Introduction
γ-aminobutyrate (GABA) is a crucial inhibitory
neurotransmitter in the mammalian peripheral nervous system (PNS)
and central nervous system (CNS) (1). GABAA receptor is a pentamer
comprised of multiple subunits (α−6, β1–3,
γ1–3, π, ε, δ and θ) with an absolute chloride ion
channel and diversiform allosteric binding sites through which
rapid inhibitory synaptic neurotransmission may be modulated
(1). The GABAA receptor
is a favorable target for therapeutic agents including steroids,
barbiturates, benzodiazepines, anesthetics and convulsants
(2). Recently, it has also been
proposed that the β-subunit has an important role in determining
the chloride ion selectivity of GABAA receptors
(3,4).
GABAA receptor antagonists niflumic acid
(NFA) and 5-nitro-2-(3-phenylpropylamino) benzoic acid (NPPB), are
the only chloride ion channel blockers able to protect cells from
excitotoxicity (5). Accumulating
evidence suggests that non-steroidal anti-inflammatory drugs
(NSAIDs) modulate GABAA receptor function in
heterologous expression systems (6).
NSAIDS at clinically relevant concentrations (low micromolar) are
sufficient to potentiate β2/3-containing
GABAA receptors (7). The
NSAID-sensitive α1β2γ2 receptor
subtype is the predominant and the largest GABAA
receptor population in mammalian PNS and CNS (7). In addition to their effect on
GABAA receptors, fenamate NSAIDs also affect a variety
of other ion channels (8,9). Several drugs that have an effect on
GABAA receptor function have been revealed to depend on
subtypes (subunits combinations), and on specific amino acids situs
of specific subunits (10).
Halliwell et al (11) revealed that the regulation of
GABAA receptors by one particular anti-inflammatory
agent, mefenamic acid, was dependent on the β-subunit. Conversely,
Sinkkonen et al (12)
reported that the potentiation of
α1β2γ2 receptors by NFA was
dependent on the presence of a γ2 subunit, which also
effects mefenamic acid modulation (11). Antagonism of the
α6β2γ2 receptor subtype by NFA has
also been reported (12), and the
substitution of an α4 subunit reduced the mefenamic
potentiation of α1β2γ2 receptors
by 50% (13). Similar observations
have been observed in electrophysiological studies regarding the
actions of mefenamic acid, pentobarbital and etomidate (11,14,15).
The aim of the present study is to use conventional
whole-cell patch-clamp recordings, immunofluorescence and NSAIDs,
including NFA and NPPB, to investigate the effect of
Ca2+-activated Cl− channels (CaCCs) on
GABA-induced currents in the dorsal root ganglion (DRG) of rats.
The present study intended to elucidate the diversity of the
modulatory effect route of NSAIDs on GABA-activated currents via
CaCCs.
Materials and methods
Isolation of DRG neurons
A total of 120 Sprague-Dawley rats (SDRs) were
provided by the Experimental Animal Center of Xinjiang Medical
University, Urumqi, China (certificate no. SCXK 2003-0001; age,
8–10 weeks; weight, 250–280 g) irrespective of gender. All
protocols were approved by the Institutional Animal Care and Use
Committee at the Medical College of Shihezi University (Shihezi,
China) and were consistent with the Guidelines for the Care and Use
of Laboratory Animals published by the US National Institutes of
Health (16). SDRs were bred in
separate specific pathogen-free cages at a relative humidity of
40–70%, (24±3°C), 100–120 lx/12-h dark:light illumination and free
access to food and water. The DRG neuron selection and the
separation process are described in our previous studies (17,18).
Rats were sacrificed by decapitation.
Electrophysiological recordings
A gap-free recording with a sampling interval of 50
msec (17,18) was performed in the present study.
Briefly, with the aid of a whole-cell patch clamp amplifier,
perforated patch-clamp recordings in the whole-cell mode were
performed. Using an Axon 700B amplifier (Axon, San Jose, CA, USA)
and pCLAMP version 0.2 hardware and software (Axon), currents were
recorded from the DRG neurons in vitro. The room temperature
was set at 22–24°C. The resistance of the recording pipette ranged
from 3 to 5 MΩ. The experimental procedures were performed
according to the Regulations for the Administration of Affairs.
Concerning Experimental Animals, formulated by the Ministry of
Science and Technology of the People's Republic of China (The
Ministry of Science and TechnoIogy of the People's Republic of
China. Guidance Suggestions for the Care and Use of Laboratory
Animals 2011).
Immunofluorescent staining of TMEM16A
and TMEM16B to determine expression in the DRG
Rats were anesthetized with 0.3% (w/v) sodium
pentobarbital [Sangon Biotech (Shanghai,) Co., Ltd., Shanghai,
China], and perfused through the aorta with 0.9% (w/v) normal
saline, followed by fresh 4% (w/v) paraformaldehyde in
phosphate-buffered saline [both purchased from Sangon Biotech
(Shanghai,) Co., Ltd.] for 10 min for tissue fixation. The lumbar
DRG at level L4–6 to the nerve injury was removed
rapidly and placed in 4% (w/v) paraformaldehyde in PBS for 24 h.
The L4–6 DRG were cut into 5-µm sections with a freezing
microtome (CM1510S; Leica Biosystems, Wetzlar, Germany).
Immunofluorescent staining was performed using rabbit anti-TMEM16A
polyclonal antibody (1:20; sc-135235) and goat anti-TMEM16B
polyclonal antibody (1:20; sc-169622) (both purchased from Cell
Signaling Technology, Inc., Danvers, MA, USA) overnight at 4°C. The
sections were incubated for 1 h in a solution containing donkey
anti-rabbit IgG-fluorescein isothiocyanate (FITC; 1:50;
711-095-152) and donkey anti-goat IgG-tetramethylrhodamine (TRITC;
1:50; 705-025-003) (both purchased from Jackson ImmunoResearch,
West Grove, PA, USA) at 37°C. Several tissue sections were selected
for double-labeling of TNEM16A and TMEM16B and were incubated in a
mixture of primary antibodies against TNEM16A and TMEM16B, followed
by donkey anti-rabbit IgG conjugated with FITC and donkey anti-goat
IgG conjugated with TRITC. Slides were then examined by confocal
microscopy (LSM710; Carl Zeiss AG, Oberkochen, Germany).
Quantitative analysis of TMEM16A and TMEM16B expression in the DRG
was performed by measuring the mean absorbance at 488 and 550 nm
(Zeiss LSM 510 System; Carl Zeiss, Jena, Germany) following laser
confocal microscopy and using analysis software (ZEN 2009 Light
Edition; Carl Zeiss).
Drug application
GABA, muscimol, bicuculline, NFA, NPPB, caffeine and
NTDP were purchased from Sigma-Aldrich, (St. Louis, MO, USA).
1,2-Bis(2-aminophenoxy)ethane-N, N,N′,N′-tetraacetic acid tetrakis
(acetoxymethyl ester; BAPTA-AM) was from Merck Millipore
(Darmstadt, Germany). Rabbit anti-TMEM16A polyclonal antibody and
donkey anti-rabbit IgG-FITC were purchased from Santa Cruz
Biotechnology, Inc., (Dallas, TX, USA). All drugs used in
electrophysiological recordings were dissolved in extracellular
fluid and applied by gravity flow from a home-made perfusion system
consisting a row of tubules connected with a series of individual
reservoirs (17). This rapid
solution exchange system was manipulated by shifting the tubules
horizontally with a micromanipulator (17,18). The
time of pre-perfusion of antagonists was 0.5–5 min, and the time of
pre-perfusion of GABA was 5–10 sec.
Statistical analysis
Statistical analysis of the data was performed using
SPSS (version 13.0; SPSS, Inc., Chicago, IL, USA) and the values of
GABA-activated currents are presented as mean ± standard error of
the mean and a Student's t test was used to assess the
significance. P<0.05 was considered to indicate a statistically
significant difference.
Results
GABA-induced inward currents
Treatment with different concentrations of GABA
(1–1,000 µmol/l) activated an inward current in the majority of
cells (94.32%, 150/159) examined. The GABA-induced response was
concentration-dependent, and displayed evidence of desensitization
at high concentrations (Fig. 1). The
activation threshold was ~1 µmol/l and the maximal response was
achieved at 300 µmol/l GABA. The value of the dissociation constant
was ~30 µmol/l, derived from a concentration-response curve
(17,18). The averaged amplitude of 100 µmol/l
GABA-evoked inward current was (1.29±0.72 nA; neurons = 52). The
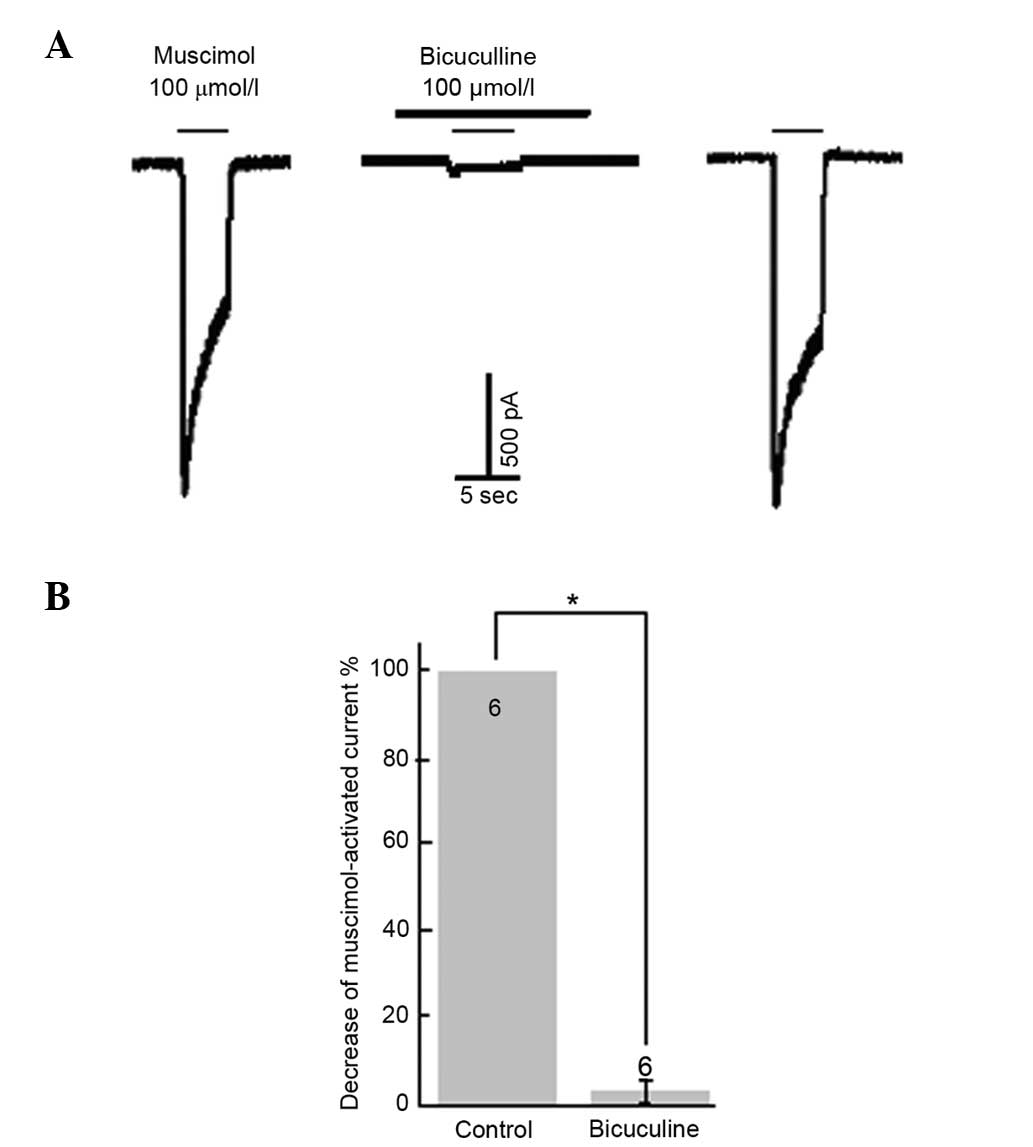
selective GABAA receptor agonist, muscimol (100 µmol/l)
mimicked the GABA-evoked response (neurons = 8). A selective
GABAA receptor antagonist, bicuculline (100 µmol/l),
suppressed GABA (neurons = 9) and muscimol-evoked (neurons = 8)
inward currents (Fig. 1).
Inhibition of GABA-induced inward
currents by NFA and NPPB
NFA and NPPB were pre-incubated for 20 sec prior to
application of GABA, resulting in the marked attenuation of the
GABA-induced inward current in the majority of the neurons examined
(96.3%, 52/54). Inhibition of GABA-induced responses by NFA and
NPPB were concentration-dependent. The inward currents induced by
100 µmol/l GABA were suppressed by (0±0.09%; neurons=4),
(5.32±3.51%; neurons=6), (21.3±4.00%; neurons=5), (33.8±5.20%;
neurons=17), (52.2±5.10%; neurons=4), (61.1±4.12%; neurons=12) and
(57.6±4.20%; neurons=4) by 0.1, 1, 3, 10, 30, 100 and 300 µmol/l
NFA, respectively (Fig. 2A). The
inhibition threshold was ~0.1 µmol/l and the maximal inhibition was
achieved at 300 µmol/l NFA. The IC50 value was ~6.7
µmol/l derived from the concentration-inhibition curve (Fig. 2B). NFA did not alter the half maximal
effective concentration EC50 value for GABA (~30 µmol/l)
(17,19), but reduced the maximal GABA currents
by ~60%. The inward current induced by 100 µmol/l GABA was
suppressed by 13.8±6.7%, neurons=6, P<0.05; 23.2±14.7%,
neurons=6, P<0.01 and (29.7±9.1%, neurons=9, P<0.01, by 3, 10
and 30 µmol/l NPPB, respectively. The inhibition threshold was ~1
µmol/l and the maximal inhibition was achieved by 30 µmol/l NPPB.
The IC50 value was ~11 µmol/l (Fig. 3).
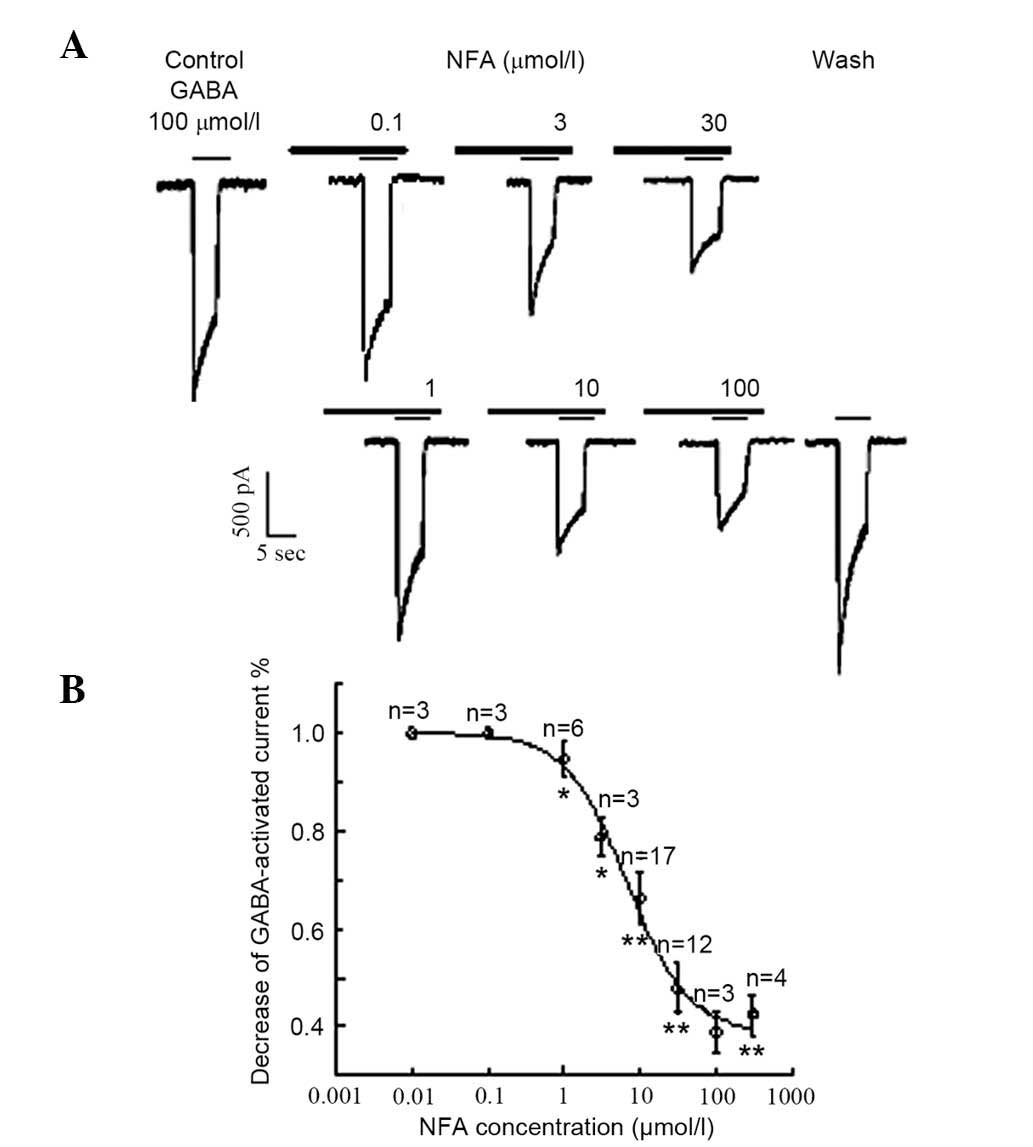 | Figure 2.Effects of different concentrations
of NFA on GABA-activated inward currents. (A) Results of 100 µmol/l
GABA-activated inward currents in the different-concentration of
NFA (0.1–100 µmol/l). (B) Inward current induced by 100 µmol/l GABA
was suppressed by 0±0.09% (neurons=4), 5.32±3.51% (neurons=6),
21.3±4.00% (neurons=5), 33.8±5.20% (neurons=17), 52.2±5.10%
(neurons=4), 61.1±4.12% (neurons=12) and 57.6±4.20% (neurons=4) by
0.1, 1, 3, 10, 30, 100 and 300 µmol/l NFA (neurons=3–17),
respectively. *P<0.05 and **P<0.01 vs. control. Data are
results of a paired t test. |
Effects of NTDP and extracellular
calcium on GABA-induced inward currents
The L-type calcium channel blocker, NTDP (0.1–30
µmol/l), inverted the inhibitory effect of 100 µmol/l NFA on 100
µmol/l GABA-induced inward current (Fig.
4). The inhibitory ratio of NFA on inward current induced by
GABA were (59.6±8.70%, neurons=4, P>0.05; 43.6±5.10%, neurons=3,
P<0.05; 32.3±6.62%, neurons=8, P<0.01; 8.7±7.6%, neurons=6,
P<0.01 and 8.6±7.4%, neurons=4, P<0.01, in the presence of
0.1, 1, 3, 10 and 30 µmol/l NTDP, respectively. To investigate the
effect of extracellular free calcium on NFA, calcium-free
extracellular fluid was utilized. The inhibitory effect of NFA on
GABA-evoked inward currents was strongly suppressed by calcium-free
extracellular fluid (P<0.01; Fig.
4). The 100 µmol/l GABA-activated current with 100 µmol/l NFA
were 1,298.8±124.4 pA; neurons = 22 and 775.9±104.9 pA; neurons =
6; P<0.01, in the presence and absence of calcium-free
extracellular solution, respectively. The inhibition ratio of NFA
on the GABA-evoked inward current was 8.4±7.2% (neurons = 6,
P<0.01) in the calcium-free extracellular solution.
Effect of intracellular calcium on the
GABA-induced inward current by intracellular calcium
To investigate the effect of intracellular free
calcium on the GABA-induced inward current, BAPTA-AM was utilized
and caffeine. The inhibitory effect of NFA on GABA-evoked inward
current was strongly suppressed by BAPTA-AM and caffeine. The
inhibition ratio of NFA on the GABA-evoked inward current was
decreased by 48.2±15.7% (neurons=9, P<0.01) and 38.7±13.2%
(neurons=7, P<0.05) with BAPTA-AM (100 µmol/l) and caffeine (30
µmol/l), respectively (Fig. 5).
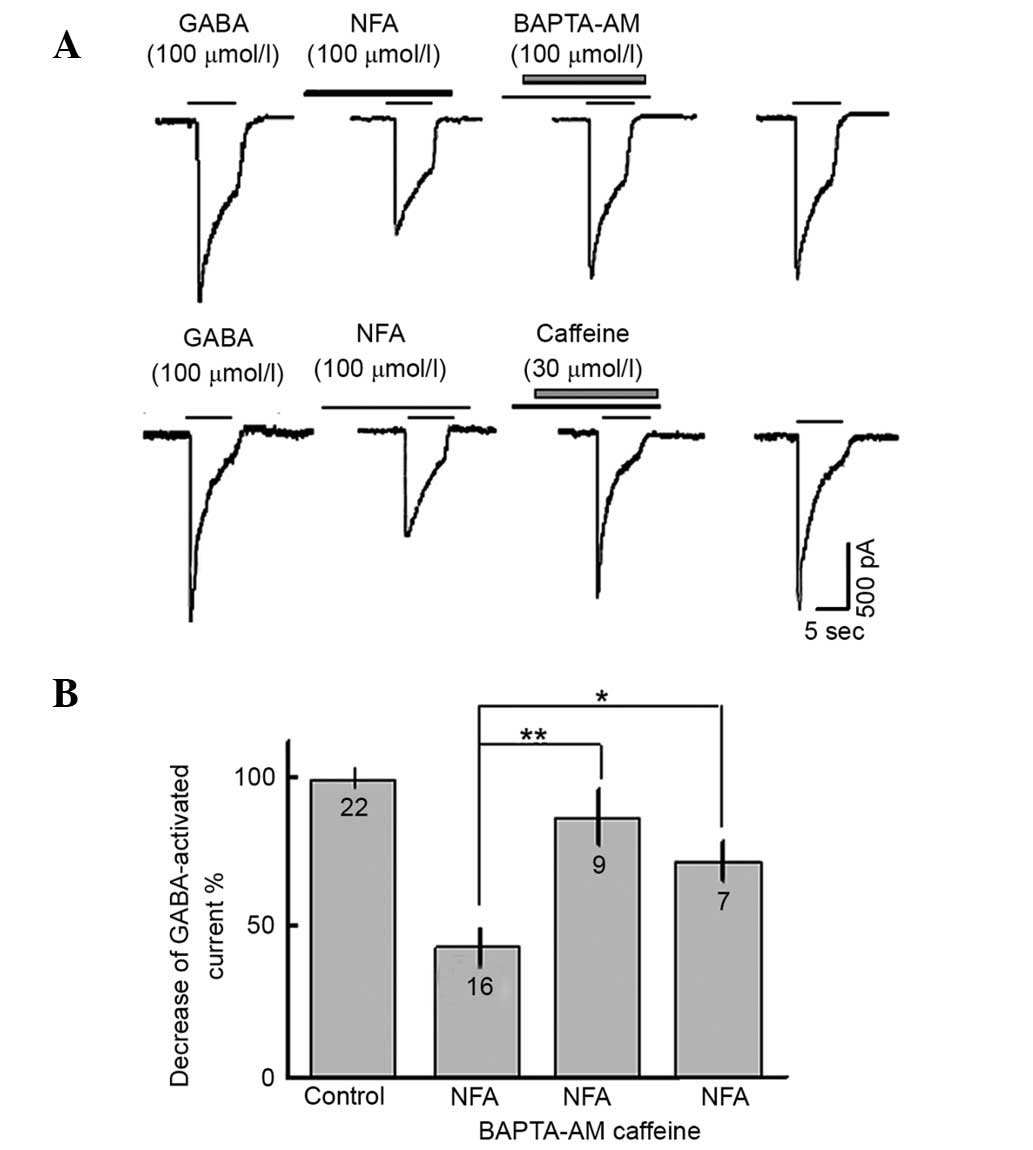 | Figure 5.Effects of intracellular
Ca2+ on GABA-activated inward currents. (A) Results of
100 µmol/l GABA-activated inward currents in the presence of 100
µmol/l NFA, 100 µmol/l NFA + 100 µmol/l BAPTA-AM and 100 µmol/l NFA
+ 30 µmol/l caffeine. (B) Inward current induced by GABA was
suppressed by 61.8±13.4, 18.9±17.6 and 28.4±13.2% NFA, NFA +
BAPTA-AM and NFA + caffeine, respectively. *P<0.05, **P<0.01.
Results as determined by paired t test. Numbers on bars = number of
neurons. NFA, niflumic acid; GABA, γ-aminobutyrate; BAPTA,
1,2-bis(2-aminophenoxy)ethane-N,N,N,N-tetraacetic acid. |
Distribution of TMEM16A and TMEM16B
subunits expressed in DRG neurons
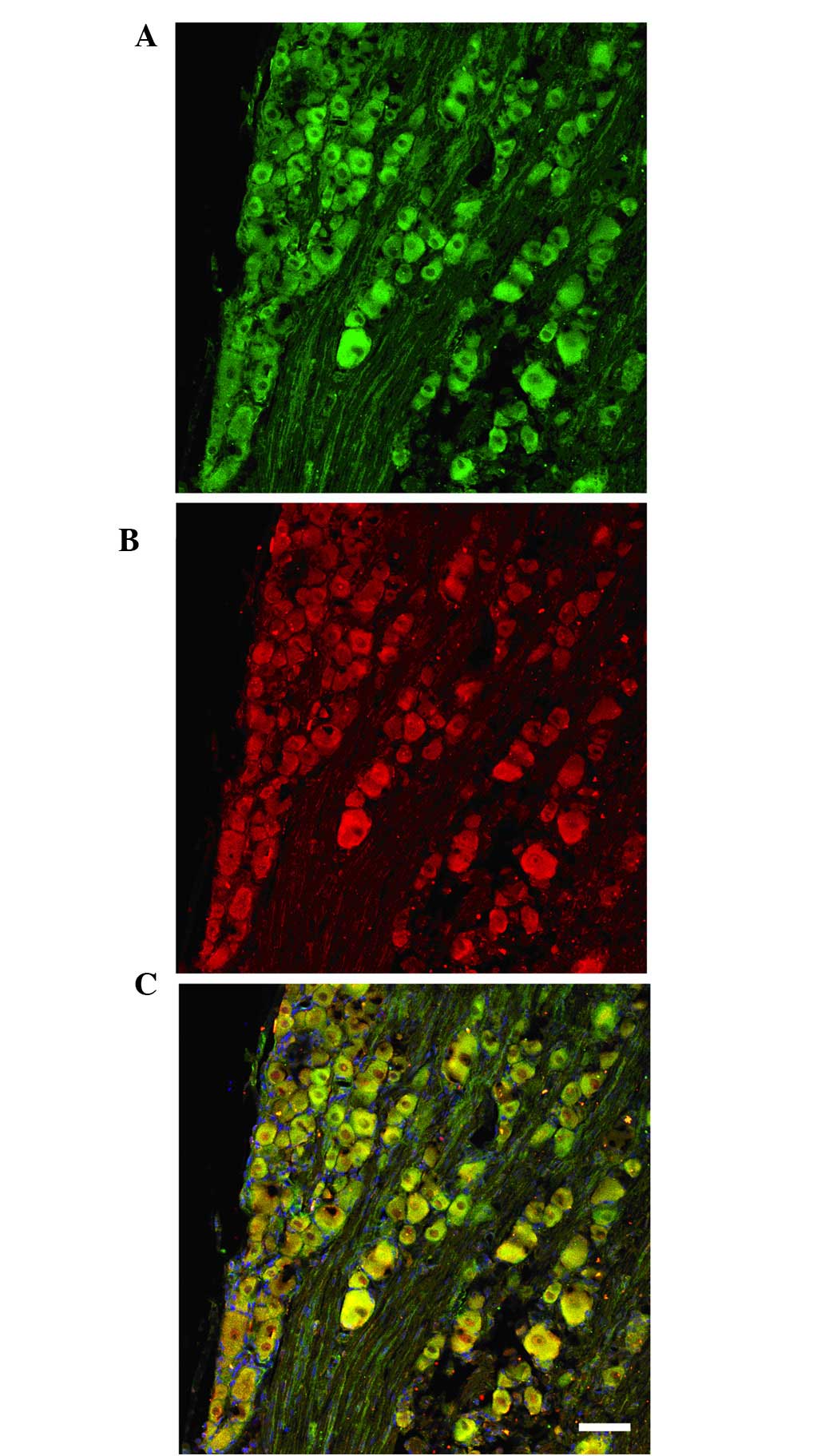
Immunofluorescence staining revealed that TMEM16A
and TMEM16B expression was widely distributed in DRG neurons, and
was predominately located in the cell membranes of various
diameters (Fig. 6A and B). Thus,
TMEM16A and TMEM16B were co-expressed in the membranes of DRG
neurons (Fig. 6C).
Discussion
GABAA receptor mediates GABA-evoked
membrane depolarization responses, or inward current, since the
selective GABAA receptor agonist muscimol mimicked
GABA-evoked responses, and the selective GABAA receptor
antagonist bicuculline blocked GABA-activated membrane responses in
rat DRG neurons (17,19). The results of the present study
indicated that NFA and NPPB, non-steroid anti-inflammatory agents,
reduced GABA-activated inward currents. The present study also
revealed that NTDP, an L-type calcium channel blocker, a
calcium-free extracellular solution, BAPTA-AM, which is a membrane
permeable Ca2+ chelator and caffeine, a Ca2+
consuming drug, also reduced the inhibitory effect of NFA on the
GABA-activated inward current. Furthermore, the current study
demonstrated that the TMEM16A and TMEM16B subunits were expressed
in rat DRG neurons.
A number of cells express CaCCs, which have several
physiological functions, including their being developmentally
adjusted with maximum peak expression in the period of peripheral
synaptogenesis in DRG neurons (20).
An association was observed between the expression of CaCCs and the
growth competence of sensory neurons (21). CaCCs activation augments after
depolarization following spike firing of action potential in
neonatal rat DRG neurons (22,23).
Axotomy upregulates the expression of CaCCs in adult sensory,
nodose and sympathetic ganglion neurons (21,24,25).
Additionally, with regard to electrical activity, CaCCs have an
important role in other basic cellular functions such as cell
adhesion, apoptosis and potentially in volume regulation (26,27). In
2008, the announcements that three labs had cloned genes that
encoded classical CaCCs generated considerable interest (28–30); the
2 genes that have been definitively shown to encode CaCCs termed
TMEM16A and TMEM16B. CaCCs are activated by an increase in
intracellular free calcium concentration following either internal
calcium release from Ca2+ stores, or external calcium
entry through Ca2+ channel (31). NFA and NPPB have been indicated to
suppress the activity of CaCCs, and NPPB inhibits chloride ion flux
through other anionic channels (32). The results of the present study show
that NFA and NPPB are able to significantly inhibit the
GABA-activated inward current, and NTDP, calcium-free extracellular
fluid and BAPTA-AM may significantly reduce the inhibitory effect
of NFA on GABA-activated inward current. The present results also
suggest that NSAIDs have an important role in the GABA-activated
inward current via CaCCs in DRG neurons in rats, and support the
hypothesis proposed by the present study. GABA activates the
GABAA receptor to open chloride ion channels, the
chloride ion efflux induces the depolarization response of the
membrane of DRG neurons (17).
Conversely, voltage dependent L-type Ca2+ channels may
be activated by depolarization, and lead to increased intracellular
Ca2+. Furthermore, the NFA-induced increase in
intracellular Ca2+ is likely due to Ca2+
release from an intracellular store (33–35).
CaCCs are activated by an increase in intracellular calcium
concentration, which in turn increases the driving force for
chloride ion efflux (28). Finally,
the synergistic action of chloride ion efflux via GABAA
receptors and NFA-sensitive CaCCs results in GABA-activated
currents or depolarization responses in rat DRG neurons. The
depolarization arising from Cl− efflux through CaCCs
indicates a mechanism of electrical amplification of the
GABA-activated currents (Fig.
7).
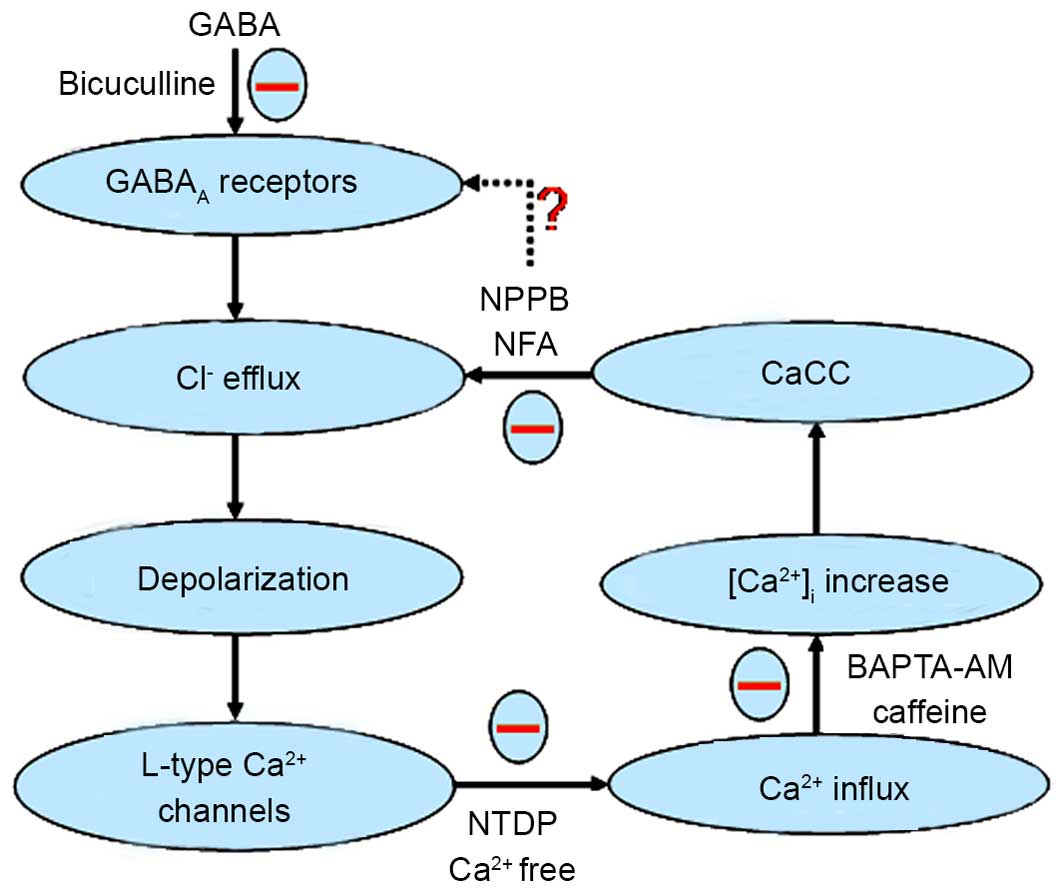 | Figure 7.Schematic displaying the effects of
CaCCs on GABA-activated inward currents and depolarization. GABA
activates the GABAA receptor to open the Cl−
channel and the Cl− efflux induces the depolarization
response (inward current) of the membrane of dorsal root ganglion
(DRG) neurons. Then, voltage dependent L-type Ca2+
channels are activated by the depolarization, and give rise to an
increase in intracellular Ca2+. CaCCs are activated by
an increase in intracellular Ca2+ concentration which,
in turn, increases the driving force for Cl− efflux.
Finally, the synergistic action of the chloride ion efflux through
GABAA receptors and NFA-sensitive CaCCs causes
GABA-activated currents or depolarization response in rat DRG
neurons. ‘−’ expressed function was inhibitory, ‘?’ expressed
function was not clear. GABA, γ-aminobutyrate; NPPB,
5-nitro-2-(3-phenylpropylamino) benzoic acid; NFA, niflumic acid;
CaCC, Ca2+-activated Cl− channels; BAPTA,
1,2-bis(2-aminophenoxy)ethane-N,N,N,N-tetraacetic acid; NTDP,
nitrendipine. |
The binding of intracellular signaling molecules or
extracellular ligands activates a conformational change that opens
or closes the pores of ligand-gated ion channels such as GABA,
glutamate, serotonin and acetylcholine, as well as the cyclic
nucleotide-gated ion channels that have important roles in sensory
biology (17). However, the
structure and function of ligand-gated ion channels and their
integral receptors have yet to be elucidated. Therefore, it is
important to develop novel tools to investigate the interactions
between various receptor subunits, which may benefit the future
designing of receptor subtype-selective therapies.
Ion channel gating is influenced by several factors,
including the binding of another ligand and intracellular
Ca2+ (36).
GABAA integrates the actions of a wide range of
therapeutic agents, including steroids, benzodiazepines,
barbiturates, convulsants and anesthetics (2). Previous studies have indicated that NFA
is able to directly act on GABAA and NMDA receptors
(5,12,37).
Notably, another chloride blocker, mefenamic acid, is also able to
directly activate GABAA receptors (38). NFA functions as a positive allosteric
modulator of α1β2γ2, and a
negative modulator of α6β2 and
α6β2γ2 (and
α1β2) GABAA receptors. Despite the
knowledge that NFA shares the same site as furosemide to mediate
its inhibitory effect, the site for the positive regulation remains
elusive, and is dependant on the presence of the γ2
subunit, yet separable from the benzodiazepine binding site
(11,12).
It has been suggested that the niflumate
potentiation of GABAA function is through a pure direct
allosteric mechanism (12).
Conversely, it has been reported that the activation of
GABAA opens NFA-sensitive anion channels (39,40).
Furthermore, GABAA-mediated chloride ion influx lowers
the magnitudes of NFA- and NPPB-sensitive chloride currents in
motorneurons. NFA and other fenamate blockers of CaCCs (41,42), and
recombinant GABAA (43).
The expression levels of rat homopentamer GABAA
receptors display GABA-independent activation (44). In addition, NFA activates single
chloride ion channels, likely to be an isoform of the
GABAA receptors in mouse sperm (45). Therefore, it is possible that
alternative mechanisms exists for NFA action on the
GABAA receptor.
In the present study, NFA did not alter the
EC50 value but reduced the maximal response of GABA
currents, which is consistent with noncompetitive antagonism.
Similar results have been reported for furosemide (46) and NFA (12) previously. Changes in the expression
and function of α2, but not α6 subunits of
GABAA, were observed in L4-L6 DRG
neurons by whole-cell patch-clamp and immunofluorescence.
Further experiments are needed to determine whether
NFA and NPPB may directly act upon the GABAA
α2 subunit in DRG neurons, and the physiological
activator of CaCCs may also provide further elucidation regarding
the contribution of CaCCs to electrical activity. As NSAIDS are
highly subtype-selective, further studies to investigate its
behavioral and cognitive effects are warranted (11). We propose that expression of CaCCs in
DRG should increase in response to peripheral nerve injury, and
enhance the responses of GABAA receptors. Therefore, the
upregulation of CaCCs may prominently enhance ‘the presynaptic
inhibition’ of GABA in the primary afferent endings, and have
involvement in pain modulation.
Acknowledgements
The authors thank Dr Hong-Zhen Hu for reading the
manuscript and for valuable suggestions. The present study was
supported by the National Natural Science Foundation of China
(grant no. 30160026 to Dr Jun-Qiang Si) and the Youth Science and
Technology Innovation Special Foundation of Xinjiang Production and
Construction Corps (China; grant no. 2010JC33 to Dr Li Li).
Glossary
Abbreviations
Abbreviations:
|
GABA
|
γ-aminobutyrate
|
|
PNS
|
periphery nervous system
|
|
CNS
|
central nervous system
|
|
NFA
|
niflumic acid
|
|
NPPB
|
5-nitro-2-(3-phenylpropylamino)
benzoic acid
|
|
NSAIDs
|
non-steroidal anti-inflammatory
drugs
|
|
CaCCs
|
Ca2+-activated
Cl− channels
|
|
DRG
|
dorsal root ganglion
|
|
SDRs
|
Sprague-Dawley rats
|
|
NTDP
|
nitrendipine
|
References
|
1
|
Whiting PJ, Bonnert TP, McKernan RM,
Farrar S, Le Bourdellès B, Heavens RP, Smith DW, Hewson L, Rigby
MR, Sirinathsinghji DJ, et al: Molecular and functional diversity
of the expanding GABA-A receptor gene family. Ann N Y Acad Sci.
868:645–653. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mehta AK and Ticku MK: An update on GABAA
receptors. Brain Res Brain Res Rev. 29:196–217. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Smith AJ, Oxley B, Malpas S, Pillai GV and
Simpson PB: Compounds exhibiting selective efficacy for different
beta subunits of human recombinant gamma-aminobutyric acid A
receptors. J Pharmacol Exp Ther. 311:601–609. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jensen ML, Timmermann DB, Johansen TH,
Schousboe A, Varming T and Ahring PK: The beta subunit determines
the ion selectivity of the GABAA receptor. J Biol Chem.
277:41438–41447. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Babot Z, Cristòfol R and Suñol C:
Excitotoxic death induced by released glutamate in depolarized
primary cultures of mouse cerebellar granule cells is dependent on
GABAA receptors and niflumic acid-sensitive chloride channels. Eur
J Neurosci. 21:103–112. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wallenstein MC: Attenuation of
epileptogenesis by nonsteroidal anti-inflammatory drugs in the rat.
Neuropharmacology. 30:657–663. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
McKernan RM and Whiting PJ: Which
GABAA-receptor subtypes really occur in the brain? Trends Neurosci.
19:139–143. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Greenwood IA and Large WA: Comparison of
the effects of fenamates on Ca-activated chloride and potassium
currents in rabbit portal vein smooth muscle cells. Br J Pharmacol.
116:2939–2948. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Malykhina AP, Shoeb F and Akbarali HI:
Fenamate-induced enhancement of heterologously expressed HERG
currents in Xenopus oocytes. Eur J Pharmacol. 452:269–277.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Korpi ER, Gründer G and Lüddens H: Drug
interactions at GABA (A) receptors. Prog Neurobiol. 67:113–159.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Halliwell RF, Thomas P, Patten D, James
CH, Martinez-Torres A, Miledi R and Smart TG: Subunit-selective
modulation of GABAA receptors by the non-steroidal
anti-inflammatory agent, mefenamic acid. Eur J Neurosci.
11:2897–2905. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sinkkonen ST, Mansikkamäki S, Möykkynen T,
Lüddens H, Uusi-Oukari M and Korpi ER: Receptor subtype-dependent
positive and negative modulation of GABA (A) receptor function by
niflumic acid, a nonsteroidal anti-inflammatory drug. Mol
Pharmacol. 64:753–763. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Whittemore ER, Yang W, Drewe JA and
Woodward RM: Pharmacology of the human gamma-aminobutyric acid A
receptor alpha 4 subunit expressed in Xenopus laevis
oocytes. Mol Pharmacol. 50:1364–1375. 1996.PubMed/NCBI
|
|
14
|
Thompson SA, Whiting PJ and Wafford KA:
Barbiturate interactions at the human GABAA receptor: Dependence on
receptor subunit combination. Br J Pharmacol. 117:521–527. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Belelli D, Lambert JJ, Peters JA, Wafford
K and Whiting PJ: The interaction of the general anesthetic
etomidate with the gamma-aminobutyric acid type A receptor is
influenced by a single amino acid. Proc Natl Acad Sci USA.
94:11031–11036. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Institute of Laboratory Animal Resources
(US). Committee on Care, Use of Laboratory Animals, and National
Institutes of Health (US). Division of Research Resources: Guide
for the care and use of laboratory animals (8th). National
Academies Press. (Washington, DC). 2011.
|
|
17
|
Si JQ, Zhang ZQ, Li CX, Wang LF, Yang YL
and Li ZW: Modulatory effect of substance P on GABA-activated
currents from rat dorsal root ganglion. Acta Pharmacol Sin.
25:623–629. 2004.PubMed/NCBI
|
|
18
|
Cheng HJ, Ma KT, Li L, Zhao L, Wang Y and
Si JQ: Differential expression of alpha-adrenoceptor subtypes in
rat dorsal root ganglion after chronic constriction injury. Hua
Zhong Ke Ji Da Xue Xue Bao. 34:322–329. 2014.(In Chinese).
|
|
19
|
Ma KT, Si JQ, Zhang ZQ, Zhao L, Fan P, Jin
JL, Li XZ and Zhu L: Modulatory effect of CCK-8S on GABA-induced
depolarization from rat dorsal root ganglion. Brain Res.
1121:66–75. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bernheim L, Bader CR, Bertrand D and
Schlichter R: Transient expression of a Ca2+-activated
Cl− current during development of quail sensory neurons.
Dev Biol. 136:129–39. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
André S, Boukhaddaoui H, Campo B,
Al-Jumaily M, Mayeux V, Greuet D, Valmier J and Scamps F:
Axotomy-induced expression of calcium-activated chloride current in
subpopulations of mouse dorsal root ganglion neurons. J
Neurophysiol. 90:3764–3773. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
De Castro F, Geijo-Barrientos E and
Gallego R: Calcium-activated chloride current in normal mouse
sympathetic ganglion cells. J Physiol. 498:397–408. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mayer ML: A calcium-activated chloride
current generates the after-depolarization of rat sensory neurones
in culture. J Physiol. 364:217–239. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sánchez-Vives MV and Gallego R:
Calcium-dependent chloride current induced by axotomy in rat
sympathetic neurons. J Physiol. 475:391–400. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lancaster E, Oh EJ, Gover T and Weinreich
D: Calcium and calcium-activated currents in vagotomized rat
primary vagal afferent neurons. J Physiol. 540:543–556. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Abdel-Ghany M, Cheng HC, Elble RC and
Pauli BU: Focal adhesion kinase activated by beta (4) integrin
ligation to mCLCA1 mediates early metastatic growth. J Biol Chem.
277:34391–34400. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Elble RC and Pauli BU: Tumor suppression
by a proapoptotic calcium-activated chloride channel in mammary
epithelium. J Biol Chem. 276:40510–40517. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang YD, Cho H, Koo JY, Tak MH, Cho Y,
Shim WS, Park SP, Lee J, Lee B, Kim BM, et al: TMEM16A confers
receptor-activated calcium-dependent chloride conductance. Nature.
455:1210–1215. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Caputo A, Caci E, Ferrera L, Pedemonte N,
Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O and
Galietta LJ: TMEM16A, a membrane protein associated with
calcium-dependent chloride channel activity. Science. 322:590–594.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Schroeder BC, Cheng T, Jan YN and Jan LY:
Expression cloning of TMEM16A as a calcium-activated chloride
channel subunit. Cell. 134:1019–1029. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Scott RH, Sutton KG, Griffin A, Stapleton
SR and Currie KP: Aspects of calcium-activated chloride currents: A
neuronal perspective. Pharmacol Ther. 66:535–565. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Frings S, Reuter D and Kleene SJ: Neuronal
Ca2+-activated Cl-channels-homing in on an elusive
channel species. Prog Neurobiol. 60:247–289. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cruickshank SF, Baxter LM and Drummond RM:
The Cl(−) channel blocker niflumic acid releases Ca(2+) from an
intracellular store in rat pulmonary artery smooth muscle cells. Br
J Pharmacol. 140:1442–1450. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li L, Ma KT, Zhao L and Si JQ: Niflumic
acid hyperpolarizes the smooth muscle cells by opening BK(Ca)
channels through ryanodine-sensitive Ca(2+) release in spiral
modiolar artery. Sheng Li Xue Bao. 60:743–750. 2008.PubMed/NCBI
|
|
35
|
Li L, Ma KT, Zhao L, Si JQ, Zhang ZS, Zhu
H and Li J: Niflumic acid hyperpolarizes smooth muscle cells via
calcium-activated potassium channel in spiral modiolar artery of
guinea pigs. Acta Pharmacol Sin. 29:789–799. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Levitan IB: Signaling protein complexes
associated with neuronal ion channels. Nat Neurosci. 9:305–310.
2006. View
Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lerma J and del Rio Martin R: Chloride
transport blockers prevent N-methyl-D-aspartate receptor-channel
complex activation. Mol Pharmacol. 41:217–222. 1992.PubMed/NCBI
|
|
38
|
Coyne L, Su J, Patten D and Halliwell RF:
Characterization of the interaction between fenamates and
hippocampal neuron GABA(A) receptors. Neurochem Int. 51:440–446.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Raiteri L, Schmid G, Prestipino S, Raiteri
M and Bonanno G: Activation of alpha 6 GABAA receptors on
depolarized cerebellar parallel fibers elicits glutamate release
through anion channels. Neuropharmacology. 41:943–951. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Van Damme P, Callewaert G, Eggermont J,
Robberecht W and Van Den Bosch L: Chloride influx aggravates
Ca2+-dependent AMPA receptor-mediated motoneuron death.
J Neurosci. 23:4942–4950. 2003.PubMed/NCBI
|
|
41
|
White MM and Aylwin M: Niflumic and
flufenamic acids are potent reversible blockers of Ca2
(+)-activated Cl-channels in Xenopus oocytes. Mol Pharmacol.
37:720–724. 1990.PubMed/NCBI
|
|
42
|
Korn SJ, Bolden A and Horn R: Control of
action potentials and Ca2+ influx by the Ca
(2+)-dependent chloride current in mouse pituitary cells. J
Physiol. 439:423–437. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Woodward RM, Polenzani L and Miledi R:
Effects of fenamates and other nonsteroidal anti-inflammatory drugs
on rat brain GABAA receptors expressed in Xenopus oocytes. J
Pharmacol Exp Ther. 268:806–817. 1994.PubMed/NCBI
|
|
44
|
Sigel E, Baur R, Malherbe P and Möhler H:
The rat beta 1-subunit of the GABAA receptor forms a
picrotoxin-sensitive anion channel open in the absence of GABA.
FEBS Lett. 257:377–379. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Espinosa F, de la Vega-Beltrán JL,
López-González I, Delgado R, Labarca P and Darszon A: Mouse sperm
patch-clamp recordings reveal single Cl-channels sensitive to
niflumic acid, a blocker of the sperm acrosome reaction. FEBS Lett.
426:47–51. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Korpi ER, Kuner T, Seeburg PH and Lüddens
H: Selective antagonist for the cerebellar granule cell-specific
gamma-aminobutyric acid type A receptor. Mol Pharmacol. 47:283–289.
1995.PubMed/NCBI
|