Introduction
Androgens, such as testosterone and
dihydrotestosterone, are essential for normal male sex
differentiation and are responsible for the normal development of
male secondary sexual characteristics at puberty (1). The physiological effects of androgens
are mediated by the androgen receptor (AR), which is a nuclear
transcription factor encoded by the AR gene (2). The AR gene, which is located on
the X chromosome (Xq11-q12) and contains 8 exons, encodes a protein
of 919 amino acids (3). Mutations in
the AR gene have been shown to cause androgen insensitivity
syndrome (AIS), which is characterized by complete or partial
resistance to the biological effects of androgens in 46,XY
karyotype individuals with normal testis determination and
production of age-appropriate androgen concentrations (4). AIS may be classified into mild (MAIS),
partial (PAIS) or complete androgen insensitivity (CAIS), based on
its phenotypic expression, which ranges from a male phenotype with
isolated infertility, to ambiguous genitalia, to a completely
female external phenotype (5).
CAIS, which has previously been termed testicular
feminization syndrome (6), is
characterized by unilateral or often bilateral inguinal hernias in
prepubertal girls and with primary amenorrhea during puberty
(7). The characteristic features of
CAIS include a normal female phenotype, normal breast development,
an absence of or sparse pubic and axillary hair, an absence of the
uterus and ovaries, and a short blind-ending vagina (8). The estimated prevalence of CAIS is
1:20,000–64,000 male births (9).
CAIS is typically diagnosed by clinical and laboratory findings and
confirmed by the detection of a defect in the AR gene. However,
only a small number of patients affected by CAIS have been
confirmed by AR gene mutational screening in Chinese hospitals
(10–12).
The present study aimed to investigate patients with
CAIS from two unrelated Chinese families by screening mutations of
the AR gene. Furthermore, in silico tools were used to predict the
potential effect of the novel mutation on the AR protein.
Patients and methods
Families
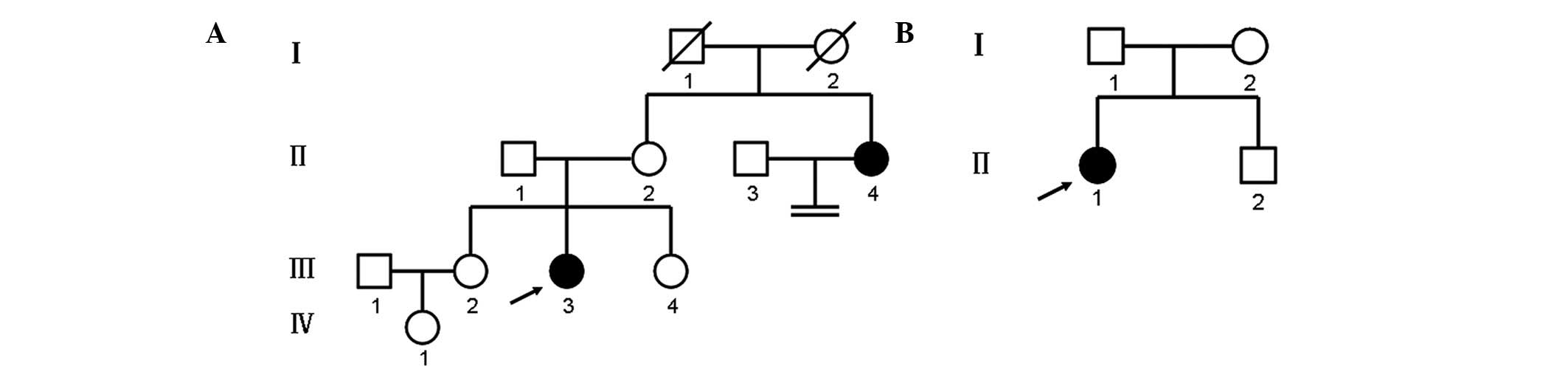
Two unrelated families affected by CAIS were
investigated in the present study. The family pedigrees and
generations are illustrated in Fig.
1. In both families, there was no history of consanguineous
marriage for three generations. To determine the karyotype of
patients, peripheral blood lymphocytes were cultured using
lymphocyte culture medium (Yishengjun; Bedi Biotechnology,
Guangzhou, China) at 37°C for 72 h. Dividing cells were arrested at
metaphase stage with 20 µg/ml colchicin (Bedi Biotechnology) for 3
h prior to culture termination. Mitotic cells were incubated in a
hypotonic solution (0.075 M KCl in H2O) for 30 min at 37°C, and
then the swollen cells were fixed with Carnoy's fixative (methanol
and acetic acid = 3:1). Mitotic cells were dropped onto pre-cleaned
glass microscope slides and allowed to dry for 1 day at room
temperature. Chromosomes were G-banded by treating the preparations
with trypsin (Gibico; Thermo Fisher Scientifc, Inc., Waltham, MA,
USA) followed by staining with Giemsa solution (Sigma-Aldrich, St.
Louis, MO, USA), according to the manufacturer's instructions. Each
patient had a 46,XY karyotype. The diagnosis of CAIS was based on
the combination of a physical examination, the patient medical
history, measurements of sex hormones and a gene mutational
analysis. Informed written consent was obtained from all
participants. The present study was approved by the ethics
committee of The First Hospital of Jilin University (Changchun,
China).
Patients
Family 1 is presented in Fig. 1A. The proband (III-3) was a
25-year-old female, who was referred to the Center for Prenatal
Diagnosis at The First Hospital of Jilin University (Changchun,
China) in January 2013 with primary amenorrhea. The patient was the
second child in her family and had two normal sisters (III-2 and
III-4). The maternal aunt (II-4) of the proband had a history of
primary amenorrhea and infertility. At the time of examination, the
proband exhibited normal female external genitalia, normal breast
development and an absence of axillary and pubic hair. An
ultrasound (GE LOG IQ9 device; GE Healthcare Life Sciences,
Chalfont, UK) revealed the absence of a uterus and ovaries, and the
presence of bilateral testes-like gonads. Immunoassays detected the
serum concentrations of luteinizing hormone (LH; cat. no. 1732234),
follicle-stimulating hormone (FSH; cat. no. 11775863), testosterone
(T; cat. no. 05200067) and estradiol (E2; cat. no. 03000079) (all
purchased from Roche Diagnostics GmbH, Mannheim, Germany). The
serum hormone concentrations were as follows: LH, 52 mIU/ml; FSH,
56.8 mIU/ml (both above the normal male range); testosterone (T),
25.8 nmol/l (within the normal male range); and estradiol (E2),
26.35 pg/ml. The normal ranges for males are as follows: LH,
1.70–8.60 mIU/ml; FSH, 1.50–12.40 mIU/ml; T, 9.9–27.8 nmol/l; and
E2, 7.63–42.60 pg/ml. The normal ranges for females are as follows:
LH follicular phase, 2.40–12.60 mIU/ml; LH ovulation phase,
14.00–95.60 mIU/ml; LH luteal phase, 1.00–11.40 mIU/ml; LH
menopause phase, 7.70–58.50 mIU/ml; FSH follicular phase,
3.50–12.50 mIU/ml; FSH ovulation phase, 4.70–21.50 mIU/ml; FSH
luteal phase, 1.70–7.70 mIU/ml; FSH menopause phase, 25.80–134.8
mIU/ml; T, 0.22–2.9 nmol/l; E2 follicular phase, 12.5–166 pg/ml; E2
ovulation phase, 85.8–498 pg/ml; E2 luteal phase, 43.8–211 pg/ml;
and E2 menopause phase, <5–54.7 pg/ml.
In family 2 (Fig.
1B), the proband (II-1) was an 18-year-old female, who was
referred to the Department of Urology at The First Hospital of
Jilin University in September 2012 with a right inguinal hernia and
primary amenorrhea. At 3 years of age, the patient had presented
with congenital bilateral inguinal hernias, which were
characterized by a mass in the groin that swelled during crying.
The patient had undergone a left hernia operation at the City
Hospital of Yushu (Yushu, China) 8 months prior to admission, and
the mass was confirmed as a testis. A further physical examination
revealed that the patient had normal female external genitalia,
normal breast development, sparse pubic hair and an absence of
axillary hair. A gynecological examination revealed the absence of
a uterus and ovaries, and the presence of a blind-ending vagina
(7.8 cm in depth). The serum hormone concentrations were as
follows: T, 11.65 nmol/l (within the normal male range); LH, 47
IU/ml; FSH, 58.89 mIU/ml; and E2, 22.85 pg/ml.
Genetic analysis
Genomic DNA was extracted from peripheral blood
leukocytes using the QIAamp DNA Blood Mini kit (51106; Qiagen GmbH,
Hilden, Germany), according to the manufacturer's protocol. All
eight exons and flanking intronic regions of the AR gene were
amplified by polymerase chain reaction (PCR) using the primers
listed in Table I. The
amplifications were performed using a final volume of 50 µl,
containing 1 µl genomic DNA, 25 µl 2X Premix Ex Taq™ (Takara
Biotechnology Co., Ltd., Dalian, China), 1 µl each of the forward
and reverse primers and 22 µl sterilized distilled water. The PCR
conditions were as follows: 95°C for 5 min, followed by 30 cycles
of denaturation at 95°C for 30 sec, annealing at 60–65°C for 45 sec
and extension at 72°C for 30 sec, and a final extension at 72°C for
10 min (GeneAmp 9700; Applied Biosystems; Thermo Fisher Scientific,
Inc.). A 5-µl aliquot of each PCR was loaded on a 2% agarose gel
and visualized by ethidium bromide (Sigma-Aldrich) staining to
confirm the presence of an appropriately sized product. Direct
sequencing of amplicons from the amplification of the AR gene from
the proband was performed on the ABI 3730 Automated Sequencer
(Applied Biosystems; Thermo Fisher Scientific, Inc.) using the
BigDye® Terminator v3.1 Cycle Sequencing kit (4337455;
Applied Biosystems; Thermo Fisher Scientific, Inc.). In the case of
a mutation, PCR and sequencing of the DNA sample were repeated
twice to confirm the finding and rule out the possibility of
PCR-generated errors. Furthermore, 150 healthy male individuals
were recruited as a normal control group for the study (age, 22–35
years).
 | Table I.PCR primers used for amplifying exons
and flanking intronic regions of the AR gene. |
Table I.
PCR primers used for amplifying exons
and flanking intronic regions of the AR gene.
| Primer name | Primer sequence
(5′-3′) | Product length
(bp) | Tm (°C) |
|---|
| AR-e1-F1 |
ACAGCCTGTTGAACTCTTCTGA | 483 | 60 |
| AR-e1-R1 |
GCTCTGGGACGCAACCTCT |
|
|
| AR-e1-F2 |
GGTTCTCCCCAAGCCCATCGTAG | 484 | 60 |
| AR-e1-R2 |
GCTCCAACGCCTCCACACCC |
|
|
| AR-e1-F3 |
AAGGACAATTACTTAGGGGGCACTT | 451 | 60 |
| AR-e1-R3 |
CCAGAGCCAGTGGAAAGTTGTAGTA |
|
|
| AR-e1-F4 |
TTGAACTGCCGTCTACCCTGTCTCT | 531 | 60 |
| AR-e1-R4 |
TGGGATAGGGCACTCTGCTCACC |
|
|
| AR-e1-F5 |
CGCTTCCTCATCCTGGCACACTCTC | 410 | 60 |
| AR-e1-R5 |
AGGTAGGAGCCGCTAGATACCCCAG |
|
|
| AR-e2-F |
CACTAACTAACTTGAGCAATGAATA | 325 | 60 |
| AR-e2-R |
TAAAGGAGAAAGGGAAAGAGAAGTG |
|
|
| AR-e3-F |
CTGGAAACTCATTATCAGGTCTATC | 257 | 60 |
| AR-e3-R |
TCAAAGAAGAAAATCTGGTCTAAAG |
|
|
| AR-e4-F |
GTTTAGAGTCTGTGACCAGGGAG | 506 | 63 |
| AR-e4-R |
GGCAGAAAAGCACCAGACAT |
|
|
| AR-e5-F |
AGCATCTCTGCCCAACAGGGACTCA | 379 | 60 |
| AR-e5-R |
CCTCATACTGGATTGGCTGGCTGGG |
|
|
| AR-e6-F |
CTCTGGGCTTATTGTAAACTTCC | 255 | 60 |
| AR-e6-R |
CAAAAGTGGTCCTCTCTGAATCTCT |
|
|
| AR-e7-F |
TGTGGTCAGAAAACTTGGTG | 290 | 65 |
| AR-e7-R |
CTCTATCAGGCTGTTCTCCC |
|
|
| AR-e8-F |
GGAGGAAACAAAAGGCTGAAAGACC | 326 | 60 |
| AR-e8-R |
AACAGGCAGAAGACATCTGAAAGGG |
|
|
Homology and structural analysis and
function prediction
Upon detection of a novel mutation, the functional
consequences of amino acid alterations were predicted using in
silico models. Three algorithms, including Polymorphism
Phenotyping (http://genetics.bwh.harvard.edu/pph2/; version 2.2.2),
Scale Invariant Feature Transform (http://sift.jcvi.org/; version 4.0) and Protein
Analysis Through Evolutionary Relationships (http://www.pantherdb.org/tools/csnpScoreForm.jsp;
version 6.1), were used to predict the functional consequences of
amino acid substitutions.
For homology studies, the human AR sequence was
compared with corresponding mammalian protein sequences from the
Ensemble database (http://www.ensembl.org/index.html) using the ClustalW
multiple sequence alignment tool (http://www.ebi.ac.uk/Tools/msa/clustalw2/.) The
structure of the mutant AR protein was predicted using the resolved
three dimensional structure of human AR (Protein Data Bank
accession #2AM9) as a template. Molecular modeling was performed
using the SWISS-MODEL web-server program (http://swissmodel.expasy.org/). The model images were
examined and edited using PyMOL (https://www.pymol.org/l; version 1.5).
Results
Genetic analysis of the AR gene
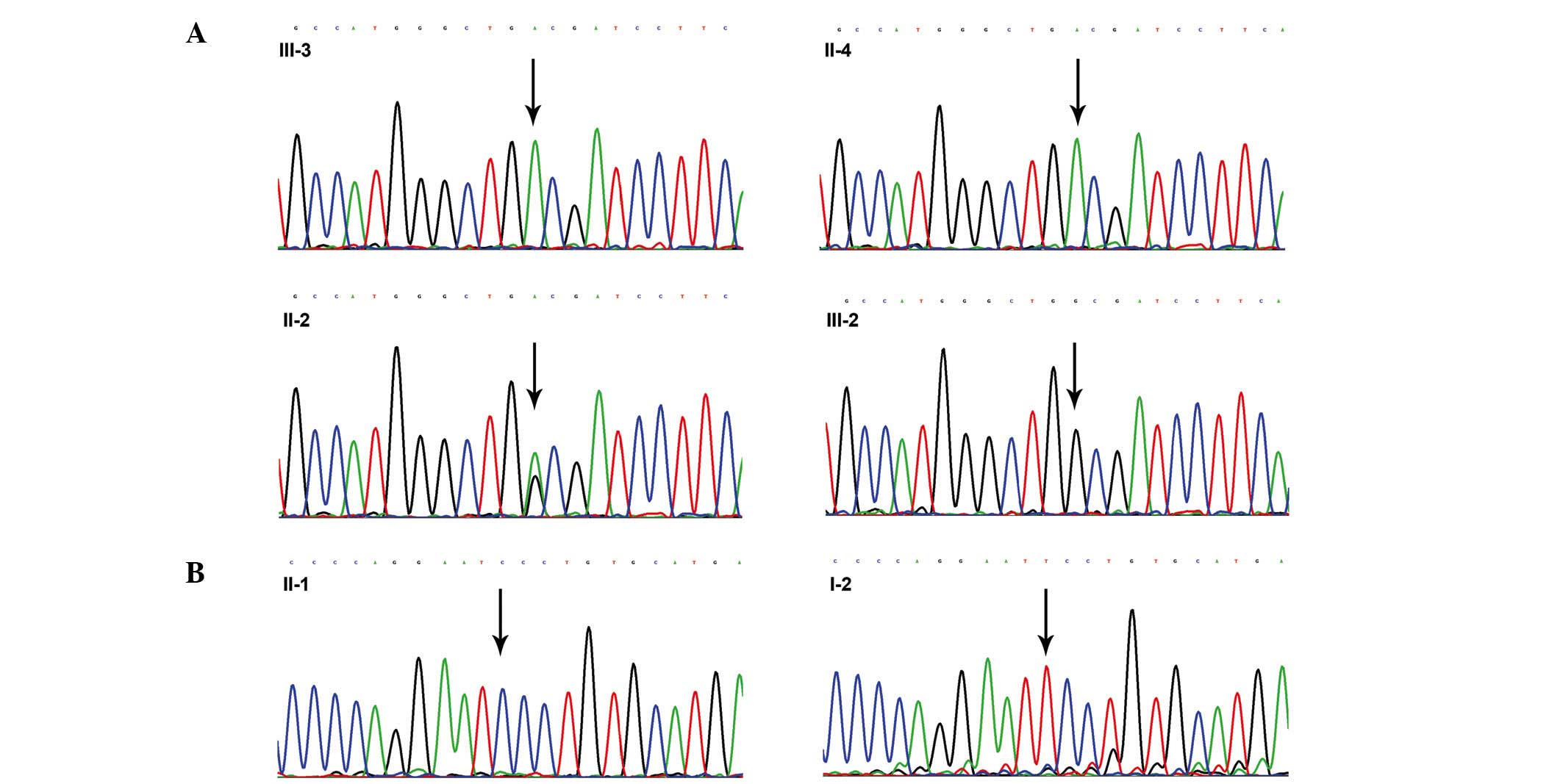
In family 1, a direct sequencing analysis of the
proband (III-3) detected a homozygous G-to-A substitution in exon 5
of the AR gene, which led to the replacement of a tryptophan
(W) codon (TGG) with a termination (X) codon (TGA) at amino acid
position 751 (p.W751X). The mother of the proband (II-2) was a
heterozygous carrier of the p.W751X mutation, and II-4 harbored the
same mutation as the proband, whereas the elder sister of the
proband (III-2) had normal alleles at the AR gene (Fig. 2A).
A T-to-C substitution at codon 804 (TTC-TCC) in exon
6 of the AR gene was identified in the proband (II-1) from
family 2; this mutation was predicted to result in an amino acid
change from phenylalanine (F) to serine (S) at amino acid position
804 (p.F804S) in the ligand-binding domain (LBD) of the AR protein.
However, a molecular analysis of the proband's mother (I-2)
revealed normal alleles, and this mutation was absent in all of the
150 normal control subjects (Fig.
2B).
Functional and structural prediction
of the AR protein with the novel mutation
To the best of our knowledge, the p.F804S mutation
has not previously been described, and was thus analyzed in
silico. All three in silico algorithms predicted that
the substitution of F with S would affect the function of the AR
protein (Table II). F804 is a
highly conserved residue, as demonstrated when comparing the human
AR protein sequence to other mammalian AR proteins by multiple
sequence alignments (Fig. 3).
| Table II.Prediction of the effect of the
p.F804S mutation on the function of the androgen receptor protein
using three algorithms. |
Table II.
Prediction of the effect of the
p.F804S mutation on the function of the androgen receptor protein
using three algorithms.
| Algorithm | Prediction |
|---|
| PolyPhen | Probably
damaging |
|
| Score = 1.000
(sensitivity, 0.00; specificity, 1.00) |
| SIFT | Affect protein
function |
|
| SIFT score =
0.00 |
| PANTHER | Probability of
deleterious effect = 0.79 |
|
| subPSEC score =
−4.31 |
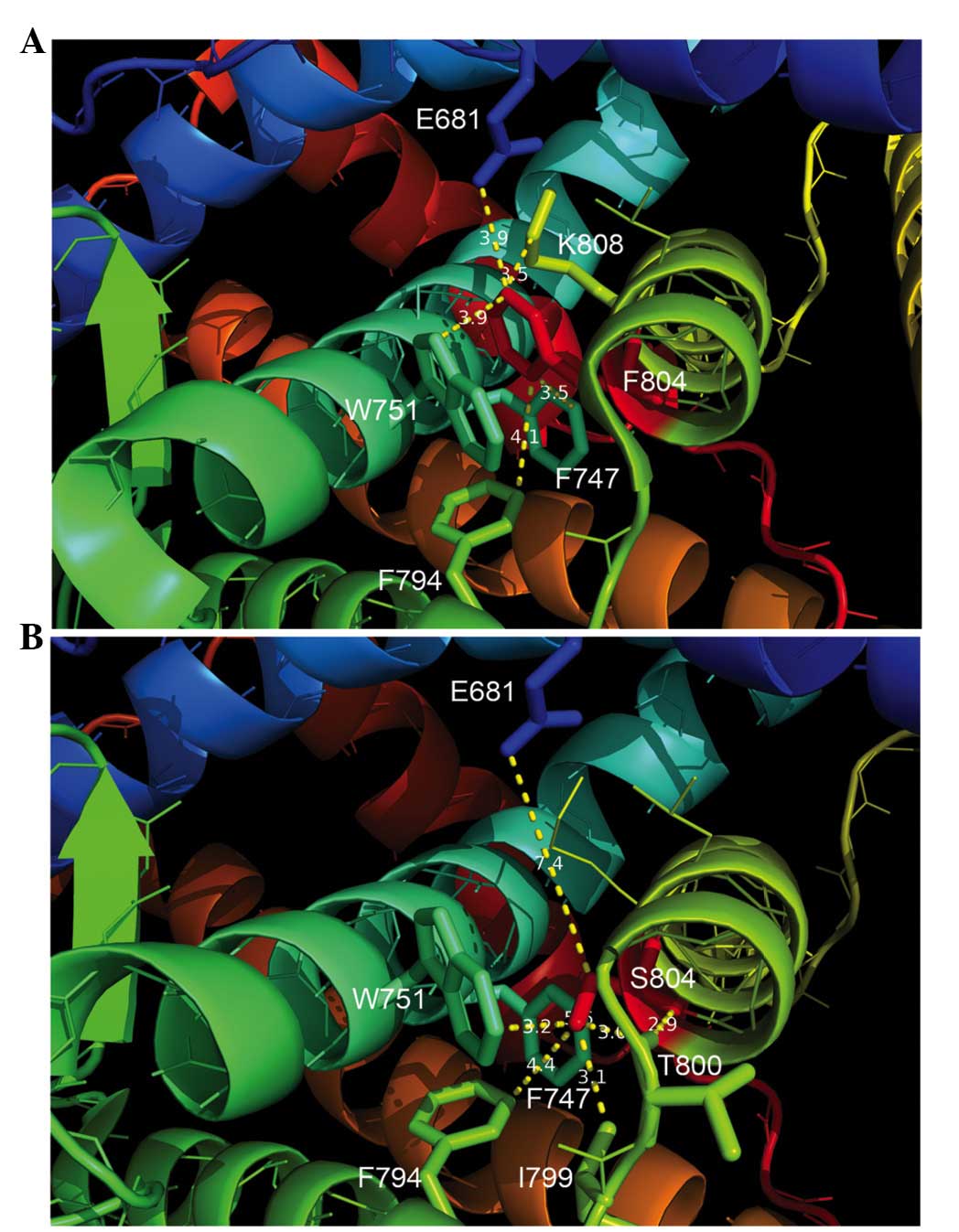
The structural analysis demonstrated that, in the
wild-type AR protein, F804 was located in a hydrophobic cage
consisting of F747, W751, F794, leucine 805 and lysine 808. At the
same time, the aromatic side chains of F804 and W751 were aligned
in an off-centered parallel orientation to form a π-stacking
structure (Fig. 4A). However, in the
mutant AR protein, the structural analysis by modeling revealed
that, as compared with the wild-type F804 reside, S804 did not form
a hydrophobic interaction with F747, and instead formed an
intramolecular hydrophobic interaction with isoleucine (I) 799. In
addition, S804 was extended from the hydrophobic cage and
established a hydrogen bond with the backbone carbonyl group of
threonine (T) 800 (Fig. 4B).
Discussion
AIS is the most common cause of male
pseudohermaphroditism, and is considered a member of the 46,XY
disorders of sex development (13).
AIS is an X-linked inherited disorder caused by mutations in the AR
gene (14). The AR is an
intracellular transcription factor belonging to the nuclear
receptor superfamily. In the absence of a ligand, the AR protein is
located in the cytoplasm; however, androgen binding induces the AR
to adopt its trans-conformation, permitting its translocation into
the nucleus and direct interaction with its target genes in order
to regulate their transcription and initiate a series of molecular
events required for male sex differentiation (15). Consistent with all nuclear receptors,
the AR consists of three major functional domains, as follows: i)
The N-terminal transactivational domain, which is encoded by exon 1
and is involved in the transcriptional activation of target genes;
ii) the DNA-binding domain, which is encoded by exons 2 and 3 and
contains two zinc finger motifs; and iii) the C-terminal LBD, which
is encoded by exons 4–8 and is involved in dimerization and
transcriptional activation (16). To
date, >800 different mutations, associated with various forms of
AIS and scattered throughout the AR gene, have been reported
worldwide (17).
The present study screened the entire coding and
intron-exon regions of the AR gene in two unrelated Chinese
families with CAIS. In family 1, one previously reported mutation
(p.W751X) in exon 5 of the AR gene was identified in two
CAIS individuals and one unaffected carrier. In family 2, a novel
mutation (p.F804S) was identified in the LBD of AR, and the
consequence of this amino acid substitution was predicted using
bioinformatics.
In family 1, the p.W751X mutation identified in the
proband (III-3) was inherited from the unaffected mother (II-2),
who was a normal heterozygous carrier of this mutation. In
addition, the aunt of the proband (II-4) was positive for the
p.W751X mutation, which may have accounted for her history of
primary amenorrhea and infertility. Conversely, the elder sister of
the proband had the wild-type allele, and the younger sister
(III-4) of the proband may have been an unaffected carrier of the
p.W751X mutation, although this individual was not analyzed due to
lack of parental consent.
Yaegashi et al (18) initially reported the p.W751X mutation
in a Japanese patient who presented with the CAIS phenotype and had
a 47,XXY karyotype in 1999. They identified two nonsense mutations
(p.G641X and p.W751X) in this patient, and a cultured genital skin
fibroblast study demonstrated that these mutations eliminated the
ligand binding capacity of AR (18).
Furthermore, they also reported that the p.G641X mutation alone was
able to cause the CAIS phenotype and eliminate the androgen binding
capacity (18). Conversely, Köhler
et al (19) detected the
p.W751X mutation in a patient with PAIS from Italy. In the present
study, the p.W751X mutation was identified in a CAIS patient with a
46,XY karyotype. Thus, the same mutation in the AR gene may
cause different clinical manifestations in patients from different
populations.
In family 2, the proband (II-1) was affected by
CAIS, which presented as a bilateral inguinal hernia in childhood
and primary amenorrhea during puberty. DNA sequencing identified
the novel p.F804S mutation in the proband (II-1), which was not
detected in the mother of the proband (I-2). These results
suggested that the mutation was either de novo or the result
of a possible gametic mosaicism. Furthermore, the mutation was
absent in the genomes of 150 normal control individuals, which
excluded the possibility of this alteration being the result of a
single nucleotide polymorphism. To the best of our knowledge, the
p.F804S mutation has not previously been reported, although two
previously reported mutations in the same codon involving different
nucleotides (TTC-CTC, p.F804l) and (TTC-ATC, p.F804L) were also
associated with CAIS (20,21). Considering that all three identified
mutations at F804 have been associated with CAIS, the present study
compared the human AR sequence with the corresponding mammalian
protein sequences using ClustalW. F804 was shown to be part of
highly conserved amino acid residues in the AR-LBD; thus suggesting
that this residue is important for the function of the protein. In
addition, all three in silico algorithms predicted that the
p.F804S mutation would affect protein function.
In a previous study, X-ray crystallography
demonstrated that the three-dimensional structure of the AR-LBD
consisted of 12 α-helices (H), which folded into a three-layered
sandwich (22,23). The outer leaves of this sandwich,
which consisted of H1/2 and 3 on one side and H6, 7 and 10/11 on
the other, enveloped a hydrophobic core. H4/5, 8 and 9 formed
one-half of this hydrophobic core, whereas the second half of the
core was an open ligand-binding pocket, which, in the presence of
androgen, was closed by repositioning of the terminal H12 (24). F804 was shown to be located in H8,
which was the central helix in the hydrophobic core of the AR-LBD.
In the present study, a structural analysis demonstrated that, in
the wild-type AR-LBD, F804 interacted with E681 in H1 and F794 in
H7 from the outer layer of the sandwich, and with F747 and W751 in
H5 from the hydrophobic core. Furthermore, F804 was shown to be
inserted into a hydrophobic cage which consisted of F747, W751,
F794, L805 and K808. In addition, the aromatic side chains of F804
and W751 were aligned in an off-centered parallel orientation to
form a π-stacking structure. These results suggested that F804 is a
focal residue that anchors the outer layer (H1/2 and 7) of the
AR-LBD to its hydrophobic core (H5 and 8). However, upon
substitution with the non-polar, aromatic F with a polar amino S,
the modeling revealed that the S804 in the mutant AR-LBD mainly
eradicated the hydrophobic interaction with F747 from H5, and lost
interaction with E681 on H1, and established a new interaction
between I799 and H7. Furthermore, due to the polar nature of S,
S804 was extended from the hydrophobic core, which enabled it to
form a hydrogen bond with the backbone carbonyl group of T800.
Therefore, it may be hypothesized that the loss of the interaction
between F747 and E681 in H1 may have altered the stability of the
AR-LBD, such that the LBD was prone to misfolding. Furthermore,
suppression of the hydrophobic interaction with residue F747 may
have destabilized the interaction between H5 and H8, which in the
wild-type protein forms an important hydrophobic core that
maintains the AR binding capacity.
In conclusion, the present study identified two
mutations in two unrelated Chinese families with CAIS by molecular
screening of the AR gene. The novel missense mutation
(p.F804S) identified in family 2 may provide insights into the
molecular mechanism underlying CAIS. In addition, the novel
mutation expanded in the AR database may aid in the
identification of mutational hot spots, which may be useful in
prenatal diagnosis and genetic counseling.
Acknowledgements
The authors of the present study would like to thank
the study participants for their involvement in this study.
References
|
1
|
Gottlieb B, Lombroso R, Beitel LK and
Trifiro MA: Molecular pathology of the androgen receptor in male
(in)fertility. Reprod Biomed Online. 10:42–48. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rosa S, Biason-Lauber A, Mongan NP,
Navratil F and Schoenle EJ: Complete androgen insensitivity
syndrome caused by a novel mutation in the ligand-binding domain of
the androgen receptor: Functional characterization. J Clin
Endocrinol Metab. 87:4378–4382. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lubahn DB, Joseph DR, Sullivan PM, Willard
HF, French FS and Wilson EM: Cloning of human androgen receptor
complementary DNA and localization to the X chromosome. Science.
240:327–330. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hughes IA, Davies JD, Bunch TI, Pasterski
V, Mastroyannopoulou K and MacDougall J: Androgen insensitivity
syndrome. Lancet. 380:1419–1428. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Brinkmann AO: Molecular basis of androgen
insensiticty. Mol Cell Endocrinol. 179:105–109. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Morris JM: The syndrome of testicular
feminization in male pseudohermaphrodites. Am J Obstet Gynecol.
65:1192–1211. 1953. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Decaestecker K, Philibert P, De Baere E,
Hoebeke P, Kaufman JM, Sultan C and T'Sjoen G: A novel mutation
c.118delA in exon 1 of the androgen receptor gene resulting in
complete androgen insensitivity syndrome within a large family.
Fertil Steril. 89:1260.e3–e7. 2008. View Article : Google Scholar
|
|
8
|
Raicu F, Giuliani R, Gatta V, Palka C,
Franchi PG, Lelli-Chiesa P, Tumini S and Stuppia L: Novel mutation
in the ligand-binding domain of the androgen receptor gene (l790p)
associated with complete androgen insensitivity syndrome. Asian J
Androl. 10:687–691. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ahmed SF, Cheng A, Dovey L, Hawkins JR,
Martin H, Rowland J, Shimura N, Tait AD and Hughes IA: Phenotypic
features, androgen receptor binding, and mutational analysis in 278
clinical cases reported as androgen insensitivity syndrome. J Clin
Endocrinol Metab. 85:658–665. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sun S, Luo F, Zhou Z and Wu W: A novel
androgen receptor gene mutation in a Chinese patient with complete
androgen insensitivity syndrome. Eur J Obstet Gynecol Reprod Biol.
153:173–175. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li BK, Ding Q, Wan XD and Wang X: Clinical
and genetic characterization of complete androgen insensitivity
syndrome in a Chinese family. Genet Mol Res. 10:1022–1031. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cong P, Ye Y, Wang Y, Lu L, Yong J, Yu P,
Joseph KK, Jin F and Qi M: A large deletion/insertion-induced
frameshift mutation of the androgen receptor gene in a family with
a familial complete androgen insensitivity syndrome. Gene.
500:220–223. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hughes IA: Disorders of sex development: A
new definition and classification. Best Pract Res Clin Endocrinol
Metab. 22:119–134. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Brown TR1, Lubahn DB, Wilson EM, Joseph
DR, French FS and Migeon CJ: Deletion of the steroid-binding domain
of the human androgen receptor gene in one family with complete
androgen insensitivity syndrome: Evidence for further genetic
heterogeneity in this syndrome. Proc Natl Acad Sci USA.
85:8151–8155. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sultan C, Paris F, Terouanne B, Balaguer
P, Georget V, Poujol N, Jeandel C, Lumbroso S and Nicolas JC:
Disorders linked to insufficient androgen action in male children.
Hum Reprod Update. 7:314–322. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Brinkmann AO, Faber PW, van Rooij HC,
Kuiper GG, Ris C, Klaassen P, van der Korput JA, Voorhorst MM, van
Laar JH, Mulder E, et al: The human androgen receptor: Domain
structure, genomic organization and regulation of expression. J
Steroid Biochem. 34:307–310. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gottlieb B, Beitel LK, Nadarajah A,
Paliouras M and Trifiro M: The androgen receptor gene mutations
database: 2012 update. Hum Mutat. 33:887–894. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yaegashi N, Uehara S, Senoo M, Sato J,
Fujiwara J, Funato T, Sasaki T and Yajima A: Point mutations in the
steroid-binding domain of the androgen receptor gene of five
Japanese patients with androgen insensitivity syndrome. Tohoku J
Exp Med. 187:263–272. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Köhler B, Lumbroso S, Leger J, Audran F,
Grau ES, Kurtz F, Pinto G, Salerno M, Semitcheva T, Czernichow P
and Sultan C: Androgen insensitivity syndrome: Somatic mosaicism of
the androgen receptor in seven families and consequences for sex
assignment and genetic counseling. J Clin Endocrinol Metab.
90:106–111. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gad YZ, Mazen I, Lumbroso S, Temtamy SA
and Sultan C: A novel point mutation of the androgen receptor
(F804L) in an Egyptian newborn with complete androgen insensitivity
associated with congenital glaucoma and hypertrophic pyloric
stenosis. Clin Genet. 63:59–63. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cheikhelard A, Morel Y, Thibaud E,
Lortat-Jacob S, Jaubert F, Polak M and Nihoul-Fekete C: Long-term
followup and comparison between genotype and phenotype in 29 cases
of complete androgen insensitivity syndrome. J Urol. 180:1496–1501.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Matias PM, Donner P, Coelho R, Thomaz M,
Peixoto C, Macedo S, Otto N, Joschko S, Scholz P, Wegg A, et al:
Structural evidence for ligand specificity in the binding domain of
the human androgen receptor. Implications for pathogenic gene
mutations. J Biol Chem. 275:26164–26171. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sack JS, Kish KF, Wang C, Attar RM, Kiefer
SE, An Y, Wu GY, Scheffler JE, Salvati ME, Krystek SR Jr, et al:
Crystallographic structures of the ligand-binding domains of the
androgen receptor and its T877A mutant complexed with the natural
agonist dihydrotestosterone. Proc Natl Acad Sci USA. 98:4904–4909.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ong YC, Kolatkar PR and Yong EL: Androgen
receptor mutations causing human androgen insensitivity syndromes
show a key role of residue M807 in Helix 8-Helix 10 interactions
and in receptor ligand-binding domain stability. Mol Hum Reprod.
8:101–108. 2002. View Article : Google Scholar : PubMed/NCBI
|