Introduction
Uniparental disomy (UPD), which is the abnormal
situation in which both copies of a chromosomal pair have been
inherited from one parent (1), may
affect either a whole chromosome or only part of a chromosome. The
latter is termed segmental UPD and has previously been associated
with rearrangements in acrocentric chromosomes that frequently
occur in Beckwith-Wiedemann syndrome (2,3). Four
mechanisms underlie chromosomal UPD formation: i) Trisomy rescue
due to trisomic zygote formation resulting from the failure of two
homolog chromosomes to segregate into two daughter cells during
parental meiosis; ii) Gamete complementation resulting from
fertilization between a nullisomic fertilization and a disomic
gamete; iii) Monosomy rescue resulting from mitotic endoduplication
in a monosomic zygote; and iv) Postfertilization error during
postzygotic mitosis (4,5). There are two types of UDP, which are
associated with various mechanisms; the first is uniparental
heterodisomy, in which the affected chromosome exhibits two
different alleles transmitted from the same parent, and the second
is uniparental isodisomy (UPiD), in which the affected individual
has two copies of the same allele (4).
Since UPD was initially reported in the 1980s
(1), studies employing chromosomal
microarray analysis (CMA) and whole exome sequencing (WES)
technologies have described UPD for nearly every chromosome in the
human genome (6,7). CMA is a first-tier clinical diagnostic
test that is used to detect genomic copy number variations (CNVs)
in patients with developmental disabilities and congenital
anomalies (8). Furthermore, CMA
permits researchers to identify UPDs in a straightforward manner
(8). WES, which is a type of
next-generation sequencing technology, is able to efficiently
identify genetic defects in patients with sporadic diseases, as
well as providing the mutant information of candidate genes
involved in UPD (9). Recently, it
has been demonstrated that WES has the underlying capacity to
detect CNVs (10,11). In addition, it may be suitable for
detecting UPD (12).
The present study reports the case of a pediatric
patient with an undiagnosed and complex medical manifestation who
was shown to have UPD at chromosome 10. WES was employed to search
for potential causal variants in the patient and to identify
connections between candidate genes and the observed
phenotypes.
Materials and methods
Patient
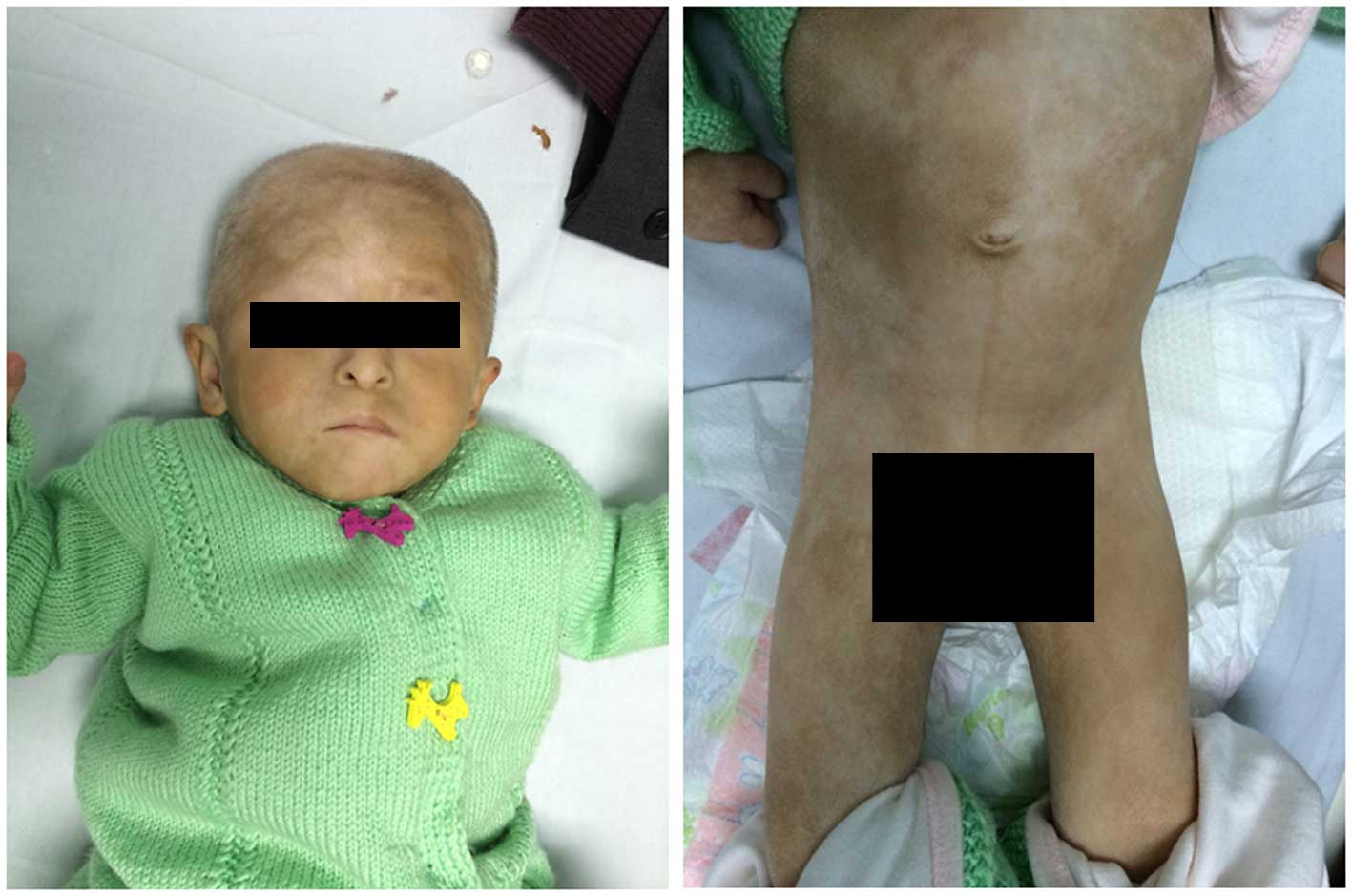
A 20-month-old female infant was referred to the
Genetics Department at Shanghai Children's Medical Center, Shanghai
Jiaotong University School of Medicine (Shanghai, China) due to
multiple severe inborn abnormalities and a severe delay in
physiological development after birth (Fig. 1). The patient's mother has had one
pregnancy and had delivered a child once, and the child was born at
37 weeks gestation by cesarean delivery due to placental
maturation. The patient had an Apgar score (13) of 7′-9′ and was comparatively small
for her gestational age, with a birth weight of 1,800 g and a
length of 40 cm. The patient's parents were well-educated, not
biologically related to each other, and were physically and
mentally healthy. The parents claimed that they had not been
exposed to any toxic materials. The conception had been natural and
the course of the pregnancy uneventful. The patient exhibited
feeding difficulties, constipation, severe malnutrition and
retardation of growth and development after birth.
A physical examination showed the weight and height
of the patient to be 4,200 g and 50 cm, respectively. The patient's
head circumference was 37 cm and anterior fontanel size was 2×2 cm.
The patient had a mild cleft palate, orbital hypertelorism, a long
philtrum and blepharophimosis, and was unable to open her right
eye. In addition, the patient had dry skin with depigmented spots
of various shapes and sizes distributed over her entire body. The
patient exhibited no heart murmur, difficultly breathing nor
hepatosplenomegaly. She had female-appearing genitalia with a
normal urethral opening and a well-proportioned upper and lower
body, anus and vaginal opening. Otoacoustic emission, acoustic
impedance and brainstem auditory evoked potentials tests
demonstrated that the patient had bilateral deafness; corneal light
reflex test demonstrated that the patient had binocular blindness.
Furthermore, the patient had muscular hypotonia and was unable to
raise her head slightly from the prone position. With the exception
of crying, the patient was unable to produce any sound.
No unusual results were detected following a
laboratory examination involving routine tests of the blood, urine,
electrolytes, glucose level and liver, renal and thyroid functions.
Neither magnetic resonance imaging (MRI) (Ingenia 1.5T; Philips
Healthcare, DA Best, The Netherlands) of the patient's brain, nor
screening for inborn metabolic disorders by tandem and
gas-chromatography mass spectrometry (micrOTOFI system; Bruker
Corporation, Billerica, MA, USA), were able to detect any
abnormalities. The study was approved by the ethics committee of
Shanghai Children's Medical Center, Shanghai Jiaotong University
School of Medicine. Written informed consent was obtained from the
child's parents.
CMA
The genomic DNA of the patient was isolated from
peripheral blood samples (2 ml) using a QIAamp® DNA
Blood Mini kit (Qiagen GmbH, Hilden, Germany). Genomic
hybridization was conducted using a CytoScan® HD Array
kit (Affymetrix, Inc., Santa Clara, CA, USA), according to the
manufacturer's protocol. The array is characterized by
>2,600,000 CNV markers, including 750,000 genotype-able single
nucleotide polymorphism (SNP) probes and >1,900,000
non-polymorphism probes. Data were visualized and analyzed using
the Chromosome Analysis Suite (ChAS 3.1; Affymetrix, Inc.) software
package with a minimal cut-off of 20 consecutive markers for CNV
calling. All reported CNVs were based on the National Center for
Biotechnology Information Human Genome Build 37 (hg19) (http://genome.ucsc.edu/cgi-bin/hgGateway).
WES
DNA (3 µg) from the patient was sheared to create
150–200 bp fragments using a Covarias M220 Ultrasonicator (Covaris,
Woburn, MA, USA). The adapter-ligated library was prepared using a
SureSelectXT Human All Exon Kit (Agilent Technologies, Santa Clara,
CA, USA) as previously described (14), with coding exons and flanking
intronic regions enriched. Subsequently, clusters were generated by
isothermal bridge amplification using an Illumina cBot system
(Illumina, San Diego, CA, USA), and sequencing was performed with
an Illumina HiSeq 2000 system.
A quality assessment of base calling and sequence
reads was conducted using an HCS 2.2.58 software (Illumina) and an
Illumina HiSeq 2000 system including new versions of HiSeq Control
Software and Real-Time Analysis. The alignment of sequence reads to
a reference human genome (Human 37.3, SNP135; http://hgdownload.soe.ucsc.edu/goldenPath/hg19/snp135Mask/)
was performed using NextGENe® software (version 2.4.1;
SoftGenetics, LLC., State College, PA, USA). All single nucleotide
variants (SNVs) and indels were saved in a VCF file format, and
upladed to the Ingenuity Variant Analysis platform (Qiagen GmbH)
for biological analysis and interpretation.
In silico analysis
The candidate variants obtained from WES were first
screened by the databases of the 1000 Genomes Project (http://www.1000genomes.org/), the National Heart, Lung
and Blood Institute Exome Sequencing Project Variant Server
(http://evs.gs.washington.edu/EVS/)
and the public Complete Genomics (http://www.completegenomics.com/public-data/).
Evaluation of the pathogenicity of the candidate variants was
performed using the PolyPhen2 online software (http://genetics.bwh.harvard.edu/pph2/).
Sanger sequencing of the
Hermansky-Pudlak syndrome 1 (HPS1) gene
Sanger sequencing was performed as previously
described (15). The primers for
amplification of the HPS1 gene (GenBank accession number,
NM_000195.3; http://www.ncbi.nlm.nih.gov/genbank/) were designed
using the UCSC ExonPrimer online software (http://genome.ucsc.edu/index.html). The primers for
exon 5 were as follows: Forward, 5′-GGCATCTTATCAAACCCGCC-3′ and
reverse, 5′-ACCAACCAGCTAGATGACCC-3′. The exons and exon-intron
boundaries were amplified by polymerase chain reaction (PCR). The
reaction mixture for each amplification contained 1X Premix Taq (Ex
Taq version 2.0; RR003; Takara Biotechnology Co., Ltd., Dalian,
China), 100 ng genomic DNA and 1 pmol forward and reverse primer in
a final volume of 25 µl. The reaction was performed in the
following PCR conditions: Initial denaturation at 95°C for 5 min,
then 19 cycles of 95°C for 30 sec, 65°C for 30 s and 72°C for 45
sec, 14 cycles of 95°C for 30 sec, 55°C for 30 sec and 72°C for 45
sec, and a final elongation step at 72°C for 5 min using a C1000TM
Thermal Cycler (Bio-Rad Laboratories, Inc. Hercules, CA, USA). PCR
products were separated by 1% agarose gel (Sangon Biotechnology
Co., Ltd., Shanghai, China) electrophoresis and purified using a
QIAquick Gel Extraction kit (Qiagen GmbH). The purified DNA was
sequenced using the ABI3730XL sequencer (Applied Biosystems; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) with forward and reverse
primers. The results were analyzed using a 3730xl DNA Analyzer
(Thermo Fisher Scientific, Inc.). The sequence data was analyzed
using the Mutation Surveyor DNA Variant Analysis software (version
4.0.4; SoftGenetics, LLC).
Results
CMA analysis
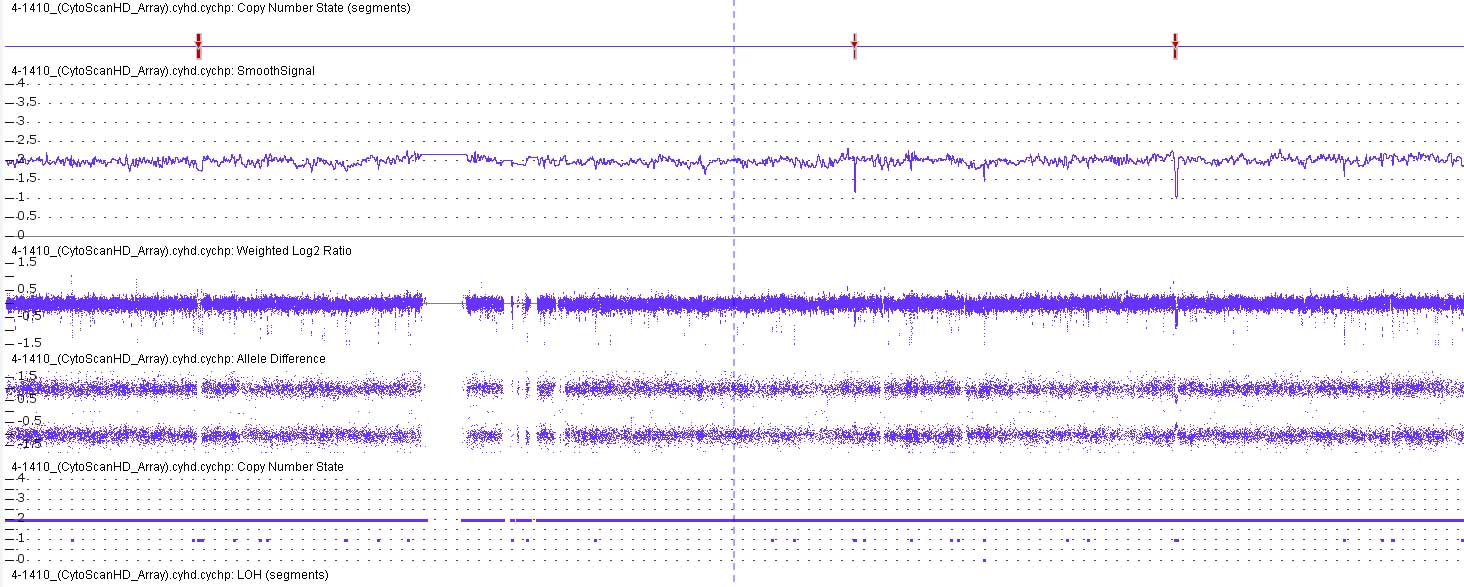
The CMA analysis using SNP probes suggested that the
genome of the patient had a loss of heterozygosity without any CNVs
in chromosome 10, which implied that the patient had UPiD of the
entire of chromosome 10 (UPiD10) (Fig.
2).
DNA sequencing
The authors of the present study hypothesized that
autosomal-recessive variants associated with UPiD10 may have
contributed to the clinical manifestations in the patient.
Therefore, WES was conducted in order to identify underlying
variants. To further characterize chromosome 10, variants in the
1000 Genomes Project, the National Heart, Lung and Blood Institute
Exome Sequencing Project Variant Server (http://evs.gs.washington.edu/EVS/) and the public
Complete Genomics (http://www.completegenomics.com/public-data/) genomes,
were screened for SNPs with a minor allele frequency (MAF) of
<3%. The area of analysis included each exon and ~20 bp at
exon-intron boundaries. A total of 427 SNVs and 43 indels were
shown to meet the filter criteria; these variants covered all
chromosomes and the majority were in the heterozygous state
(451/470; Table I). For chromosome
10, there were 13 SNVs present in the homozygous state and no SNVs
in the heterozygous state, which was consistent with the results of
the CMA. Among these, two candidate genes with intron variations
were predicted to be silent and not alter protein translation and
expression (PFKFB3, c.873+13C>T; MMS19,
c.2776–15G>A).
 | Table I.Summary of variants detected by whole
exome sequencing (minor allele frequency <0.03). |
Table I.
Summary of variants detected by whole
exome sequencing (minor allele frequency <0.03).
| Parameter | Homozygous (Chr.
10) | Heterozygous |
|---|
| Total variants | 19 (13) | 451 |
| Missense | 14 (11) | 311 |
| Stop gain | 0 | 9 |
| Frameshift | 0 | 14 |
| Splicing | 1 (1) | 1 |
| In-frame | 0 | 10 |
| Intron | 4 (1) | 106 |
Among the remaining 11 homozygous variants on
chromosome 10 (Table II), the
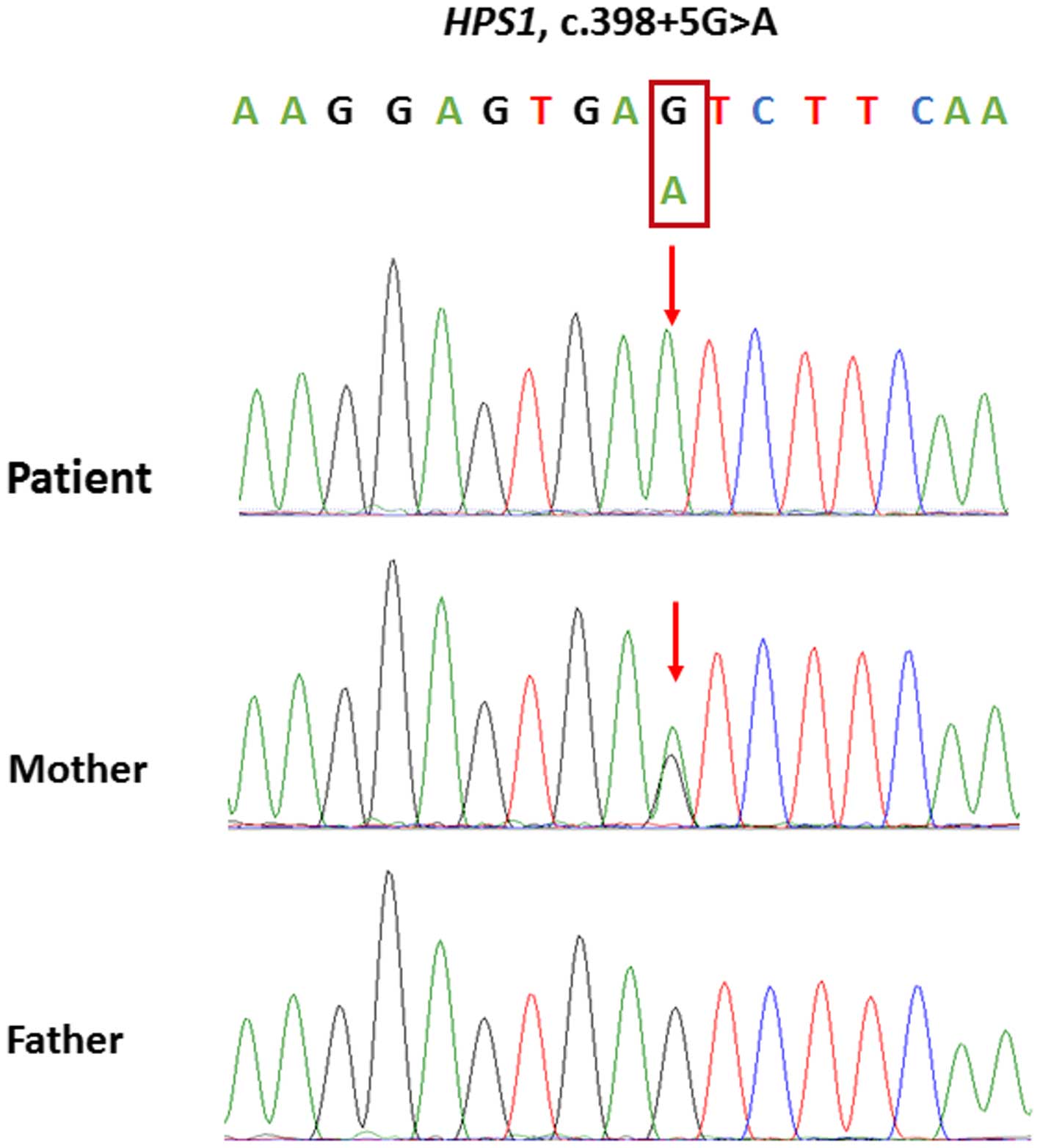
classical splice variant in the HPS1 gene (c.398+5G>A),
which results in the skipping of exon 5 in HPS1 mRNA
(16), was hypothesized to be
associated with the ocular and dermal disorders of the patient. An
in-depth analysis of the remaining 10 homozygous variants was
conducted, and the results did not show any direct association with
the clinical manifestations of the patient. Subsequently, the
HPS1 gene in the parents of the patient was evaluated using
Sanger sequencing. The sequencing results demonstrated that the
patient's mother was heterozygous for c.398+5G>A, whereas the
patient's father was wild-type (Fig.
3). These results suggested that the patient has maternal
UPiD10.
| Table II.Rare homozygous variants on
chromosome 10. |
Table II.
Rare homozygous variants on
chromosome 10.
| Gene | Position
(hg19) | Transcript
variant | Translation
impact | PolyPhen-2 function
prediction | Frequency (1,000
Genome) | Associated Disease
(OMIM) | Function |
|---|
| HPS1 | 10:100195024 | c.398+5G>A | Splice site
loss |
| NA | Hermansky-Pudlak
syndrome 1 | Involved in
organelle biogenesis associated with melanosomes, platelet dense
granules and lysosomes |
| ITGA8 | 10:15726034 | c.537C>A | Missense
p.S179R | Probably
damaging | NA | Renal
hypodysplasia | Has an important
role in wound-healing and organogenesis |
| HACD1 | 10:17659338 | c.1A>G | Start loss
p.M1V | Benign | 0.0004 |
| Regulation of
cardiac development and differentiation |
| SEC31B | 10:102275940 | c.116C>G | Missense
p.T39R | Probably
damaging | NA |
| Unknown |
| PRDX3 | 10:120928705 | c.701C>T | Missense
p.T234I | Probably
damaging | 0.0040 |
| Regulation of
cellular proliferation and differentiation; antioxidant |
| CACNB2 | 10:18828169 | c.1343G>A | Missense
p.G462D | Benign | NA | Brugada syndrome
4 | A member of the
voltage-gated calcium channel gene |
| IFIT1 | 10:91162664 | c.632G>A | Missense
p.R211H | Benign | 0.0008 |
| May inhibit viral
replication and translation initiation |
| HOGA1 | 10:99358604 | c.284G>A | Missense
p.R95H | Benign | NA | Primary
hyperoxalurea type III | Catalyzes the final
step in the metabolic pathway of hydroxyproline, releasing
glyoxylate and pyruvate |
| PAX2 | 10:102586802 | c.1127A>C | Missense
p.Q376P | Benign | 0.0004 | Papillorenal
syndrome; renal hypoplasia; focal segmental glomerulosclerosis
7 | Regulation of
mesenchyme-to-epithelium transition in renal development |
| WDR11 | 10:122618246 | c.290T>C | Missense
p.V97A | Benign | NA | Hypogonadotropic
hypogonadism 14 | Regulation of cell
cycle progression, signal transduction, apoptosis and gene
expression |
| CFAP46 | 10:134660611 | c.6092G>A | Missense
p.R2031K |
| NA |
| Unknown |
None of the candidate genes could be associated with
the deafness of the patient; thus this feature was used to filter
the WES results. All symptom-associated SNVs were filtered using
the MAF criterion. In addition, they were predicted by the
PolyPhen2 software and compared to the database established by the
Department of Laboratory Medicine, Shanghai Children's Medical
Center, Shanghai Jiaotong University School of Medicine (containing
the WES results of >200 Chinese individuals). An autosomal
dominant deafness causal gene, MYO1A, was shown to have a
heterozygous mutation (c.1630C>T, p.R544 W), which PolyPhen2
predicted to be the likely cause of deafness in the patient.
Furthermore, an analysis using ocular symptoms identified five
heterozygous variants and one homozygous variant (Table III), of which the NR2E3 gene
had a heterozygous mutation (c.1127C>T, p.P376L), which may have
been associated with the autosomal dominant retinitis pigmentosa
and may also have been the cause of the patient's ocular symptoms.
The other three candidate genes (NEB, HMX1 and KCNV2)
were autosomal recessive, indicating that heterozygous mutations
were not the cause of the disease.
| Table III.Probable pathogenic variants
associated with the ocular and aural disorders of the patient. |
Table III.
Probable pathogenic variants
associated with the ocular and aural disorders of the patient.
| Classification of
variants | Gene | Position
(hg19) | Transcript
variant | Translation
impact | Het/Hom | Mode of
inheritance | Associated disease
(OMIM) |
|---|
| Ocular genes |
|
| 1 | NEB | 2:152506779 | c.7342C>T | Missense
p.R2448C | Het | Recessive | Nemaline myopathy
2 |
| 2 | HMX1 | 4:8869775 | c.691G>A | Missense
p.A231T | Het | Recessive | Oculoauricular
syndrome |
| 3 | KCNV2 | 9:2718925 | c.1186G>A | Missense
p.G396R | Het | Recessive | Retinal cone
dystrophy 3B |
| 4 | HPS1 | 10:100195024 | c.398+5G>A | Splice site
loss | Hom | Recessive | Hermansky-pudlak
syndrome type 1 |
| 5 | NR2E3 | 15:72109919 | c.1127C>T | Missense
p.P376L | Het | Dominant or
recessive | Enhanced S-cone
syndrome; retinitis pigmentosa 37 |
| 6 | HPS4 | 22:26879985 | c.129_142del | Frameshift
p.R44fsx10 | Het | Recessive | Hermansky-pudlak
syndrome type 4 |
| Aural genes |
|
| 1 | MOY1A | 12:57432326 | c.1630C>T | p.R544W | Het | Dominant | Autosomal dominant
deafness |
Discussion
The first reported case of a clinical manifestation
attributable to UPD was cystic fibrosis, which was caused by
maternal UPD of chromosome 7 (17).
Numerous pathogenic chromosome UPDs have since been reported,
including those of chromosomes 6, 11, 14, 15 and 20 (4). In the majority of cases, the UPD leads
to imprinting disorders (ID), where the UPD event involves genomic
imprinting, which alters epigenetic regulation and DNA methylation
and histone modifications (18).
Angelman syndrome is a well known ID [Online Mendelian Inheritance
in Man (OMIM), #105830], which is caused by paternal UPD of
chromosome 15, leading to the lack of expression of the maternally
inherited UBE3A gene (19).
With the exception of IDs, UPDs may cause disease if there is a
mutation in a recessive gene, where two identical mutant alleles in
the proband are inherited from a heterozygous father or mother. The
best example of this is cystic fibrosis, in which the underlying
molecular mechanism is the CFTR gene mutation as a result of
UPiD7 (17,20).
The present study reports a case of a pediatric
patient with UPiD10 associated with numerous severe medical
problems, including bilateral deafness, binocular blindness,
stunted growth and leukoderma. To the best of our knowledge, such
complicated clinical features have not previously been reported in
the literature. CMA was used to diagnose the patient with UPiD10.
To date, there are only five cases of UPD10 reported in the
literature and all UPD10s have been maternal; however, none of the
previously described cases have involved genomic imprinting
(21–25). The CMA did not reveal any CNVs on
chromosome 10; therefore, the authors of the present study
predicted that the etiology of the patient may be caused by rare
recessive mutations on chromosome 10. Chromosome 10 of the patient
was analyzed by WES, and 11 candidate genes were shown to have
homozygous variants. Unlike prior studies of UPD10, in the present
study, no single candidate gene was able to fully explain the
patient's complex clinical manifestations (26).
The HPS1 gene encodes a protein that may have
a role in the biogenesis of organelles, including melanosomes,
platelet dense granules and lysosomes (27). In addition, mutations in HPS1
lead to Hermansky-Pudlak syndrome type 1 (OMIM, #203300), which is
characterized by oculocutaneous albinism, hemorrhagic diathesis,
ceroid-lipofuscin accumulation and pulmonary fibrosis (16,28).
Thus, this may in part explain the phenotypes associated with the
present patient's eyes and skin. A number of the other identified
variants, including ITGA8, SEC31B and PRDX3,
were predicted to be potentially damaging using PolyPhen2 software;
however, they could not be associated with the clinical
manifestations of the patient. Whether these genes exacerbated
symptom caused by the HPS1 gene mutation is unclear and
requires further study. Direct sequencing of the HPS1 genes
in the parents revealed that the mother had heterozygous loci; thus
suggesting that the UPiD10 of the patient was maternal. The authors
of the present study hypothesize that UPD10 may always involve a
maternal chromosome; however, the underlying mechanism requires
further investigation.
In order to improve our understanding of the
pathogenesis of the patient, the WES data was filtered using the
clinical features, including symptoms of the ears and eyes. Using
this strategy, one homozygous variant of the HPS1 gene, and
six heterozygous variants located on other chromosomes, were
identified. Among these heterozygous variants, the MYO1A
gene encodes a member of the myosin superfamily and has a role in
actin-based molecular motors (29).
Mutations in this gene have previously been associated with
autosomal dominant deafness (OMIM, #607841) (29). The NR2E3 gene encodes a
retinal nuclear receptor whose activity is essential to proper rod
and cone photoreceptor development and maintenance (30). Defects in this gene are one cause of
retinitis pigmentosa 37 (OMIM, #611131), which is characterized by
retinal pigment deposits, rod and cone degeneration and loss of
vision (31). Various types of
retinitis pigmentosa are autosomal dominant (31,32).
Therefore, in the present case, the NR2E3 variant may have a
gene dosage effect on the occurrence and development of ocular
disorders and may have rendered the illness more complicated.
WES and CMA permit efficient identification of
genetic variations. However, they pose significant challenges in
terms of data analysis; in particular in the determination of
associations between genotypes and complex phenotypes. Despite
this, as the use of sequencing technologies and CMA becomes
increasingly widespread, the associations between genotypes and
phenotypes may become better understood and cases resembling the
present report may become more common.
In conclusion, the present study aimed to establish
the connection between the patient's phenotype and genotype. The
HPS1 gene provided the clearest explanation of this
patient's ocular and dermal disorders, while the MOY1A gene
may have a role in her deafness and the NR2E3 gene may have
exacerbated her ocular symptoms. At present, little is known
regarding the functions of several of the candidate genes
(CFAP46 and SEC31B), and their roles in the clinical
manifestations of the patient remain unclear.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81201353 and
81472051) and the Project of Shanghai Municipal Science and
Technology Commission (grant no. 12411950402). The authors of the
present study would like to thank the patient and her family for
participating in the present study.
References
|
1
|
Engel E: A new genetic concept:
Uniparental disomy and its potential effect, isodisomy. Am J Med
Genet. 6:137–143. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kim SR and Shaffer LG: Robertsonian
translocations: Mechanisms of formation, aneuploidy, and
uniparental disomy and diagnostic considerations. Genet Test.
6:163–168. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Keren B, Chantot-Bastaraud S, Brioude F,
Mach C, Fonteneau E, Azzi S, Depienne C, Brice A, Netchine I, Le
Bouc Y, et al: SNP arrays in Beckwith-Wiedemann syndrome: An
improved diagnostic strategy. Eur J Med Genet. 56:546–550. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eggermann T, Soellner L, Buiting K and
Kotzot D: Mosaicism and uniparental disomy in prenatal diagnosis.
Trends Mol Med. 21:77–87. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Grati FR, Grimi B, Frascoli G, Di Meco AM,
Liuti R, Milani S, Trotta A, Dulcetti F, Grosso E, Miozzo M, et al:
Confirmation of mosaicism and uniparental disomy in amniocytes,
after detection of mosaic chromosome abnormalities in chorionic
villi. Eur J Hum Genet. 14:282–288. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yamazawa K, Ogata T and Ferguson-Smith AC:
Uniparental disomy and human disease: An overview. Am J Med Genet C
Semin Med Genet. 154C:329–334. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li W, Xia Y, Wang C, Tang YT, Guo W, Li J,
Zhao X, Sun Y, Hu J, Zhen H, et al: Identifying human genome-wide
CNV, LOH and UPD by targeted sequencing of selected regions. PLoS
One. 10:e01230812015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Miller DT, Adam MP, Aradhya S, Biesecker
LG, Brothman AR, Carter NP, Church DM, Crolla JA, Eichler EE,
Epstein CJ, et al: Consensus statement: Chromosomal microarray is a
first-tier clinical diagnostic test for individuals with
developmental disabilities or congenital anomalies. Am J Hum Genet.
86:749–764. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ng SB, Buckingham KJ, Lee C, Bigham AW,
Tabor HK, Dent KM, Huff CD, Shannon PT, Jabs EW, Nickerson DA, et
al: Exome sequencing identifies the cause of a mendelian disorder.
Nat Genet. 42:30–35. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang WB, Wang W, Sun W, Crowley JJ and
Szatkiewicz JP: Allele-specific copy-number discovery from
whole-genome and whole-exome sequencing. Nucleic Acids Res.
43:e902015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tan R, Wang Y, Kleinstein SE, Liu Y, Zhu
X, Guo H, Jiang Q, Allen AS and Zhu M: An evaluation of copy number
variation detection tools from whole-exome sequencing data. Hum
Mutat. 35:899–907. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Martin HC, Kim GE, Pagnamenta AT, Murakami
Y, Carvill GL, Meyer E, Copley RR, Rimmer A, Barcia G, Fleming MR,
et al: Clinical whole-genome sequencing in severe early-onset
epilepsy reveals new genes and improves molecular diagnosis. Hum
Mol Genet. 23:3200–3211. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
American Academy of Pediatrics Committee
on Fetus and Newborn; American College of Obstetricians and
Gynecologists Committee on Obstetric Practice: The Apgar score.
Pediatrics. 136:819–822. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen R, Im H and Snyder M: Whole-exome
enrichment with the agilent SureSelect Human All Exon platform.
Cold Spring Harb Protoc. 2015:626–633. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lee HK, Tang JW, Kong DHL and Koay ES:
Simplified large-scale Sanger genome sequencing for influenza
A/H3N2 virus. PLoS One. 8:e647852013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Oh J, Ho L, Ala-Mello S, Amato D,
Armstrong L, Bellucci S, Carakushansky G, Ellis JP, Fong CT, Green
JS, et al: Mutation analysis of patients with Hermansky-Pudlak
syndrome: A frameshift hot spot in the HPS gene and apparent locus
heterogeneity. Am J Hum Genet. 62:593–598. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Spence JE, Perciaccante RG, Greig GM,
Willard HF, Ledbetter DH, Hejtmancik JF, Pollack MS, O'Brien WE and
Beaudet AL: Uniparental disomy as a mechanism for human genetic
disease. Am J Hum Genet. 42:217–226. 1988.PubMed/NCBI
|
|
18
|
Girardot M, Feil R and Llères D:
Epigenetic deregulation of genomic imprinting in humans: causal
mechanisms and clinical implications. Epigenomics. 5:715–728. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sadikovic B, Fernandes P, Zhang VW, Ward
PA, Miloslavskaya I, Rhead W, Rosenbaum R, Gin R, Roa B and Fang P:
Mutation Update for UBE3A variants in Angelman syndrome. Hum Mutat.
35:1407–1417. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Le Caignec C, Isidor B, de Pontbriand U,
David V, Audrezet MP, Ferec C and David A: Third case of paternal
isodisomy for chromosome 7 with cystic fibrosis: A new patient
presenting with normal growth. Am J Med Genet A. 143A:2696–2699.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nogueira C, Marques JS, Nesti C, Azevedo
L, Di Lullo M, Meschini MC, Orlacchio A, Santorelli FM and
Vilarinho L: Identification of maternal uniparental isodisomy of
chromosome 10 in a patient with mitochondrial DNA depletion
syndrome. Mol Genet Metab. 110:493–494. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Al-Jasmi F, Abdelhaleem M, Stockley T, Lee
KS and Clarke JT: Novel mutation of the perforin gene and maternal
uniparental disomy 10 in a patient with familial hemophagocytic
lymphohistiocytosis. J Pediatr Hematol Oncol. 30:621–624. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hahnemann JM, Nir M, Friberg M, Engel U
and Bugge M: Trisomy 10 mosaicism and maternal uniparental disomy
10 in a liveborn infant with severe congenital malformations. Am J
Med Genet A. 138A:150–154. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Schlegel M, Baumer A, Riegel M, Wiedemann
U and Schinzel A: Maternal uniparental isodisomy 10 and mosaicism
for an additional marker chromosome derived from the paternal
chromosome 10 in a fetus. Prenat Diagn. 22:418–421. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jones C, Booth C, Rita D, Jazmines L,
Spiro R, McCulloch B, McCaskill C and Shaffer LG: Identification of
a case of maternal uniparental disomy of chromosome 10 associated
with confined placental mosaicism. Prenat Diagn. 15:843–848. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kotzot D and Utermann G: Uniparental
disomy (UPD) other than 15: Phenotypes and bibliography updated. Am
J Med Genet A. 136:287–305. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Carmona-Rivera C, Simeonov DR, Cardillo
ND, Gahl WA and Cadilla CL: A divalent interaction between HPS1 and
HPS4 is required for the formation of the biogenesis of
lysosome-related organelle complex-3 (BLOC-3). Biochim Biophys
Acta. 1833:468–478. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Carmona-Rivera C, Hess RA, O'Brien K,
Golas G, Tsilou E, White JG, Gahl WA and Huizing M: Novel mutations
in the HPS1 gene among Puerto Rican patients. Clin Genet.
79:561–567. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Eisenberger T, Di Donato N, Baig SM,
Neuhaus C, Beyer A, Decker E, Mürbe D, Decker C, Bergmann C and
Bolz HJ: Targeted and genomewide NGS data disqualify mutations in
MYO1A, the ‘DFNA48 gene’, as a cause of deafness. Hum Mutat.
35:565–570. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cheng H, Khanna H, Oh EC, Hicks D, Mitton
KP and Swaroop A: Photoreceptor-specific nuclear receptor NR2E3
functions as a transcriptional activator in rod photoreceptors. Hum
Mol Genet. 13:1563–1575. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Coppieters F, Leroy BP, Beysen D,
Hellemans J, De Bosscher K, Haegeman G, Robberecht K, Wuyts W,
Coucke PJ and De Baere E: Recurrent mutation in the first zinc
finger of the orphan nuclear receptor NR2E3 causes autosomal
dominant retinitis pigmentosa. Am J Hum Genet. 81:147–157. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Milam AH, Rose L, Cideciyan AV, Barakat
MR, Tang WX, Gupta N, Aleman TS, Wright AF, Stone EM, Sheffield VC
and Jacobson SG: The nuclear receptor NR2E3 plays a role in human
retinal photoreceptor differentiation and degeneration. Proc Natl
Acad Sci USA. 99:473–478. 2002. View Article : Google Scholar : PubMed/NCBI
|