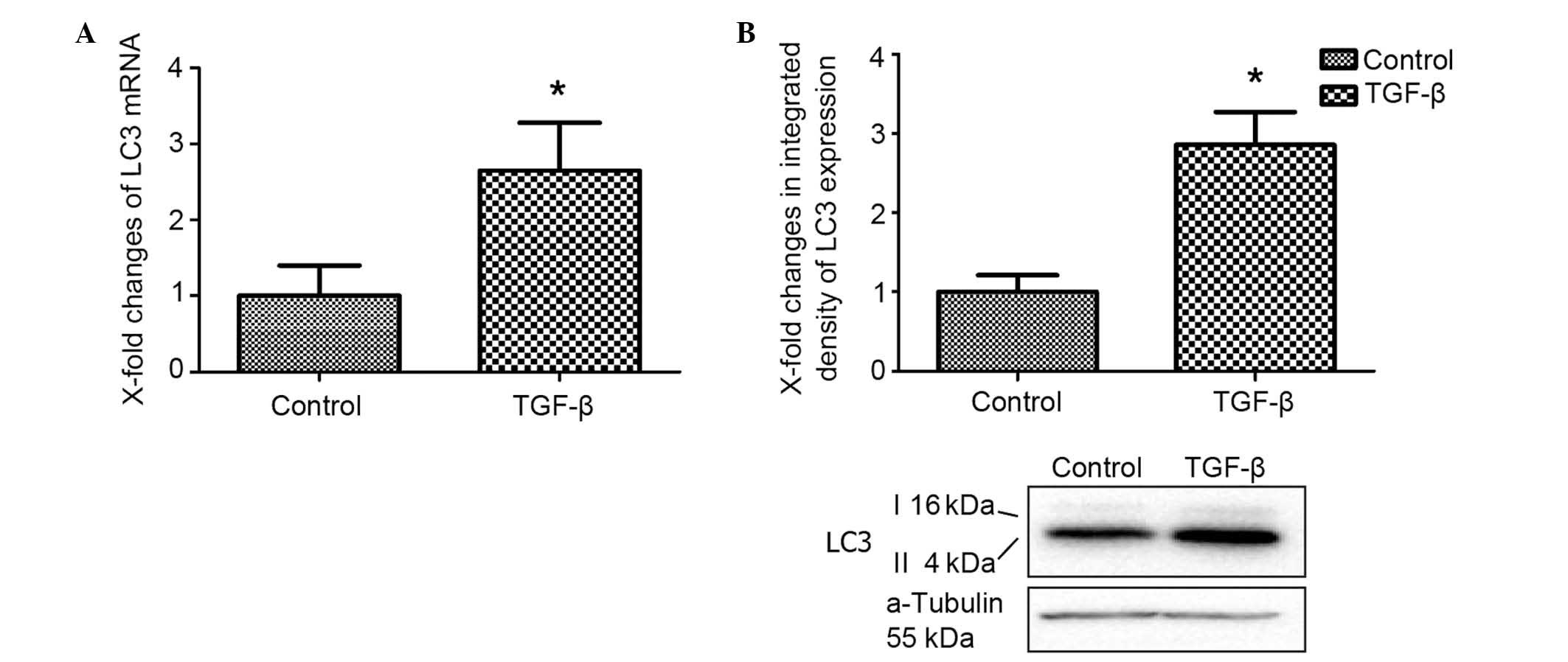
TGF-β in cultured fibroblasts induced the overexpression of LC3
mRNA (Fig. 2A). In addition, the
LC3-II protein levels were found to be significantly higher than
those in the control group.
LC3, also known as MAP1LC3, is commonly used to
monitor autophagy in cultured cells and animal tissues (16). The cytosolic form of LC3 (termed
LC3-I) is represented by the upper band of the immunoblot, and the
autophagosomal membrane lipidated form of LC3 (termed LC3-II) is
represented by the lower band (17).
In combination, these data indicated that TGF-β induced autophagy
in cultured fibroblasts.
Inhibition of autophagy decreased
critical fibrogenic gene (Col1A2) expression in fibroblasts
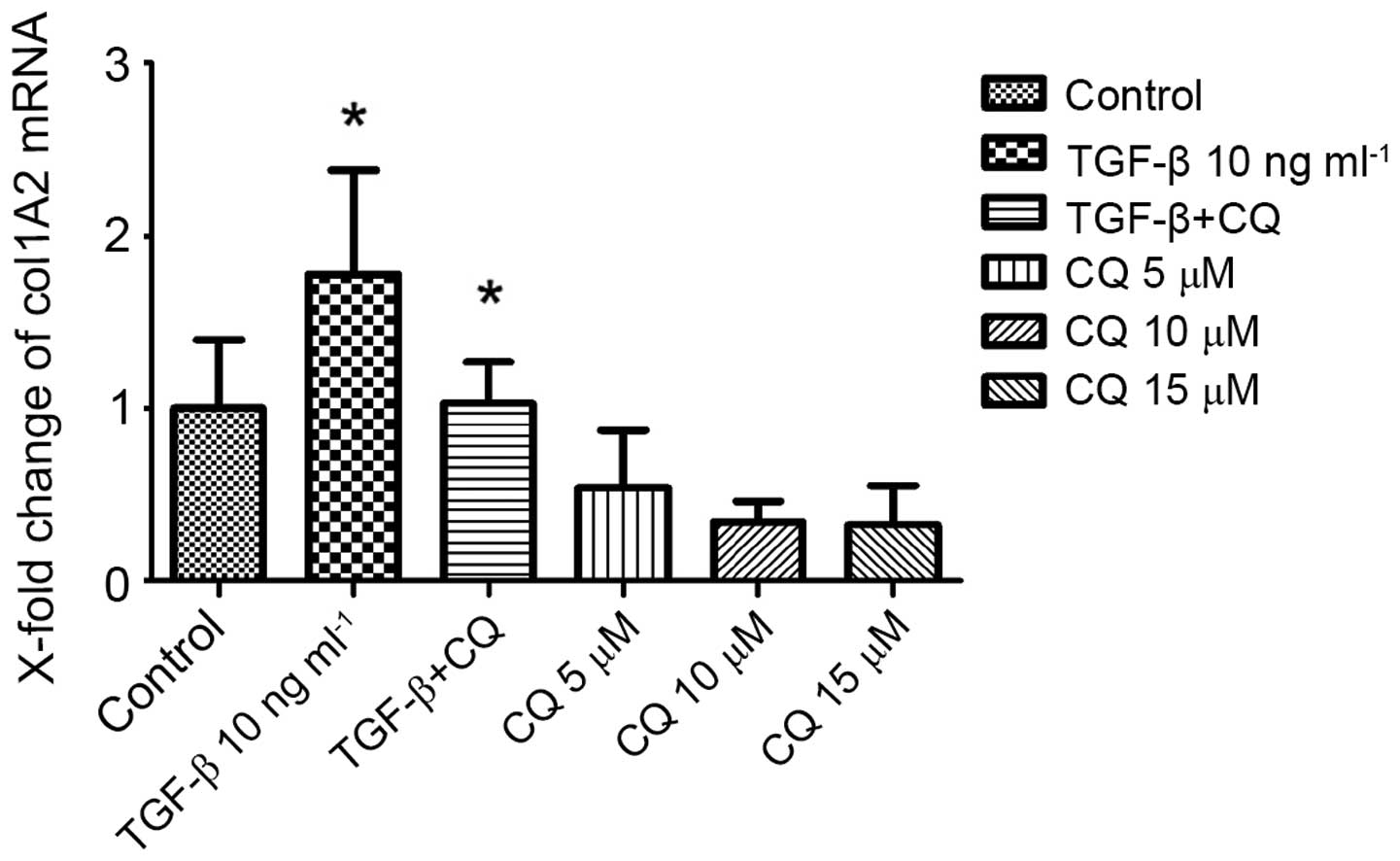
To investigate the functional role of the autophagy
in OSF, human oral fibroblasts were incubated with CQ, the only
inhibitor of autophagy available for use in clinical practice
(18). CQ suppresses the
transcriptional activity in Col1A2 reporter assays. Consistently, a
dose-dependent reduction in collagen release was observed in the
supernatants of fibroblasts stimulated with CQ. Notably, the
stimulatory effects of CQ were comparable to those of TGF-β, which
is considered to be a potent profibrotic mediator (Fig. 3).
Suppressing autophagy promotes
apoptosis in fibroblasts while suppressing proliferation
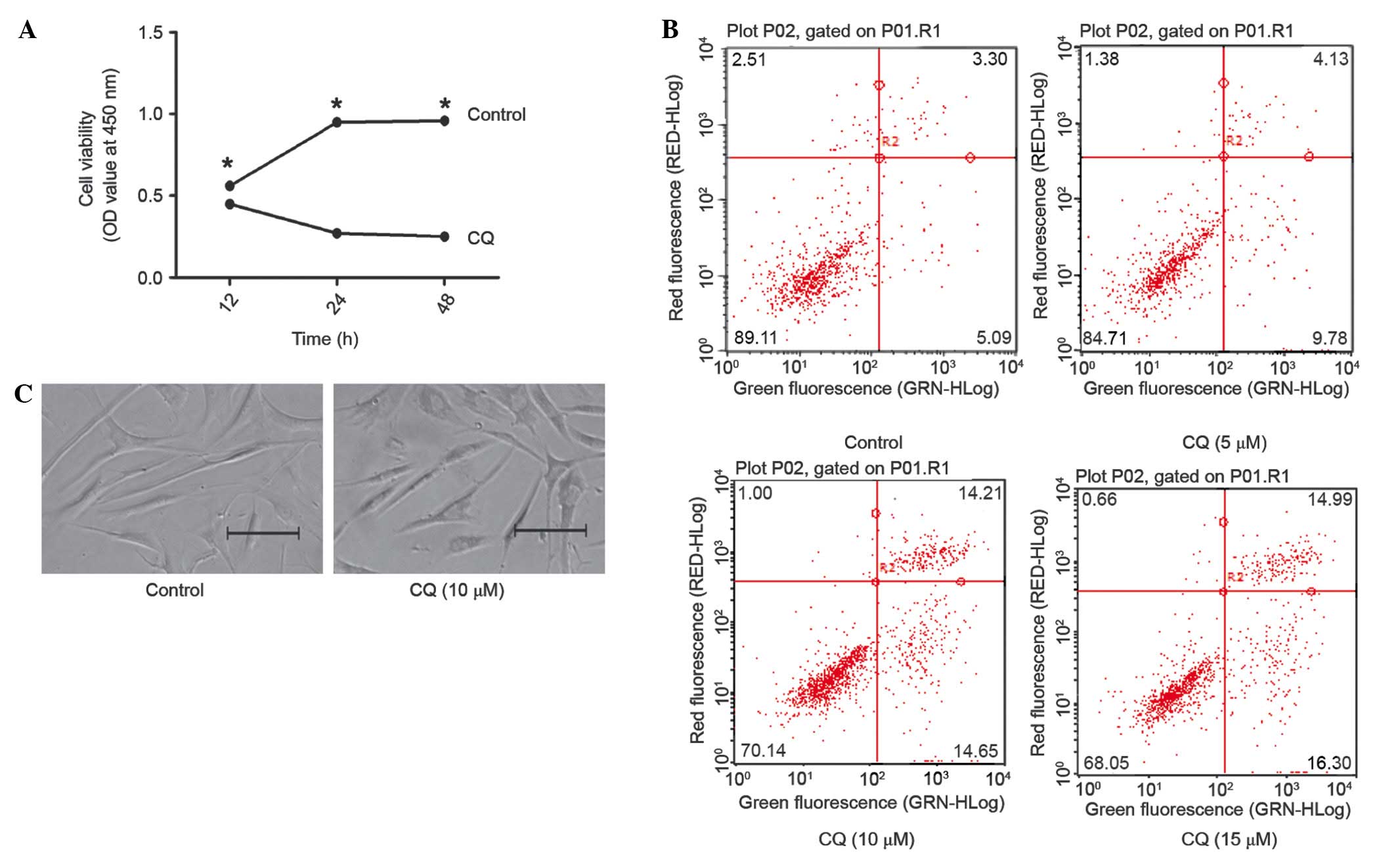
Fibroblasts were incubated with CQ and subjected to
MTS assay. A significant reduction was observed in the growth rate
of 5 µm CQ-incubated cells, as compared with those incubated with
the control (Fig. 4A). In order to
determine the mechanism of growth inhibition in CQ-incubated
fibroblasts, the apoptotic rate of the cells was analyzed. FCM
showed that CQ increased the apoptotic rate, as compared with the
control group (Fig 4B). In addition,
the inhibition of autophagy was shown to disrupt the physiological
tissue architecture (Fig. 4C). These
data suggested that CQ inhibited the growth of fibroblasts by
inducing cell apoptosis via the suppression of autophagy.
Discussion
Activation of the autophagy appears to be a general
feature of OSF, well-described elevations in endoplasmic reticulum
stress (19,20), oxidative stress (21), and HIF-1α (22), all of which are known to induce
autophagy. Indeed, pathologically activated autophagy has been
associated with various fibrotic diseases (23–27). An
overexpression of the autophagy marker LC3 was observed in human
samples from a number of patients with OSF. These changes resulted
in the activation of the autophagy pathway and increased
transcription of target genes in OSF.
Autophagy is crucially involved in collagen release
in OSF (28). In the present study,
CQ inhibits the release of collagen in fibroblasts, indicating that
the inhibition of autophagy may be effective in acute and chronic
fibrotic diseases. The activation of the autophagy pathway and its
potent profibrotic effects suggest that autophagy may be a
potential target for novel antifibrotic approaches. In the present
study the autophagy inhibitor CQ was selected for the inhibition of
the autophagy pathway for the following reasons: Firstly, it is
able to diffuse across cell membranes, undergo protonation and
accumulate in acidic organelles, such as lysosomes (29); secondly, it is a 4-aminoquinoline
drug used in the treatment of numerous diseases; and thirdly, this
approach allows broader inhibition of autophagy compared with
targeting single autophagy proteins (30).
Activated TGF-β signaling is considered to be a
common characteristic of fibrotic diseases (31). The present results highlighted the
crosstalk between TGF-β signaling and autophagy, while suggesting
that TGF-β activates autophagy. Stimulation with TGF-β in cultured
fibroblasts induced the overexpression of LC3; a TGF-β-mediated
increase of LC3 was identified as a potential molecular mechanism.
Furthermore, autophagy inhibition by CQ significantly reduced the
stimulatory effects of TGF-β on fibroblasts. These findings
suggested that the interaction between autophagy and TGF-β is a
crucial mechanism in fibrotic diseases. TGF-β has been shown to
trigger the activation of numerous intracellular signaling cascades
(32); however, the molecular
mediators of the profibrotic effects of TGF-β are not yet fully
understood, and the optimal targets for antifibrotic treatments
have not yet been identified. Although the central role of TGF-β
signaling in the fibrotic process has been confirmed (33), the first attempts of targeting TGF-β
signaling in humans failed (34).
CAT-192, a neutralizing antibody against TGF-β1, was inefficient in
this regard due to the low-affinity binding of TGF-β1 (34). Furthermore, an attempt to target the
downstream mediator c-Abl, in combination with platelet-derived
growth factor receptor, did not prove particularly successful
(35). The inhibition of the
autophagy pathway may provide a novel approach for the prevention
of the profibrotic effects of TGF-β signalling.
It was observed in the present study that
suppressing autophagy promoted apoptosis in fibroblasts while
suppressing proliferation, demonstrated that autophagy promoted
fibroblast proliferation and anti-apoptosis. In fibrogenic cells,
the lipophagy of lipid droplets (LDs) by autophagy provides
cellular energy critical to fuel the catabolic pathways of cellular
activation. The inhibition of autophagy leads to an increase in
triglyceride-containing LDs, which is associated with a reduction
in the total adenosine triphosphate levels and may be partially
reversed by the addition of the free fatty acid oleate (9). The crucial role of autophagy in cell
survival was demonstrated in previous studies using Atg-knockout
mice. Mice deficient in Atg3, Atg5, Atg7, Atg9 or Atg16L1 failed to
induce autophagy and died on the day of birth, due to the
starvation that followed the disruption of the trans-placental
nutrient supply (36). Furthermore,
mice with neuron-specific Atg5 or Atg7 knockout suffer from
neurodegeneration and apoptotic neuronal death, and T-cell-specific
Atg5 deficiency leads to an increase in the peripheral T-cell
apoptosis upon T-cell activation (37–39). In
addition, it has been reported that autophagy is not induced in
idiopathic pulmonary fibrosis, despite the upregulation of several
activators of autophagy (40).
In conclusion, autophagy is a complex process and
the exact mechanism remains to be elucidated. It was demonstrated
in the present study that the inhibition of autophagy can decrease
the expression of Col1A2 and protect from TGF-β-induced fibrosis,
indicating one mechanism by which autophagy may mediate
fibrogenesis.
Acknowledgements
This study was supported by the Hunan Provincial
Natural Science Foundation of China (grant no. 2015JJ4074).
References
|
1
|
Tilakaratne WM, Iqbal Z, Teh MT,
Ariyawardana A, Pitiyage G, Cruchley A, Stewart JE, Hagi-Pavli E,
Lalli A, Waseem A, et al: Upregulation of HIF-1alpha in malignant
transformation of oral submucous fibrosis. J Oral Pathol Med.
37:372–377. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rajalalitha P and Vali S: Molecular
pathogenesis of oral submucous fibrosis - a collagen metabolic
disorder. J Oral Pathol Med. 34:321–328. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shieh TM, Tu HF, Ku TH, Chang SS, Chang KW
and Liu CJ: Association between lysyl oxidase polymorphisms and
oral submucous fibrosis in older male areca chewers. J Oral Pathol
Med. 38:109–113. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pindborg JJ and Sirsat SM: Oral submucous
fibrosis. Oral Surg Oral Med Oral Pathol. 22:764–779. 1966.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wirawan E, Vanden Berghe T, Lippens S,
Agostinis P and Vandenabeele P: Autophagy: For better or for worse.
Cell Res. 22:43–61. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Boya P and Codogno P: Micronucleophagy: A
new mechanism to protect against chromosomal instability? Cell
Cycle. 11:645–646. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Ashford TP and Porter KR: Cytoplasmic
components in hepatic cell lysosomes. J Cell Biol. 12:198–202.
1962. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hernández-Gea V and Friedman SL: Autophagy
fuels tissue fibrogenesis. Autophagy. 8:849–850. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang CC, Liu TY, Wey SP, Wang FI and Jan
TR: Areca nut extract suppresses T-cell activation and
interferon-gamma production via the induction of oxidative stress.
Food Chem Toxicol. 45:1410–1418. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kiffin R, Bandyopadhyay U and Cuervo AM:
Oxidative stress and autophagy. Antioxid Redox Signal. 8:152–162.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang H, Bosch-Marce M, Shimoda LA, Tan
YS, Baek JH, Wesley JB, Gonzalez FJ and Semenza GL: Mitochondrial
autophagy is an HIF-1-dependent adaptive metabolic response to
hypoxia. J Biol Chem. 283:10892–10903. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2−ΔΔCt method. Methods. 25:402–408. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Akhmetshina A, Dees C, Pileckyte M, Szucs
G, Spriewald BM, Zwerina J, Distler O, Schett G and Distler JH:
Rho-associated kinases are crucial for myofibroblast
differentiation and production of extracellular matrix in
scleroderma fibroblasts. Arthritis Rheum. 58:2553–2564. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mizushima N, Yoshimori T and Levine B:
Methods in mammalian autophagy research. Cell. 140:313–326. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee E, Koo Y, Ng A, Wei Y, Luby-Phelps K,
Juraszek A, Xavier RJ, Cleaver O, Levine B and Amatruda JF:
Autophagy is essential for cardiac morphogenesis during vertebrate
development. Autophagy. 10:572–587. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mizushima N, Yamamoto A, Matsui M,
Yoshimori T and Ohsumi Y: In vivo analysis of autophagy in response
to nutrient starvation using transgenic mice expressing a
fluorescent autophagosome marker. Mol Biol Cell. 15:1101–1111.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Maycotte P, Aryal S, Cummings CT, Thorburn
J, Morgan MJ and Thorburn A: Chloroquine sensitizes breast cancer
cells to chemotherapy independent of autophagy. Autophagy.
8:200–212. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Korfei M, Ruppert C, Mahavadi P, Henneke
I, Markart P, Koch M, Lang G, Fink L, Bohle RM, Seeger W, et al:
Epithelial endoplasmic reticulum stress and apoptosis in sporadic
idiopathic pulmonary fibrosis. Am J Respir Crit Care Med.
178:838–846. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lawson WE, Cheng D, Degryse AL, Tanjore H,
Polosukhin VV, Xu XC, Newcombe DC, Jones BR, Roldan J, Lane KB, et
al: Endoplasmic reticulum stress enhances fibrotic remodeling in
the lungs. Proc Natl Acad Sci USA. 108:10562–10567. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kliment CR and Oury TD: Oxidative stress,
extracellular matrix targets, and idiopathic pulmonary fibrosis.
Free Radic Biol Med. 49:707–717. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tzouvelekis A, Harokopos V, Paparountas T,
Oikonomou N, Chatziioannou A, Vilaras G, Tsiambas E, Karameris A,
Bouros D and Aidinis V: Comparative expression profiling in
pulmonary fibrosis suggests a role of hypoxia-inducible
factor-1alpha in disease pathogenesis. Am J Respir Crit Care Med.
176:1108–1119. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
He Y, Jin L, Wang J, Yan Z, Chen T and
Zhao Y: Mechanisms of fibrosis in acute liver failure. Liver Int.
35:1877–1885. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
De Stefano D, Villella VR, Esposito S,
Tosco A, Sepe A, De Gregorio F, Salvadori L, Grassia R, Leone CA,
De Rosa G, Maiuri MC, et al: Restoration of CFTR function in
patients with cystic fibrosis carrying the F508del-CFTR mutation.
Autophagy. 10:2053–2074. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
He L, Livingston MJ and Dong Z: Autophagy
in acute kidney injury and repair. Nephron Clin Pract. 127:56–60.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gomez-Arroyo J, Sakagami M, Syed AA,
Farkas L, Van Tassell B, Kraskauskas D, Mizuno S, Abbate A, Bogaard
HJ, Byron PR and Voelkel NF: Iloprost reverses established fibrosis
in experimental right ventricular failure. Eur Respir J.
45:449–462. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lee JH, Jang EJ, Seo HL, Ku SK, Lee JR,
Shin SS, Park SD, Kim SC and Kim YW: Sauchinone attenuates liver
fibrosis and hepatic stellate cell activation through TGF-β/Smad
signaling pathway. Chem Biol Interact. 224C:58–67. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Junkins RD, McCormick C and Lin TJ: The
emerging potential of autophagy-based therapies in the treatment of
cystic fibrosis lung infections. Autophagy. 10:538–547. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Solomon VR and Lee H: Chloroquine and its
analogs: A new promise of an old drug for effective and safe cancer
therapies. Eur J Pharmacol. 625:220–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Morgan MJ, Gamez G, Menke C, Hernandez A,
Thorburn J, Gidan F, Staskiewicz L, Morgan S, Cummings C, Maycotte
P and Thorburn A: Regulation of autophagy and chloroquine
sensitivity by oncogenic RAS in vitro is context-dependent.
Autophagy. 10:1814–1826. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Verrecchia F and Mauviel A: Transforming
growth factor-beta and fibrosis. World J Gastroenterol.
13:3056–3062. 2007.PubMed/NCBI
|
|
32
|
Shi Y and Massagué J: Mechanisms of
TGF-beta signaling from cell membrane to the nucleus. Cell.
113:685–700. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Akhmetshina A, Palumbo K, Dees C, Bergmann
C, Venalis P, Zerr P, Horn A, Kireva T, Beyer C, Zwerina J, et al:
Activation of canonical Wnt signalling is required for
TGF-β-mediated fibrosis. Nat Commun. 3:7352012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Denton CP, Merkel PA, Furst DE, Khanna D,
Emery P, Hsu VM, Silliman N, Streisand J, Powell J, Akesson A, et
al: Cat-192 Study Group; Scleroderma Clinical Trials Consortium:
Recombinant human anti-transforming growth factor beta 1 antibody
therapy in systemic sclerosis: A multicenter, randomized,
placebo-controlled phase I/II trial of CAT-192. Arthritis Rheum.
56:323–333. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gordon J, Mersten J, Lyman S, Kloiber SA,
Wildman HF, Crow MK, Kirou KA and Spiera RF: Imatinib mesylate
(Gleevec) in the treatment of systemic sclerosis: Interim results
of a phase IIa, one year, open label clinical trial. American
College of Rheumatology (ACR)/Association of Rheumatology Health
Professionals (ARHP) Scientific Meeting 2009 (Pennsylvania).
2009.
|
|
36
|
Kuma A and Mizushima N: Physiological role
of autophagy as an intracellular recycling system: With an emphasis
on nutrient metabolism. Semin Cell Dev Biol. 21:683–690. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Komatsu M, Waguri S, Chiba T, Murata S,
Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E and
Tanaka K: Loss of autophagy in the central nervous system causes
neurodegeneration in mice. Nature. 441:880–884. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hara T, Nakamura K, Matsui M, Yamamoto A,
Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I,
Okano H and Mizushima N: Suppression of basal autophagy in neural
cells causes neurodegenerative disease in mice. Nature.
441:885–889. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Pua HH, Dzhagalov I, Chuck M, Mizushima
Nand and He Y: A critical role for the autophagy gene Atg5 in T
cell survival and proliferation. J Exp Med. 204:25–31. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Patel AS, Lin L, Geyer A, Haspel JA, An
CH, Cao J, Rosas IO and Morse D: Autophagy in idiopathic pulmonary
fibrosis. PLoS One. 7:e413942012. View Article : Google Scholar : PubMed/NCBI
|