Introduction
X-linked inhibitor of apoptosis (XIAP) deficiency,
also known as X-linked lymphoproliferative syndrome type 2 (XLP2),
is a rare X-linked inherited primary immunodeficiency caused by
mutations in the XIAP (also known as BIRC4) gene, and
mainly presents with familial hemophagocytic lymphohistiocytosis
(HLH) phenotypes (1,2). The mutations of
XIAP/BIRC4 were initially demonstrated by Rigaud
et al to be associated with XLP phenotypes (1). Since then, limited case reports and
clinical information have been published, and <30 male patients
have been summarized. These patients typically presented with HLH
in infancy or early childhood (1–6). This
disorder is characterized by splenomegaly, hypogammaglobulinemia
and hemorrhagic colitis. Patients with XIAP deficiency are
susceptible to Epstein-Barr virus (EBV) and cytomegalovirus (CMV)
infection (1–5); however, to the best of our knowledge,
no cases of common variable imunodeficiency and lymphoma have been
previously observed in patients with XIAP deficiency (7). Clinical presentation and laboratory
tests facilitate the diagnosis of XIAP deficiency. Positive family
history, clinical presentation (including hemorrhagic colitis,
recurrent HLH, fever and hepatosplenomegaly), and laboratory tests
(such as hemophagocytosis in the bone marrow, elevated ferritin
levels (>500 ng/ml), cytopenia, hypertriglyceridemia and
hypofibrinogenemia) facilitate the diagnosis of XIAP deficiency
(8). However, genetic testing for
XIAP/BIRC4 genes is crucial for establishing a
definite diagnosis (5). Although
allogeneic hematopoietic stem cell transplantation (HSCT) is the
only strategy for radical treatment of this condition, there is
only a limited number of published studies concerning the outcomes
of HSCT in patients with XIAP deficiency (6).
The current study presents the case of an XIAP
deficiency patient resulting from a two-nucleotide deletion and
frameshift mutation. Subsequently, successful allogeneic HSCT was
performed in the patient, with good intermediate follow-up results
obtained.
Case report
A 5.8-year-old boy, who had been experiencing
abdominal distention, fever (temperature of >38.5°C) and
pancytopenia [hemoglobin level, 81 g/l (normal range, 110–146 g/l);
platelets, 80×109/l (normal range,
100–450×109/l); neutrophils, 0.78×109/l
(normal range, 0.88–5.7×109/l) with 14% atypical
lymphocytes] for 14 days was admitted to the Department of
Pediatric Hematology and Oncology, West China Second University
Hospital of Sichuan University (Chengdu, China) in February 2013.
Physical examination showed that the patient's spleen was 6 cm
below the left costal margin in the mid-clavicular line, with a
soft and sharp margin, whereas the liver was 7 cm below the right
costal margin in the mid-clavicular line. The patient also
presented with hypertriglyceridemia (fasting triglyceride level,
4.11 mmol/l; normal range, <2.83 mmol/l) and hypofibrinogenemia
(fibrinogen level, 125 mg/dl; normal range, 200–400 mg/dl).
Laboratory tests, including Wright's staining of bone marrow
smears, revealed hemophagocytosis in the bone marrow, with a
ferritin level of >16,500 ng/ml. EBV DNA and CMV DNA serological
tests, as well as blood culture, were negative. A bone marrow smear
indicated the presence of hemophagocytosis. Therefore, based on the
aforementioned findings, the patient was clinically diagnosed with
HLH.
Familial HLH associated genetic testing (including
the following genes: PRF1, UNC13D, STX11,
STXBP2, XIAP, SH2D1A, Rab27a,
AP3B1 and LYST) was performed on the patient
subsequent to obtaining written informed consent from his parents
and the approbation of the Ethics Committee of West China Second
University Hospital. Genomic DNA was extracted from peripheral
blood using a commercially available kit according to the
manufacturer's protocol (Tiangen Biotech, Co., Ltd., China). The
quality and quantity of the extracted DNA samples were determined
by UV spectrophotometry. Mutation of XIAP was detected by
polymerase chain reaction (PCR) analysis. Specific primers were
used for exon 3 of XIAP: Forward, 5′-ACTGAAAAGCAAGTTAATGG-3′ and
reverse, 5′-ACTGTGAATATCACATGAAG-3′. The total reaction volume of
25 µl contained 150 ng DNA, 12.5 µl PCR buffer (2X; Tiangen Biotech
Co., Ltd.) and 1 µl of specific primers (final PCR concentration
0.4 µM). Amplifications were performed using a programmable PCR
thermal cycler (Bio-Rad Laboratories, Inc., Hercules, CA, USA)
using the following thermal cycling parameters: 5 min at 94°C, 30
cycles of 30 sec at 94°C, 30 sec at 60°C, 1° min at 72°C, followed
by final extension for 7 min at 72°C. Each PCR included a negative
and positive control. The 309 bp PCR products were separated by
2.5% agarose gel electrophoresis and subsequently sequenced to
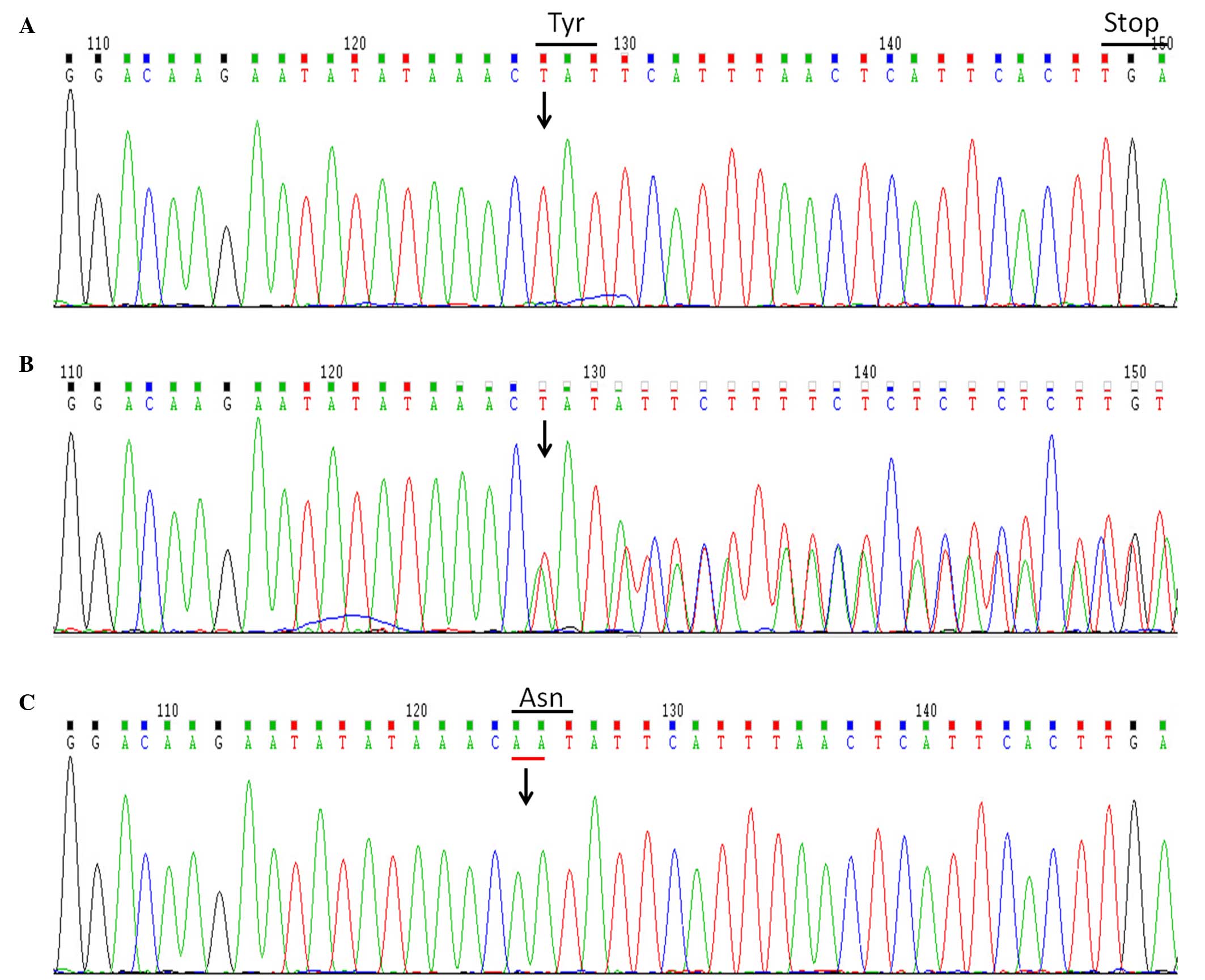
investigate the mutation of XIAP. The results indicated a
two-nucleotide deletion in BIRC4 gene (c.1021_1022delAA),
which resulted in a frameshift mutation and premature stop codon
(p.N341fsX348), while the XIAP protein lost 156 amino acids
(Fig. 1). Subsequently, BIRC4
genetic testing was performed on the patient's parents following
informed consent. The mother was found to be a heterozygote carrier
of the mutation; thus, the mutation was considered to be disease
causing, and the patient was confirmed with XIAP deficiency. Next,
the patient was treated with dexamethasone alone (initial dose of
10 mg/m2, which was subsequently tapered) for 3 months.
The treatment improved the general condition of the patient, and
resulted in a decreased spleen size and complete remission of HLH,
which was demonstrated by a reduction in ferritin levels to within
the normal range, and the recovery of blood cell count and
fibrinogen levels to within the normal range.
The patient remained in full remission of HLH at the
time of HSCT. A reduced intensity myeloablative conditioning (MAC)
regimen was performed at the West China Second University Hospital
in Octorber 2013. The transplantation procedures were performed
according to the institutional standard practices. The conditioning
regimens prior to HSCT consisted of 1 mg/kg busulfan (Otsuka
Pharmaceutical Co., Ltd., Tokushima, Japan) for 4 days (between
days −8 and −5), 60 mg/kg cyclophosphamide (Heng Rui Medicine Co.,
Ltd., Jiangsu, China) for 1 day (on day −3), 5 mg/kg antithymocyte
globulin (Fresenius Biotech GmbH, Gräfelfing, Germany) for 3 days
(between days −3 and −1) and 8 mg/kg etoposide (Heng Rui Medicine
Co., Ltd.) for 1 day (on day −4). Next, the patient received fully
matched unrelated peripheral blood stem cells based on typing 10
human leukocyte antigens (HLAs), including HLA-A, HLA-B, HLA-C,
HLA-DRB1, and HLA-DQB1, with 6.9×106/kg CD34+
cells and 4.7×108/kg mononuclear cells. Furthermore, he
received: Graft-verus-host disease (GVHD) prophylaxis, which
included 3–6 mg/kg oral cyclosporin A (North China Pharmaceutical
Group Corp., Shijiazhuang, China) for 6 months and 15 mg/kg oral
mycophenolate mofetil (Roche Pharmaceuticals Ltd., Shanghai, China)
for 3 months; other routine transplantation care, which included
antimicrobial prophylaxis with 100 mg/day oral mebendazole (Xian
Janssen Pharmaceutical Ltd., Xi'an, China) for 3 days, 50 mg/day
oral fluconazole (Pfizer, Inc., New York, NY, USA) for ~2 months, 5
mg/kg twice daily ganciclovir (North China Pharmaceutical Group
Corp.) by intravenous infusion for 10 days, 75 mg/kg twice daily
mezlocillin sodium and sulbactam sodium (Hainan General Sanyang
Pharmaceutical Co., Ltd., Haikou, China); hepatic venoocclusive
disease (VOD) prophylaxis with alprostadil, which included 10
µg/day prostaglandin E1 (Beijing Tide Pharmaceutical Co., Ltd.,
Beijing, China) by intravenous infusion for 35 days; intravenous
immunoglobulin replacement (400 mg/kg once a week; Rongsheng
Pharmaceutical Co., Ltd., Chengdu, China); and fluid and nutrition
supplementation per institutional standard practices. The patient
suffered from drug-associated enteritis while receiving the
preparative regimen. In addition, he developed engraftment syndrome
at day +6 after HSCT, CMV viremia at day +26, autoimmune hemolytic
anemia at day +35, hemorrhagic cystitis at day +42; however, no
acute GVHD, VOD, pulmonary hemorrhage, bacteremia, fungal infection
and cardiac toxicity were reported. The patient developed complete
donor chimerism (>95% host-derived cells in the whole blood) at
day +20. The recipient's blood type converted from group AB to the
donor's blood type, group B. The patient received cyclosporin A,
mycophenolate mofetil, and methylprednisolone for ~6, 3 and 6
months respectively for GVHD prophylaxis following HSCT. At an
intermediate follow-up performed 528 days after HSCT, the patient
was alive and remained free of disease. At the latest follow-up
performed in April 2016, the patient remained free of HLH,
exhibited normal cellular immunity and had successfully withdrawn
from all therapeutic agents for GVHD, VOD and antimicrobial
prophylaxis.
Discussion
XIAP deficiency, also known as XLP2, is a rare
X-linked inherited primary immunodeficiency resulting from
XIAP/BIRC4 mutations (1), and is mainly associated with familial
HLH phenotypes (8). The present
study reported the case of a patient presenting with typical
clinical features of HLH, including fever, hepatosplenomegaly,
pancytopenia, hypertriglyceridemia, coagulopathy with
hypofibrinogenemia, hemophagocytosis in the bone marrow and
elevated levels of ferritin. Although the current patient presented
negative results in EBV serological tests, EBV infection remains
one of the most frequent pathogens detected in HLH patients. Other
symptoms of XLP, such as hemorrhagic colitis, have been reported in
patients with XIAP deficiency, but malignant B-cell lymphoma has
not been reported (1–3,5). In
addition, recurrent splenomegaly frequently occurs in XIAP
deficiency, and it may occur even in the absence of systemic HLH
(3,8).
Gene testing is essential for the diagnosis of XIAP
deficiency, particularly in patients who present an atypical
phenotype or have no positive family history. XIAP is
located in Xq25, adjacent to the SH2D1A gene, and comprises
6 exons (9). XIAP protein, as an
anti-apoptotic molecule, consists of 497 amino acids and contains
three baculovirus IAP repeat domains and one RING domain; these
bind to caspases 3, 7, and 9 together with flanking residues,
thereby inhibiting the proteolytic activity of caspases 3, 7, and 9
(10). To date, at least two small
deletions and three large deletions of XIAP have been
identified, resulting in frameshifts and premature stop codons
(6,11). In particular, a deletion of cytidine
291 (291delC) resulted in a frameshift leading to a stop codon at
position 387 (G99K/X129) (1). A
deletion of 2,606 nucleotides encompassing exon 2 (1), and deletions of exons 1–5 or exon 6
resulted the expression of XIAP decreased significantly (2). More specifically, 2 brothers from Japan
have been reported to have the same two-nucleotide deletion
(c.1021_1022delAA) in exon 3 as that reported in the present case,
but their mother was not a carrier of an XIAP mutation
(6). In the present study, a direct
sequencing method was used to identify not only mutations in the
Chinese boy, but also heterozygous mutation in his mother. This
mutation resulted in a frameshift and premature termination
(p.N341fsX348). In addition, other XIAP mutations have been
reported in previous studies, including exonic nonsense mutations,
missense mutations, large deletions and gross deletion exons
(6,11).
Although allogeneic HSCT is the only strategy for
the complete treatment of XIAP deficiency, limited studies have
been published concerning the outcomes of HSCT in patients with
this syndrome. As reported in an international survey (11), 19 patients with XIAP deficiency
received HSCT. Among them, 8 patients received MAC regimens, while
11 patients received reduced intensity conditioning (RIC) regimens
(consisting of fludarabine, alemtuzumab and melphalan) (11). The comparison between the two groups
revealed that the conditioning regimens affected the HSCT outcome.
There was a high incidence of conditioning-associated toxicity
among MAC regimen-treated patients, with a higher mortality
observed in these patients compared with patients receiving the RIC
regimens (11). The main causes of
mortality included pulmonary hemorrhage, VOD and multiple organ
failure (MOF). Therefore, it is possible that the loss of XIAP and
its antiapoptotic functions contributes to the high incidence of
toxicity observed in patients receiving the MAC regimen (11).
Although it is suggested that RIC regimens should be
administered to patients with XIAP deficiency, this regimen was not
selected in the present study, due to inability to obtain
alemtuzumab (a CD52 monoclonal antibody) in our hospital. Given the
lack of alemtuzumab, the current patient underwent HSCT with
reduced intensity MAC regimen (including etoposide, which may
contribute to controlling HLH) and still presented good
intermediate follow-up results. Furthermore, the results of the
current study indicated that the full-matched HLA donor (12), the effective control of HLH activity
prior to HSCT (13) and the
successful prevention of implications (such as VOD, pulmonary
hemorrhage and infections) may also contribute to the success of
HSCT in patients with XIAP deficiency in developing countries,
where alemtuzumab cannot be obtained.
In conclusion, the current study detected a two-base
deletion mutation of XIAP/BIRC4 in a Chinese family,
and successfully performed allogeneic HSCT in the patient.
Identifying XIAP mutations is important for the diagnosis of
affected families, and more evidence is required prior to making
transplantation decisions. Thus, conditioning regimens should be
administered with caution in patients with an available matched
stem cell donor. In addition, where possible, efforts should be
made to ensure HLH remission prior to HSCT and successful
prevention of implications following HSCT in these patients.
Acknowledgements
The present study was supported by the Chengdu
Scientific and Technologic Bureau (project no. 11DXYB086JH-027),
the Doctoral Scientific Fund Project of the Ministry of Education
of China (project no. 20120181120050), and the University Program
for Changjiang Scholars and Innovative-Research Team (project no.
IRT0935).
References
|
1
|
Rigaud S, Fondanèche MC, Lambert N,
Pasquier B, Mateo V, Soulas P, Galicier L, Le Deist F, Rieux-Laucat
F, Revy P, et al: XIAP deficiency in humans causes an X-linked
lymphoproliferative syndrome. Nature. 444:110–114. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marsh RA, Madden L, Kitchen BJ, Mody R,
McClimon B, Jordan MB, Bleesing JJ, Zhang K and Filipovich AH: XIAP
deficiency: A unique primary immunodeficiency best classified as
X-linked familial hemophagocytic lymphohistiocytosis and not as
X-linked lymphoproliferative disease. Blood. 116:1079–1082. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Pachlopnik Schmid J, Canioni D, Moshous D,
Touzot F, Mahlaoui N, Hauck F, Kanegane H, Lopez-Granados E,
Mejstrikova E, Pellier I, et al: Clinical similarities and
differences of patients with X-linked lymphoproliferative syndrome
type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP
deficiency). Blood. 117:1522–1529. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhao M, Kanegane H, Ouchi K, Imamura T,
Latour S and Miyawaki T: A novel XIAP mutation in a Japanese boy
with recurrent pancytopenia and splenomegaly. Haematologica.
95:688–689. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Filipovich AH, Zhang K, Snow AL and Marsh
RA: X-linked lymphoproliferative syndromes: Brothers or distant
cousins? Blood. 116:3398–3408. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang X, Kanegane H, Nishida N, Imamura T,
Hamamoto K, Miyashita R, Imai K, Nonoyama S, Sanayama K, Yamaide A,
et al: Clinical and genetic characteristics of XIAP deficiency in
Japan. J Clin Immunol. 32:411–420. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Salzer U, Hagena T, Webster DB and
Grimbacher B: Sequence analysis of BIRC4/XIAP in male patients with
common variable immunodeficiency. Int Arch Allergy Immunol.
147:147–151. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang X, Miyawaki T and Kanegane H: SAP and
XIAP deficiency in hemophagocytic lymphohistiocytosis. Pediatr Int.
54:447–454. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liston P, Roy N, Tamai K, Lefebvre C,
Baird S, Cherton-Horvat G, Farahani R, McLean M, Ikeda JE,
MacKenzie A and Korneluk RG: Suppression of apoptosis in mammalian
cells by NAIP and a related family of IAP genes. Nature.
379:349–353. 1996. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Galbán S and Duckett CS: XIAP as a
ubiquitin ligase in cellular signaling. Cell Death Differ.
17:54–60. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Marsh RA, Rao K, Satwani P, Lehmberg K,
Müller I, Li D, Kim MO, Fischer A, Latour S, Sedlacek P, et al:
Allogeneic hematopoietic cell transplantation for XIAP deficiency:
An international survey reveals poor outcomes. Blood. 121:877–883.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Shaw BE, Arguello R, Garcia-Sepulveda CA
and Madrigal JA: The impact of HLA genotyping on survival following
unrelated donor haematopoietic stem cell transplantation. Br J
Haematol. 150:251–258. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Baker KS, Filipovich AH, Gross TG,
Grossman WJ, Hale GA, Hayashi RJ, Kamani NR, Kurian S, Kapoor N,
Ringdén O and Eapen M: Unrelated donor hematopoietic cell
transplantation for hemophagocytic lymphohistiocytosis. Bone Marrow
Transplant. 42:175–180. 2008. View Article : Google Scholar : PubMed/NCBI
|